2.1. In-Silico Mutagenesis
Residues 71 and 204 of the NBD protein mutated from lysine (basic polar) to leucine (non-polar) and threonine (polar) to valine (non-polar) respectively. These mutations do not change the overall electronic nature of the side chains. The classification of amino acid chemical properties was based on the research done by Biro
et al. (2003) [
11]. Proteins that contain changes in residues may have some effects on the overall structure or function of the protein. Therefore, all mutants were chosen for the subsequent
in-silico based modeling.
The conserved LYS71 is a catalytically important residue that affects ATP hydrolysis [
12]. The proposed mechanism of ATP hydrolysis suggested that the role of LYS71 in accepting a proton from the hydroxide ion or water molecule involved is in-line with a nucleophilic attack [
12–
15]. The inorganic phosphate group (Pi) is coordinated by a salt bridge with LYS71, hydrogen bonds to THR13 and THR204 and interacts directly with a calcium ion. A water molecule mediates additional interactions with the protein’s main chain at positions 202, 203 and 204. The Pi-binding site is on the protein face opposite the highly conserved GLY32 loop that has been implicated in the binding of nucleotide release factor (GrpE) to the ATPase domain of Hsp40 (DNAK) [
16]. Therefore, there are potential channels for Pi exit to the protein surface. However, release of the inorganic phosphate group has been implicated in the conformational transition of Hsp70 molecular chaperone [
17]. Phospho-threonine was postulated as an intermediate of ATP hydrolysis. In addition, ATPase activity of Hsp70 initiates viral DNA replication. This has been demonstrated for bacterial DNAJ which stimulates ATPase activity of Hsp70 to start DNA replication of SV40 [
18]. Thus, mutational study of these two residues which are important for ATPase activity was carried out.
2.2. Physiochemical Characterization
The computed pI value for NBD, K71L and T204V (pI < 7) indicated their acidic character. The extinction coefficient was calculated as 20,525–20,625 M
−1·cm
−1 based on the molar extinction coefficient of TYR, TRP and CYS residues. This measure indicates how much light is absorbed by a protein at a particular wave length. On the basis of instability index, Expasy’s ProtParam [
19] classified NBD, K71L and T204V proteins as stable (instability index < 40). Instability index relies upon the occurrence of certain dipeptides along the length of the enzyme. The aliphatic index is defined as the relative volume of a protein that is occupied by an aliphatic side chain. An increase in the aliphatic index increases the thermo stability of globular proteins. The very high aliphatic index of all NBD and mutant proteins infers that these proteins may be stable for a wide range of temperatures. The very low grand average of hydropathicity (GRAVY) index (a negative value GRAVY) of NBD and all the mutant proteins infers that these proteins could result in a better interaction with water (hydrophilic in nature) (
Table 1). The secondary structure indicates whether a given amino acid lies in a helix, strand or coil. The results from the SOPMA server [
20] revealed that alpha helix dominated among secondary structure elements followed by random coils, extended strand and beta turns (
Table 2).
2.3. Model Simulation and Evaluation
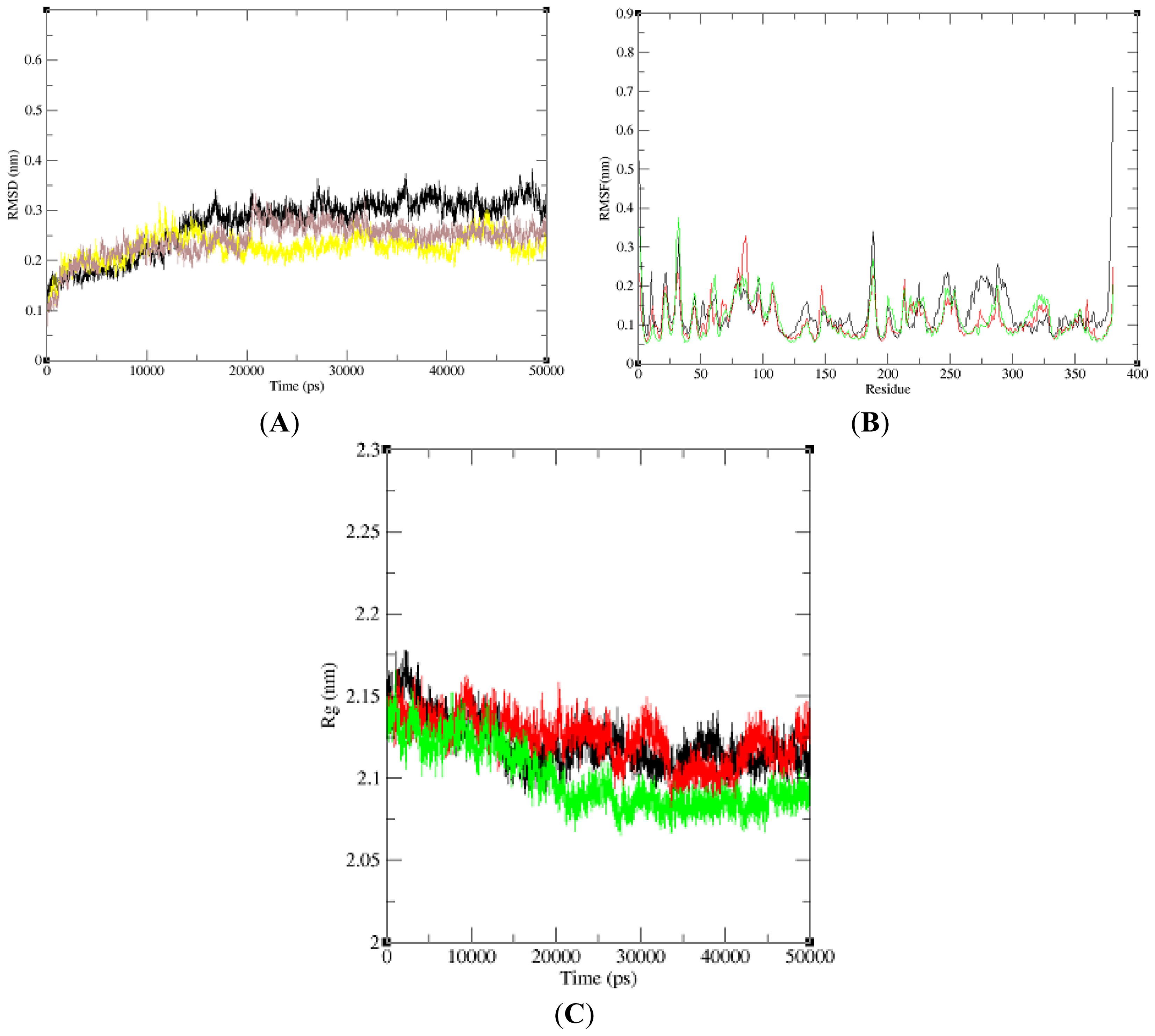
In this paper, we performed three 50 ns (50,000 ps) MD simulations of NBD, K71L and T204V to explore and compare the protein internal dynamics. To analyze the global behavior of the studied systems, the root mean square deviations (RMSDs) of the protein backbone with respect to the initial conformation were plotted
versus simulation time (
Figure 1A). RMSD played an important role in protein stability. The RMSD of the NBD model increased slightly and stabilized at 16,000 ps. K71L RMSD value increased gradually and stabilized at 5000 ps. It decreased slightly and then increased until it stabilized at 12,000 ps. At 40,000 ps, the NBD increased until 45,000 ps and dropped. In T204V’s simulation, the RMSD increased and stabilized at 5000 ps. T204V then increased slightly until it attained a constant level at 21,000 ps; and decreased at 31,000 ps until it stabilized at 35,000 ps. From the RMSD analysis, the T204V structure seemed more stable compared to the K71L and NBD structures due to lower fluctuation of RMSD values from 25,000 to 50,000 ps compared to those of the NBD and K71L. The root mean square fluctuation (RMSF) of the Cα atom of NBD, K71L and T204V as a function of residue number was plotted to evaluate the average fluctuation of each residue during the simulation (
Figure 1B). All the residues in mutant models fluctuated around the NBD value which was approximately 0.2 nm. The RMSF of the Cα value of T204V exhibited a higher fluctuation than NBD and K71L at residue 56 (0.88 nm). In addition, NBD and mutant models stabilized at a gyration distance of about 2.11 nm at 20,000 ps (
Figure 1C).
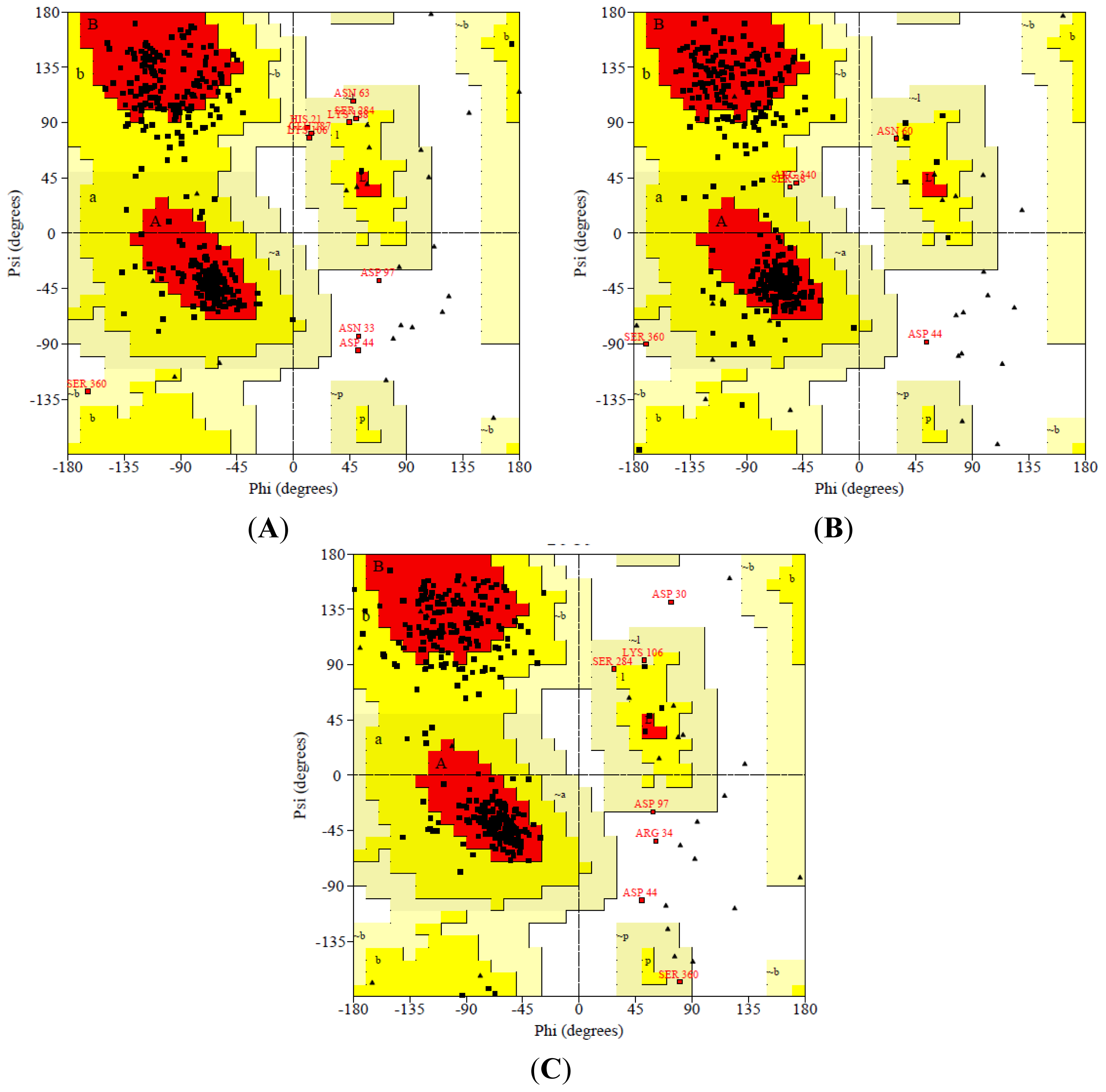
After 50,000 ps MD simulation, the geometry of three dimensional protein models was carried out with Ramachandran’s plot calculations using PROCHECK [
21]. In the current study, the stereo-chemical evaluation of backbone psi and Phi dihedral angles of the NBD revealed that 81.7%, 15.4%, 2.1% and 0.9% of residues were falling within the most favored regions, additionally allowed regions, generously allowed regions and disallowed regions (ASN33, ASP44 and ASP97) respectively. In general, a score close to 100% implies good stereo-chemical quality of the model [
22]. Therefore, these PROCHECK results suggest that the predicted model was of good quality (
Figure 2).
The results also show that residues of the NBD model in the most favorable region were more than 80% except K71L and T204V mutants which scored slightly less than 80% for the most favored region (
Table 3). However, the stereo chemical quality of the predicted models were found to be satisfactory and a low percentage of residues having phi/psi angles in the outlier region. The analysis explored that no bad contacts and no bad scores for main-chain or side-chain parameters. In spite of that, the overall G-factor values of NBD and mutants were slightly out of range because the values were lower than −0.5 but higher than −1.0. The acceptable values of the G-factor in PROCHECK are between 0 and −0.5, with the best quality models displaying values close to zero [
23]. The quality of the protein structures were checked using ProQ [
24]. The results show that the predicted LG score (>4: extremely good model) and predicted MaxSub score (>0.5 good model) for all protein models were in an acceptable range of a good model (
Table 3).
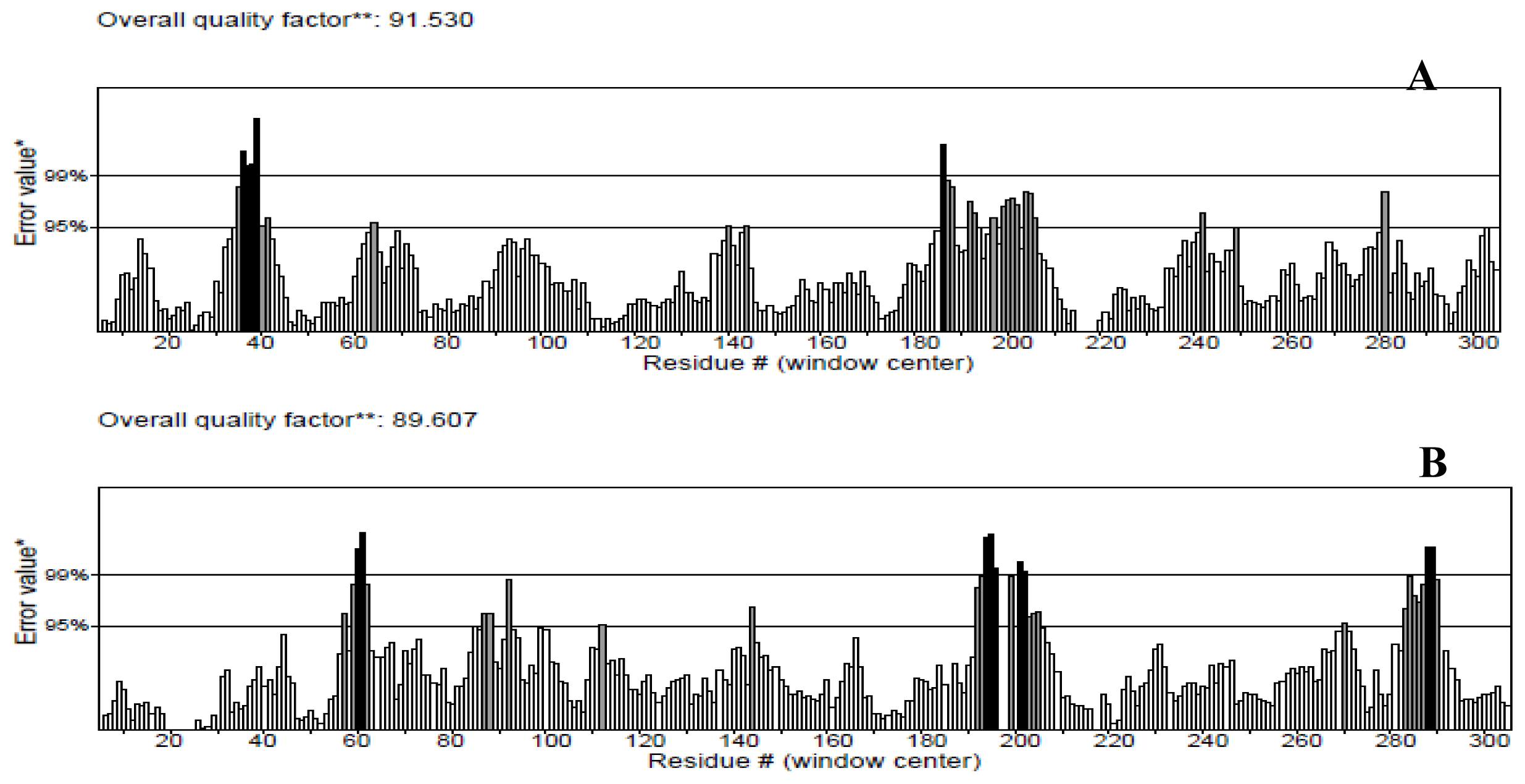
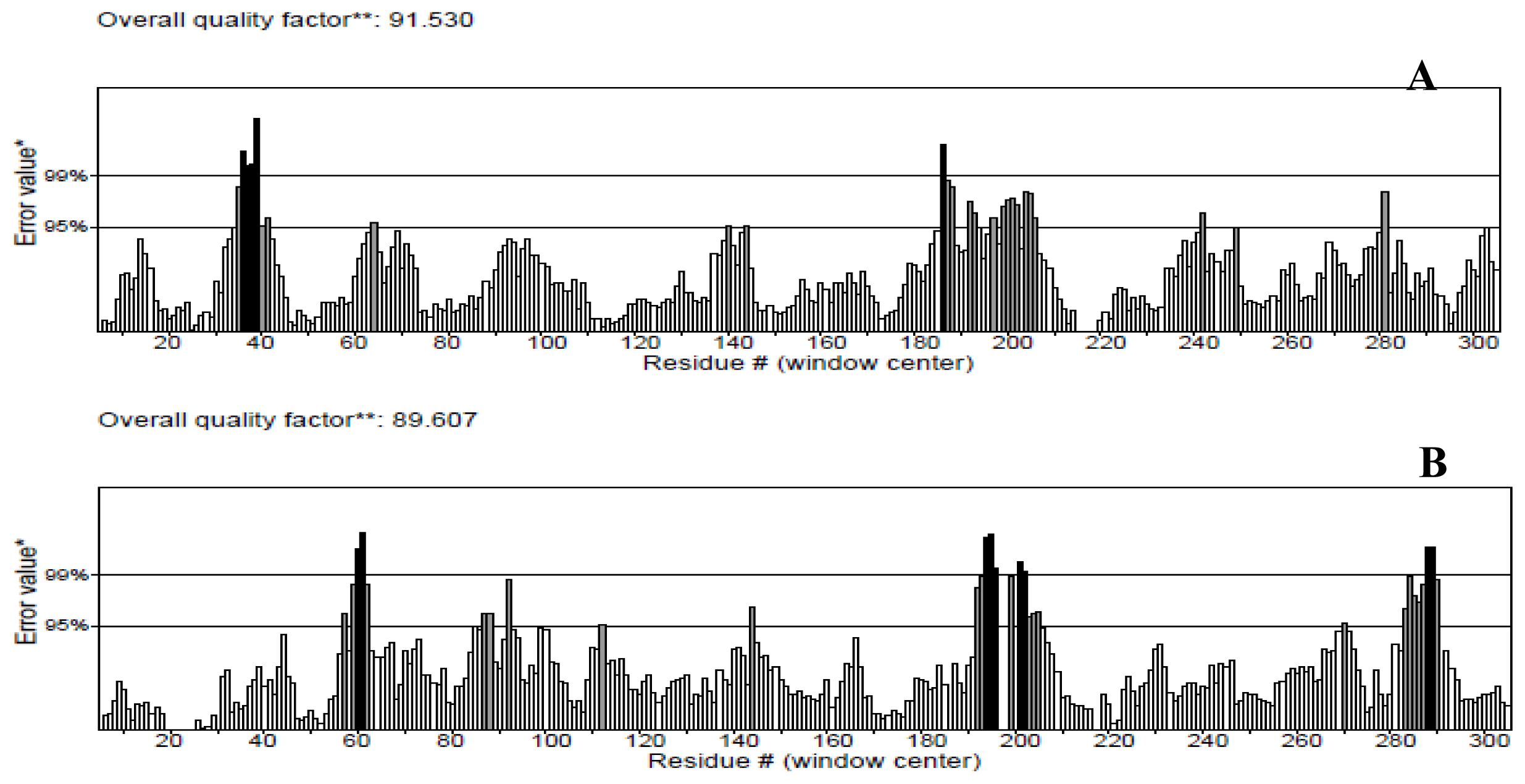
The protein structures were also validated by other structure verification servers such as Verify 3D and ERRAT to check the quality of the models. ERRAT works by analyzing the statistics of non-bonded interactions between different atom types, with higher scores indicating higher quality [
25]. The ERRAT score for T204V was the highest (94.536%). Nevertheless, this score was close with score values of the NBD (91.530%) and K71L (89.607%) proteins (
Figure 3). None of the residues were above the 99% cut off of error-value. However, the generally accepted range is >50 for a high quality model [
21]. Thus, this analysis revealed that the backbone conformation and non-bonded interactions of the NBD and mutant models fit well within the range of a high quality model.
In the Verify 3D analysis, it was found that none of the amino acids had a negative score (
Figure 4). Therefore, the predicted models were compatible with its amino acid sequence. It should be noted that compatibility scores above zero correspond to an acceptable side chain environment [
26].
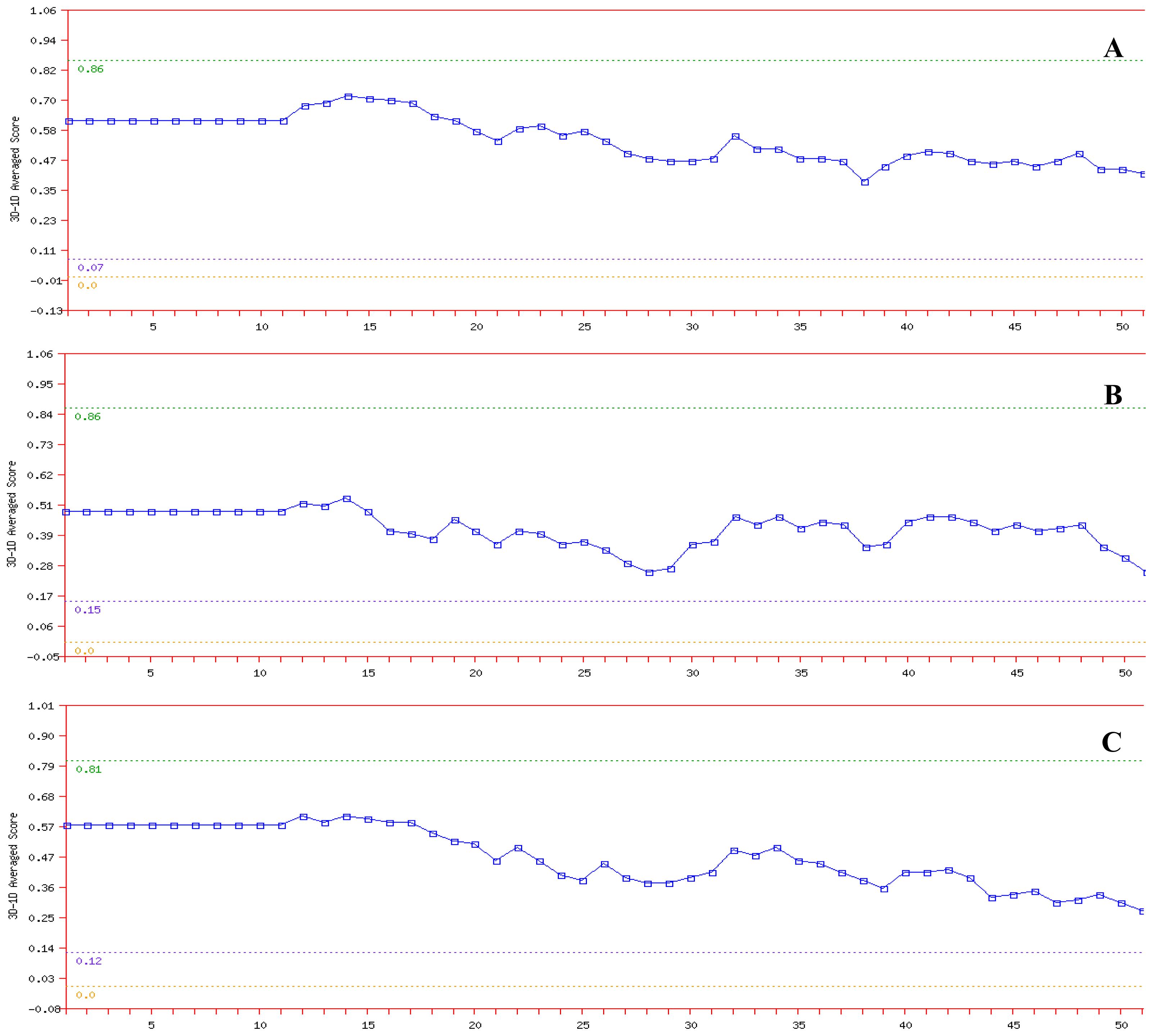
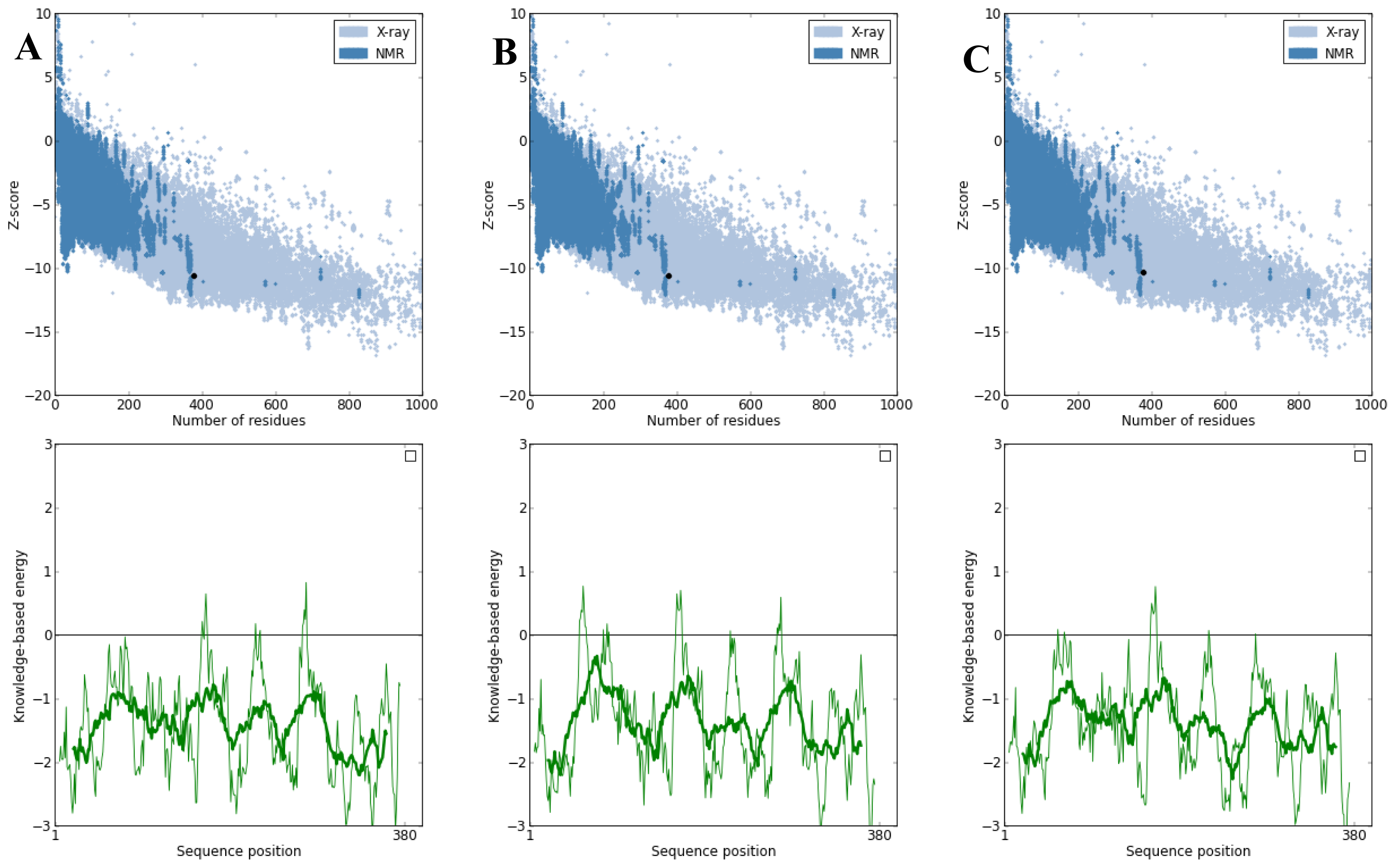
PROSA was used to check three dimensional models of proteins under this study for potential errors [
27]. The program displays two characteristics of the input structure: its Z-score and a plot of its residue energies. The Z-score indicates overall model quality and measures the deviation of the total energy of the structure with respect to an energy distribution derived from random conformations. Analysis of the K71L and T204V mutants with PROSA showed a Z-Score of −10.38 and −10.63 respectively, indicating no significant deviation from typical native structures of a similar size as the target protein’s Z-Score which was −10.64 (
Figure 5). The quality of the protein folds of NBD, K71L and T204V were also evaluated in terms of energy function of amino acid residues. In general, folding energy of the protein showed minimum value as this accounts for the stability and nativity of the molecules. The energy profiles of mutant models in comparison to that of the X-ray structure of the NBD is presented in
Figure 5. The energy profile of the K71L and T204V mutant models were consistent with a reliable conformation based on its similarity to that of the NBD.
2.5. Molecular Docking
The negative and low value of Δ
Gbind (−9.09 kcal/mol) indicated strong bonds between T204V and the E1A32 kDa motif, and demonstrated that the protein was in a favorable conformation. Sum of intermolecular energy and torsion energy was the binding energy. Furthermore, the total intermolecular energy of T204V (−12.97 kcal/mol) was found to be lower than the NBD and K71L (−11.93 and −10.64 kcal/mol), stating that the mutant model (T204V) had a better binding affinity than the NBD and K71L in this analysis (
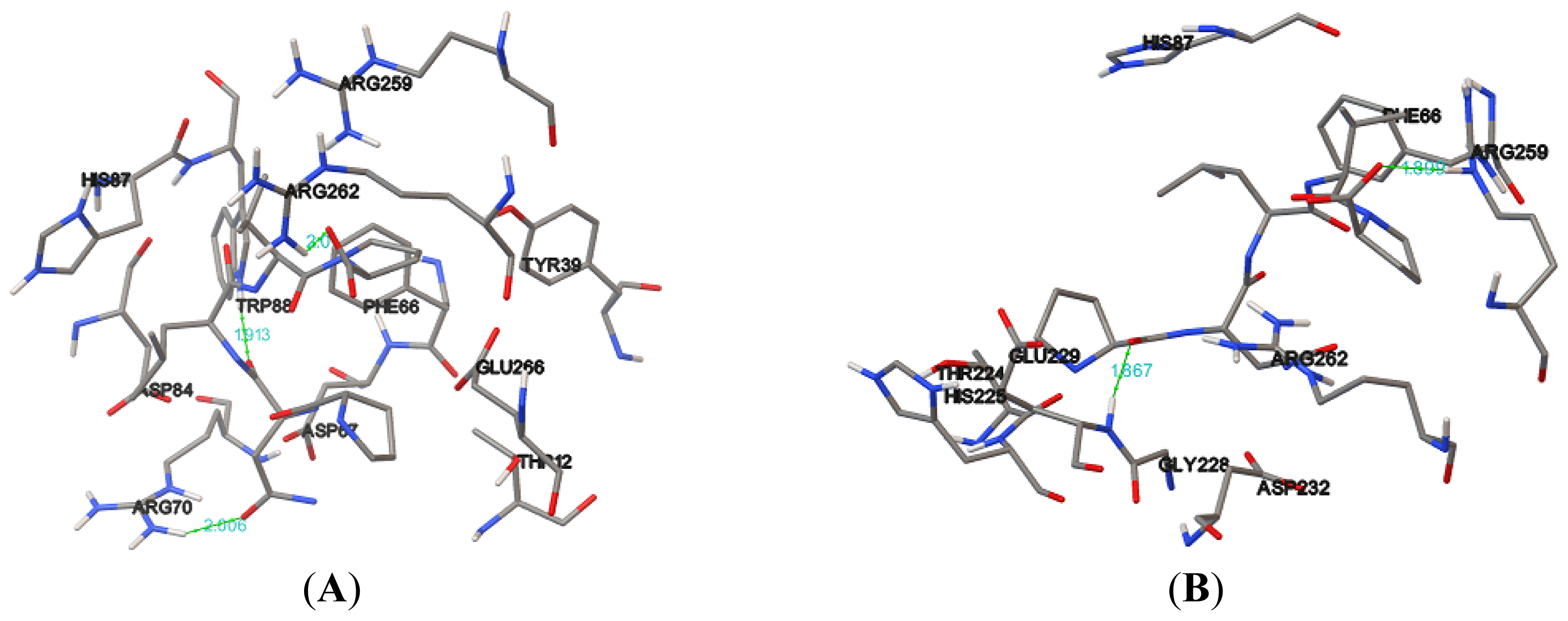
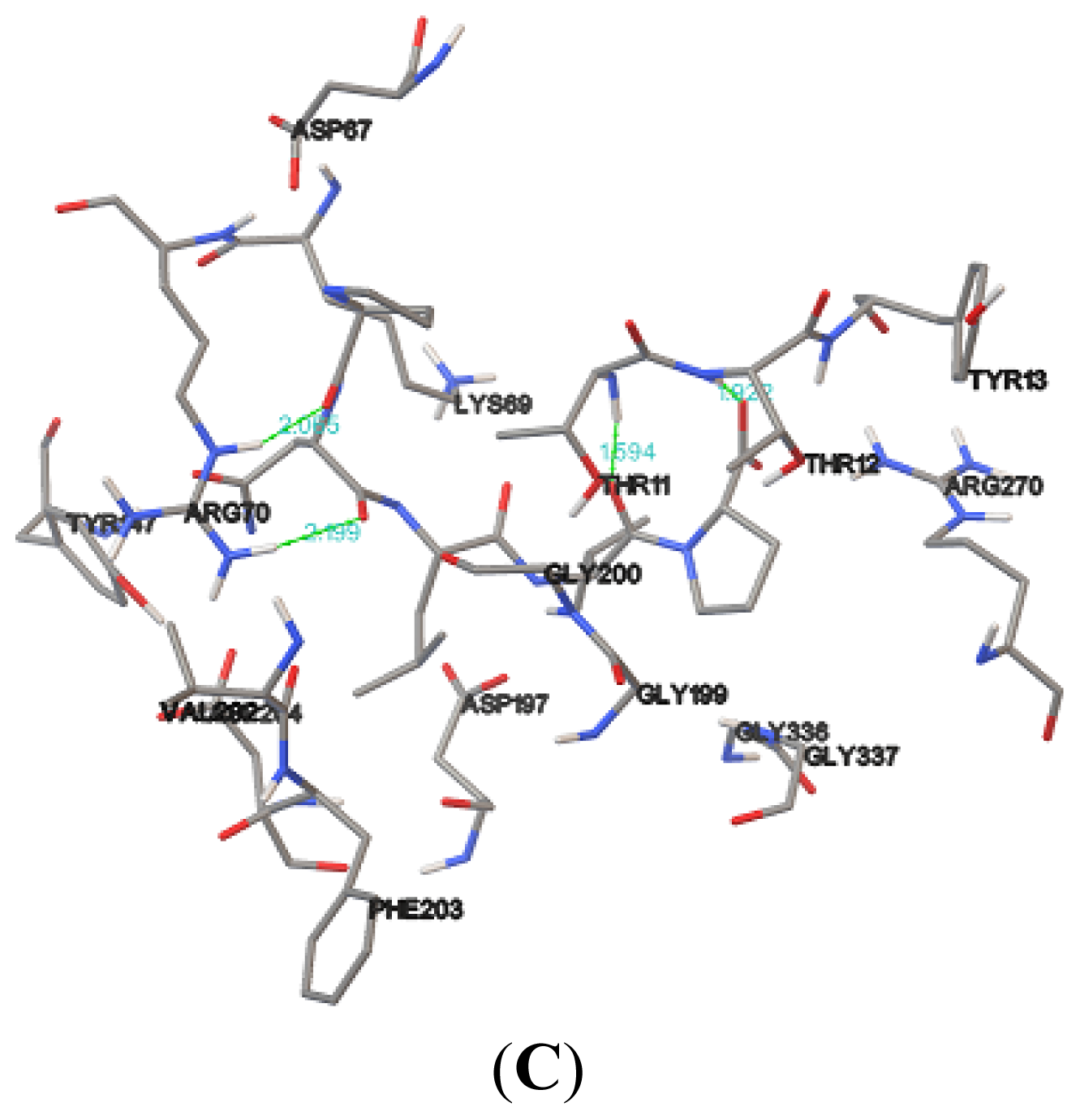
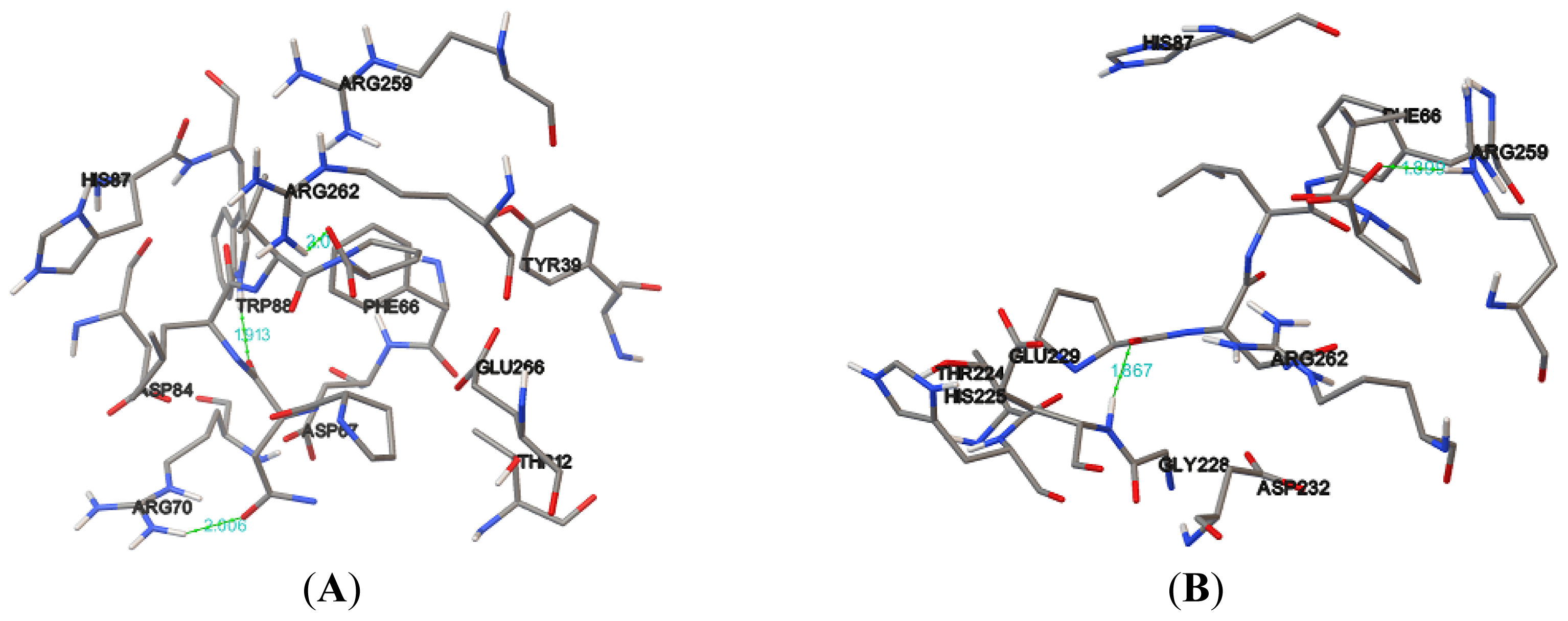
Table 5). Hydrogen bonds formed between the compound and the protein usually contribute to the stability of the protein-ligand complexes; a large number of hydrogen bonds form more stable complexes [
28,
29]. The results show that the NBD, K71L and T204V were stabilized by three, two and four hydrogen bonds with E1A32 kDa motif, respectively (
Table 6 and
Figure 7). The active residues of the T204V mutant (THR13, THR14 and ARG72) were also involved in the formation of hydrogen bonds, suggesting the protein (T204V) forms a more stable complex than the NBD and K71L (
Table 6). Therefore, in the study of protein-ligand binding mechanism, it was revealed that the novel T204V mutant has stronger interaction energy with the E1A32 kDa motif than other protein models.
2.6. Model Simulation and Evaluation of Protein-Ligand Complex
In this study, three 50 ns (50,000 ps) molecular dynamics simulation were run with NBD, K71L and T204V-E1A32 kDa motif complexes.
Figure 8A shows the RMSD value of the protein-ligand complex structures over the simulation time. The RMSD value of 0.15 nm for NBD, K71L and T204V-E1A32 kDa motif complex structures were less deviated until 50 ps from their starting structure. NBD-E1A32 kDa motif complex reached stabilization at 35,000 ps (0.17 nm) while the RMSD attained a stable value of 0.20 nm at 35,000 ps for the K71L-E1A32 kDa complex. The T204V-E1A32 kDa complex attained 0.15 nm of RMSD backbone at 35,000 ps during simulation. From these results, it can be concluded that T204V-E1A32 kDa complex deviated less compared to the NBD and K71L-E1A32 kDa complexes indicating that the T204V-E1A32 kDa complex was more stable than the other two complex structures. The RMSF values of carbon alpha for each amino acid residue were obtained from the trajectory data of NBD, K71L and T204V-E1A32 kDa motif complexes shown in
Figure 8B. In RMSF analysis, all the residues in the protein model fluctuated between 0.05 and 0.20 nm throughout the simulation period. The NBD-E1A32 kDa motif complex exhibited a high fluctuation up to 0.37 nm at residue 86.
History independent hydrogen bond autocorrelation function was calculated between the protein and the ligand (
Figure 8C). The independent autocorrelation function measured the probability of a hydrogen bond present (broken or reformed between a time interval allowed) at time (
t), given that it was present at time zero. The hydrogen bond analysis revealed that the life-time of a hydrogen bond between protein and ligand is highest for T204V followed by K71L and NBD; the results imply that the association between the protein and the ligand T204V is stronger.
Salt bridges formed between the amino acid side chains at positive ions in NBD, K71L and T204V, and negative ions in the E1A32 kDa motif. The salt bridge is important for stabilizing the protein’s structure. The presence of salt-bridges was a proof of the close proximity in the structure [
30]. Salt-bridges occurring between the NBD, K71L and T204V-E1A32 kDa motif were calculated (
Figure 8D). All three complexes attained the stable distance of 2.70, 2.60 and 2.40 nm in the entire simulation period. This suggested that the salt bridge with the shortest distance stabilizes the protein the most.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}