Signaling Pathways in Cartilage Repair

Abstract
:1. Introduction
2. Transforming Growth Factor-β (TGF-β), Bone Morphogenetic Protein (BMP), Insulin Growth Factor (IGF) and Fibroblast Growth Factor (FGF) Pathways
3. Hypoxia-Inducible Factor (HIF) Pathway
4. Wnt/β-Catenin Pathway
5. Nuclear Factor-Kappa B (NF-κB) Pathway
6. Mitogen-Activated Protein Kinase (MAPK) Pathway
7. Hedgehog (Hh)/Smoothened (Smo) Pathway
8. Signaling Cross-Talk and Conclusions
Acknowledgments
Conflicts of Interest
References
- Goldring, M.B.; Marcu, K.B. Cartilage homeostasis in health and rheumatic diseases. Arthritis Res. Ther 2009, 11. [Google Scholar] [CrossRef]
- Poole, A.R.; Kojima, T.; Yasuda, T.; Mwale, F.; Kobayashi, M.; Laverty, S. Composition and structure of articular cartilage: A template for tissue repair. Clin. Orthop. Relat. Res 2001, 391, S26–S33. [Google Scholar]
- Wang, M.; Shen, J.; Jin, H.; Im, H.J.; Sandy, J.; Chen, D. Recent progress in understanding molecular mechanisms of cartilage degeneration during osteoarthritis. Ann. N. Y. Acad. Sci 2011, 1240, 61–69. [Google Scholar]
- Goldring, M.B. Osteoarthritis and cartilage: The role of cytokines. Curr. Rheumatol. Rep 2000, 2, 459–465. [Google Scholar]
- Melchiorri, C.; Meliconi, R.; Frizziero, L.; Silvestri, T.; Pulsatelli, L.; Mazzetti, I.; Borzi, R.M.; Uguccioni, M.; Facchini, A. Enhanced and coordinated in vivo expression of inflammatory cytokines and nitric oxide synthase by chondrocytes from patients with osteoarthritis. Arthritis Rheum 1998, 41, 2165–2174. [Google Scholar]
- Kapoor, M.; Martel-Pelletier, J.; Lajeunesse, D.; Pelletier, J.P.; Fahmi, H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol 2011, 7, 33–42. [Google Scholar]
- Borzi, R.M.; Mazzetti, I.; Marcu, K.B.; Facchini, A. Chemokines in cartilage degradation. Clin. Orthop. Relat. Res 2004, S53–S61. [Google Scholar]
- Wei, L.; Kanbe, K.; Lee, M.; Wei, X.; Pei, M.; Sun, X.; Terek, R.; Chen, Q. Stimulation of chondrocyte hypertrophy by chemokine stromal cell-derived factor 1 in the chondro-osseous junction during endochondral bone formation. Dev. Biol 2010, 341, 236–245. [Google Scholar]
- Rasheed, Z.; Akhtar, N.; Haqqi, T.M. Advanced glycation end products induce the expression of interleukin-6 and interleukin-8 by receptor for advanced glycation end product-mediated activation of mitogen-activated protein kinases and nuclear factor-kappaB in human osteoarthritis chondrocytes. Rheumatology 2011, 50, 838–851. [Google Scholar]
- Wenke, A.K.; Niebler, S.; Grassel, S.; Bosserhoff, A.K. The transcription factor AP-2varepsilon regulates CXCL1 during cartilage development and in osteoarthritis. Osteoarthr. Cartil 2011, 19, 206–212. [Google Scholar]
- Chao, P.Z.; Hsieh, M.S.; Cheng, C.W.; Lin, Y.F.; Chen, C.H. Regulation of MMP-3 expression and secretion by the chemokine eotaxin-1 in human chondrocytes. J. Biomed. Sci 2011, 18. [Google Scholar] [CrossRef]
- Fosang, A.J.; Beier, F. Emerging Frontiers in cartilage and chondrocyte biology. Best Pract. Res. Clin. Rheumatol 2011, 25, 751–766. [Google Scholar]
- Van der Kraan, P.M.; van den Berg, W.B. Chondrocyte hypertrophy and osteoarthritis: Role in initiation and progression of cartilage degeneration? Osteoarthr. Cartil 2012, 20, 223–232. [Google Scholar]
- Fuerst, M.; Bertrand, J.; Lammers, L.; Dreier, R.; Echtermeyer, F.; Nitschke, Y.; Rutsch, F.; Schafer, F.K.; Niggemeyer, O.; Steinhagen, J.; et al. Calcification of articular cartilage in human osteoarthritis. Arthritis Rheum 2009, 60, 2694–2703. [Google Scholar]
- Little, C.B.; Barai, A.; Burkhardt, D.; Smith, S.M.; Fosang, A.J.; Werb, Z.; Shah, M.; Thompson, E.W. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum 2009, 60, 3723–3733. [Google Scholar]
- Von der Mark, K.; Kirsch, T.; Nerlich, A.; Kuss, A.; Weseloh, G.; Gluckert, K.; Stoss, H. Type X collagen synthesis in human osteoarthritic cartilage. Indication of chondrocyte hypertrophy. Arthritis Rheum 1992, 35, 806–811. [Google Scholar]
- Gelse, K.; Ekici, A.B.; Cipa, F.; Swoboda, B.; Carl, H.D.; Olk, A.; Hennig, F.F.; Klinger, P. Molecular differentiation between osteophytic and articular cartilage—Clues for a transient and permanent chondrocyte phenotype. Osteoarthr. Cartil 2012, 20, 162–171. [Google Scholar]
- Mueller, M.B.; Tuan, R.S. Functional characterization of hypertrophy in chondrogenesis of human mesenchymal stem cells. Arthritis Rheum 2008, 58, 1377–1388. [Google Scholar]
- Van der Kraan, P.M.; Blaney Davidson, E.N.; van den Berg, W.B. A role for age-related changes in TGFβ signaling in aberrant chondrocyte differentiation and osteoarthritis. Arthritis Res. Ther 2010, 12, 201. [Google Scholar]
- Leijten, J.C.; Emons, J.; Sticht, C.; van Gool, S.; Decker, E.; Uitterlinden, A.; Rappold, G.; Hofman, A.; Rivadeneira, F.; Scherjon, S.; et al. Gremlin 1, frizzled-related protein, and Dkk-1 are key regulators of human articular cartilage homeostasis. Arthritis Rheum 2012, 64, 3302–3312. [Google Scholar]
- Minina, E.; Kreschel, C.; Naski, M.C.; Ornitz, D.M.; Vortkamp, A. Interaction of FGF, Ihh/Pthlh, and BMP signaling integrates chondrocyte proliferation and hypertrophic differentiation. Dev. Cell 2002, 3, 439–449. [Google Scholar]
- Caplan, A.I.; Elyaderani, M.; Mochizuki, Y.; Wakitani, S.; Goldberg, V.M. Principles of cartilage repair and regeneration. Clin. Orthop. Relat. Res 1997, 342, 254–269. [Google Scholar]
- Hunziker, E.B. Articular cartilage repair: Basic science and clinical progress A review of the current status and prospects. Osteoarthr. Cartil 2002, 10, 432–463. [Google Scholar]
- Johnstone, B.; Alini, M.; Cucchiarini, M.; Dodge, G.R.; Eglin, D.; Guilak, F.; Madry, H.; Mata, A.; Mauck, R.L.; Semino, C.E.; et al. Tissue engineering for articular cartilage repair—The state of the art. Eur. Cell Mater 2013, 25, 248–267. [Google Scholar]
- Loeser, R.F.; Pacione, C.A.; Chubinskaya, S. The combination of insulin-like growth factor 1 and osteogenic protein 1 promotes increased survival of and matrix synthesis by normal and osteoarthritic human articular chondrocytes. Arthritis Rheum 2003, 48, 2188–2196. [Google Scholar]
- Shi, S.; Mercer, S.; Eckert, G.J.; Trippel, S.B. Growth factor regulation of growth factors in articular chondrocytes. J. Biol. Chem 2009, 284, 6697–6704. [Google Scholar]
- Finnson, K.W.; Chi, Y.; Bou-Gharios, G.; Leask, A.; Philip, A. TGF-b signaling in cartilage homeostasis and osteoarthritis. Front. Biosci 2012, 4, 251–268. [Google Scholar]
- Guo, X.; Wang, X.F. Signaling cross-talk between TGF-β/BMP and other pathways. Cell Res 2009, 19, 71–88. [Google Scholar]
- Serra, R.; Johnson, M.; Filvaroff, E.H.; LaBorde, J.; Sheehan, D.M.; Derynck, R.; Moses, H.L. Expression of a truncated, kinase-defective TGF-β type II receptor in mouse skeletal tissue promotes terminal chondrocyte differentiation and osteoarthritis. J. Cell Biol 1997, 139, 541–552. [Google Scholar]
- Yang, X.; Chen, L.; Xu, X.; Li, C.; Huang, C.; Deng, C.X. TGF-beta/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J. Cell Biol 2001, 153, 35–46. [Google Scholar]
- Van Beuningen, H.M.; van der Kraan, P.M.; Arntz, O.J.; van den Berg, W.B. Protection from interleukin 1 induced destruction of articular cartilage by transforming growth factor beta: Studies in anatomically intact cartilage in vitro and in vivo. Ann. Rheum. Dis 1993, 52, 185–191. [Google Scholar]
- Madry, H.; Kaul, G.; Cucchiarini, M.; Stein, U.; Zurakowski, D.; Remberger, K.; Menger, M.D.; Kohn, D.; Trippel, S.B. Enhanced repair of articular cartilage defects in vivo by transplanted chondrocytes overexpressing insulin-like growth factor I (IGF-I). Gene. Ther 2005, 12, 1171–1179. [Google Scholar]
- Chia, S.L.; Sawaji, Y.; Burleigh, A.; McLean, C.; Inglis, J.; Saklatvala, J.; Vincent, T. Fibroblast growth factor 2 is an intrinsic chondroprotective agent that suppresses ADAMTS-5 and delays cartilage degradation in murine osteoarthritis. Arthritis Rheum 2009, 60, 2019–2027. [Google Scholar]
- Hunter, D.J.; Pike, M.C.; Jonas, B.L.; Kissin, E.; Krop, J.; McAlindon, T. Phase 1 safety and tolerability study of BMP-7 in symptomatic knee osteoarthritis. BMC Musculoskelet. Disord 2010, 11. [Google Scholar] [CrossRef]
- Blaney Davidson, E.N.; Remst, D.F.; Vitters, E.L.; van Beuningen, H.M.; Blom, A.B.; Goumans, M.J.; van den Berg, W.B.; van der Kraan, P.M. Increase in ALK1/ALK5 ratio as a cause for elevated MMP-13 expression in osteoarthritis in humans and mice. J. Immunol 2009, 182, 7937–7945. [Google Scholar]
- Plaas, A.; Velasco, J.; Gorski, D.J.; Li, J.; Cole, A.; Christopherson, K.; Sandy, J.D. The relationship between fibrogenic TGFbeta1 signaling in the joint and cartilage degradation in post-injury osteoarthritis. Osteoarthr. Cartil 2011, 19, 1081–1090. [Google Scholar]
- Zhen, G.; Wen, C.; Jia, X.; Li, Y.; Crane, J.L.; Mears, S.C.; Askin, F.B.; Frassica, F.J.; Chang, W.; Yao, J.; et al. Inhibition of TGF-β signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat. Med 2013, 19, 704–712. [Google Scholar]
- Konig, H.G.; Kogel, D.; Rami, A.; Prehn, J.H. TGF-{beta}1 activates two distinct type I receptors in neurons: Implications for neuronal NF-{kappa}B signaling. J. Cell Biol 2005, 168, 1077–1086. [Google Scholar]
- Finnson, K.W.; Parker, W.L.; ten Dijke, P.; Thorikay, M.; Philip, A. ALK1 opposes ALK5/Smad3 signaling and expression of extracellular matrix components in human chondrocytes. J. Bone Miner. Res 2008, 23, 896–906. [Google Scholar]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Lebrin, F.; Larsson, J.; Mummery, C.; Karlsson, S.; ten Dijke, P. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol. Cell 2003, 12, 817–828. [Google Scholar]
- Blaney Davidson, E.N.; Vitters, E.L.; van der Kraan, P.M.; van den Berg, W.B. Expression of transforming growth factor-beta (TGFbeta) and the TGFbeta signalling molecule SMAD-2P in spontaneous and instability-induced osteoarthritis: Role in cartilage degradation, chondrogenesis and osteophyte formation. Ann. Rheum. Dis 2006, 65, 1414–1421. [Google Scholar]
- Hellingman, C.A.; Davidson, E.N.; Koevoet, W.; Vitters, E.L.; van den Berg, W.B.; van Osch, G.J.; van der Kraan, P.M. Smad signaling determines chondrogenic differentiation of bone-marrow-derived mesenchymal stem cells: Inhibition of Smad1/5/8P prevents terminal differentiation and calcification. Tissue Eng. Part A 2011, 17, 1157–1167. [Google Scholar]
- Li, T.F.; Darowish, M.; Zuscik, M.J.; Chen, D.; Schwarz, E.M.; Rosier, R.N.; Drissi, H.; O'Keefe, R.J. Smad3-deficient chondrocytes have enhanced BMP signaling and accelerated differentiation. J. Bone Miner. Res 2006, 21, 4–16. [Google Scholar]
- Bertrand, J.; Cromme, C.; Umlauf, D.; Frank, S.; Pap, T. Molecular mechanisms of cartilage remodelling in osteoarthritis. Int. J. Biochem. Cell Biol 2010, 42, 1594–1601. [Google Scholar]
- Van Donkelaar, C.C.; Wilson, W. Mechanics of chondrocyte hypertrophy. Biomech. Model. Mechanobiol 2011, 11, 655–664. [Google Scholar]
- Van der Kraan, P.M.; Blaney Davidson, E.N.; van den Berg, W.B. Bone morphogenetic proteins and articular cartilage: To serve and protect or a wolf in sheep clothing’s? Osteoarthr. Cartil 2010, 18, 735–741. [Google Scholar]
- Nishimura, R.; Hata, K.; Matsubara, T.; Wakabayashi, M.; Yoneda, T. Regulation of bone and cartilage development by network between BMP signalling and transcription factors. J. Biochem 2012, 151, 247–254. [Google Scholar]
- Chen, A.L.; Fang, C.; Liu, C.; Leslie, M.P.; Chang, E.; di Cesare, P.E. Expression of bone morphogenetic proteins, receptors, and tissue inhibitors in human fetal, adult, and osteoarthritic articular cartilage. J. Orthop. Res 2004, 22, 1188–1192. [Google Scholar]
- Minina, E.; Wenzel, H.M.; Kreschel, C.; Karp, S.; Gaffield, W.; McMahon, A.P.; Vortkamp, A. BMP and Ihh/PTHrP signaling interact to coordinate chondrocyte proliferation and differentiation. Development 2001, 128, 4523–4534. [Google Scholar]
- Chubinskaya, S.; Segalite, D.; Pikovsky, D.; Hakimiyan, A.A.; Rueger, D.C. Effects induced by BMPS in cultures of human articular chondrocytes: Comparative studies. Growth Factors 2008, 26, 275–283. [Google Scholar]
- Goldring, M.B.; Tsuchimochi, K.; Ijiri, K. The control of chondrogenesis. J. Cell Biochem 2006, 97, 33–44. [Google Scholar]
- Blaney Davidson, E.N.; Vitters, E.L.; van Lent, P.L.; van de Loo, F.A.; van den Berg, W.B.; van der Kraan, P.M. Elevated extracellular matrix production and degradation upon bone morphogenetic protein-2 (BMP-2) stimulation point toward a role for BMP-2 in cartilage repair and remodeling. Arthritis Res. Ther 2007, 9, R102. [Google Scholar]
- Zhao, L.; Li, G.; Zhou, G.Q. SOX9 directly binds CREB as a novel synergism with the PKA pathway in BMP-2-induced osteochondrogenic differentiation. J. Bone Miner. Res 2009, 24, 826–836. [Google Scholar]
- Pan, Q.; Yu, Y.; Chen, Q.; Li, C.; Wu, H.; Wan, Y.; Ma, J.; Sun, F. Sox9, a key transcription factor of bone morphogenetic protein-2-induced chondrogenesis, is activated through BMP pathway and a CCAAT box in the proximal promoter. J. Cell. Physiol 2008, 217, 228–241. [Google Scholar]
- Chimal-Monroy, J.; Rodriguez-Leon, J.; Montero, J.A.; Ganan, Y.; Macias, D.; Merino, R.; Hurle, J.M. Analysis of the molecular cascade responsible for mesodermal limb chondrogenesis: Sox genes and BMP signaling. Dev. Biol 2003, 257, 292–301. [Google Scholar]
- Lories, R.J.; Daans, M.; Derese, I.; Matthys, P.; Kasran, A.; Tylzanowski, P.; Ceuppens, J.L.; Luyten, F.P. Noggin haploinsufficiency differentially affects tissue responses in destructive and remodeling arthritis. Arthritis Rheum 2006, 54, 1736–1746. [Google Scholar]
- Papathanasiou, I.; Malizos, K.N.; Tsezou, A. Bone morphogenetic protein-2-induced Wnt/beta-catenin signaling pathway activation through enhanced low-density-lipoprotein receptor-related protein 5 catabolic activity contributes to hypertrophy in osteoarthritic chondrocytes. Arthritis Res. Ther 2012, 14, R82. [Google Scholar]
- Caron, M.M.; Emans, P.J.; Cremers, A.; Surtel, D.A.; Coolsen, M.M.; van Rhijn, L.W.; Welting, T.J. Hypertrophic differentiation during chondrogenic differentiation of progenitor cells is stimulated by BMP-2 but suppressed by BMP-7. Osteoarthr. Cartil 2013, 21, 604–613. [Google Scholar]
- Horiki, M.; Imamura, T.; Okamoto, M.; Hayashi, M.; Murai, J.; Myoui, A.; Ochi, T.; Miyazono, K.; Yoshikawa, H.; Tsumaki, N. Smad6/Smurf1 overexpression in cartilage delays chondrocyte hypertrophy and causes dwarfism with osteopenia. J. Cell Biol 2004, 165, 433–445. [Google Scholar]
- Retting, K.N.; Song, B.; Yoon, B.S.; Lyons, K.M. BMP canonical Smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development 2009, 136, 1093–1104. [Google Scholar]
- Fan, Z.; Chubinskaya, S.; Rueger, D.C.; Bau, B.; Haag, J.; Aigner, T. Regulation of anabolic and catabolic gene expression in normal and osteoarthritic adult human articular chondrocytes by osteogenic protein-1. Clin. Exp. Rheumatol 2004, 22, 103–106. [Google Scholar]
- Flechtenmacher, J.; Huch, K.; Thonar, E.J.; Mollenhauer, J.A.; Davies, S.R.; Schmid, T.M.; Puhl, W.; Sampath, T.K.; Aydelotte, M.B.; Kuettner, K.E. Recombinant human osteogenic protein 1 is a potent stimulator of the synthesis of cartilage proteoglycans and collagens by human articular chondrocytes. Arthritis Rheum 1996, 39, 1896–1904. [Google Scholar]
- Nishida, Y.; Knudson, C.B.; Knudson, W. Osteogenic Protein-1 inhibits matrix depletion in a hyaluronan hexasaccharide-induced model of osteoarthritis. Osteoarthr. Cartil 2004, 12, 374–382. [Google Scholar]
- Huch, K.; Wilbrink, B.; Flechtenmacher, J.; Koepp, H.E.; Aydelotte, M.B.; Sampath, T.K.; Kuettner, K.E.; Mollenhauer, J.; Thonar, E.J. Effects of recombinant human osteogenic protein 1 on the production of proteoglycan, prostaglandin E2, and interleukin-1 receptor antagonist by human articular chondrocytes cultured in the presence of interleukin-1beta. Arthritis Rheum 1997, 40, 2157–2161. [Google Scholar]
- Koepp, H.E.; Sampath, K.T.; Kuettner, K.E.; Homandberg, G.A. Osteogenic protein-1 (OP-1) blocks cartilage damage caused by fibronectin fragments and promotes repair by enhancing proteoglycan synthesis. Inflamm. Res 1999, 48, 199–204. [Google Scholar]
- Umulis, D.; O’Connor, M.B.; Blair, S.S. The extracellular regulation of bone morphogenetic protein signaling. Development 2009, 136, 3715–3728. [Google Scholar]
- Miyazono, K.; Kamiya, Y.; Morikawa, M. Bone morphogenetic protein receptors and signal transduction. J. Biochem 2010, 147, 35–51. [Google Scholar]
- Andhare, R.A.; Takahashi, N.; Knudson, W.; Knudson, C.B. Hyaluronan promotes the chondrocyte response to BMP-7. Osteoarthr. Cartil 2009, 17, 906–916. [Google Scholar]
- Luo, N.; Knudson, W.; Askew, E.B.; Veluci, R.; Knudson, C.B. CD44 and hyaluronan promote the BMP7 signaling response in chondrocytes. Arthritis Rheumatol 2014. [Google Scholar] [CrossRef]
- Kanakaris, N.K.; Calori, G.M.; Verdonk, R.; Burssens, P.; de Biase, P.; Capanna, R.; Vangosa, L.B.; Cherubino, P.; Baldo, F.; Ristiniemi, J.; et al. Application of BMP-7 to tibial non-unions: A 3-year multicenter experience. Injury 2008, 39, S83–S90. [Google Scholar]
- Carreira, A.C.; Lojudice, F.H.; Halcsik, E.; Navarro, R.D.; Sogayar, M.C.; Granjeiro, J.M. Bone Morphogenetic Proteins: Facts, Challenges, and Future Perspectives. J. Dent. Res 2014, 93, 335–345. [Google Scholar]
- Bessa, P.C.; Casal, M.; Reis, R.L. Bone morphogenetic proteins in tissue engineering: The road from laboratory to clinic, part II (BMP delivery). J. Tissue Eng. Regen. Med 2008, 2, 81–96. [Google Scholar]
- Posever, J.; Phillips, F.M.; Pottenger, L.A. Effects of basic fibroblast growth factor, transforming growth factor-beta 1, insulin-like growth factor-1, and insulin on human osteoarthritic articular cartilage explants. J. Orthop. Res 1995, 13, 832–837. [Google Scholar]
- Wang, E.; Wang, J.; Chin, E.; Zhou, J.; Bondy, C.A. Cellular patterns of insulin-like growth factor system gene expression in murine chondrogenesis and osteogenesis. Endocrinology 1995, 136, 2741–2751. [Google Scholar]
- Middleton, J.; Manthey, A.; Tyler, J. Insulin-like growth factor (IGF) receptor, IGF-I, interleukin-1 beta (IL-1 beta), and IL-6 mRNA expression in osteoarthritic and normal human cartilage. J. Histochem. Cytochem 1996, 44, 133–141. [Google Scholar]
- Sah, R.L.; Chen, A.C.; Grodzinsky, A.J.; Trippel, S.B. Differential effects of bFGF and IGF-I on matrix metabolism in calf and adult bovine cartilage explants. Arch. Biochem. Biophys 1994, 308, 137–147. [Google Scholar]
- Fukumoto, T.; Sperling, J.W.; Sanyal, A.; Fitzsimmons, J.S.; Reinholz, G.G.; Conover, C.A.; O’Driscoll, S.W. Combined effects of insulin-like growth factor-1 and transforming growth factor-beta1 on periosteal mesenchymal cells during chondrogenesis in vitro. Osteoarthr. Cartil. 2003, 11, 55–64. [Google Scholar]
- McQuillan, D.J.; Handley, C.J.; Campbell, M.A.; Bolis, S.; Milway, V.E.; Herington, A.C. Stimulation of proteoglycan biosynthesis by serum and insulin-like growth factor-I in cultured bovine articular cartilage. Biochem. J 1986, 240, 423–430. [Google Scholar]
- Luyten, F.P.; Hascall, V.C.; Nissley, S.P.; Morales, T.I.; Reddi, A.H. Insulin-like growth factors maintain steady-state metabolism of proteoglycans in bovine articular cartilage explants. Arch. Biochem. Biophys 1988, 267, 416–425. [Google Scholar]
- Maor, G.; Hochberg, Z.; Silbermann, M. Insulin-like growth factor I accelerates proliferation and differentiation of cartilage progenitor cells in cultures of neonatal mandibular condyles. Acta Endocrinol 1993, 128, 56–64. [Google Scholar]
- Bohme, K.; Conscience-Egli, M.; Tschan, T.; Winterhalter, K.H.; Bruckner, P. Induction of proliferation or hypertrophy of chondrocytes in serum-free culture: The role of insulin-like growth factor-I, insulin, or thyroxine. J. Cell Biol 1992, 116, 1035–1042. [Google Scholar]
- Ekenstedt, K.J.; Sonntag, W.E.; Loeser, R.F.; Lindgren, B.R.; Carlson, C.S. Effects of chronic growth hormone and insulin-like growth factor 1 deficiency on osteoarthritis severity in rat knee joints. Arthritis Rheum 2006, 54, 3850–3858. [Google Scholar]
- Fortier, L.A.; Mohammed, H.O.; Lust, G.; Nixon, A.J. Insulin-like growth factor-I enhances cell-based repair of articular cartilage. J. Bone Jt. Surg. Br 2002, 84, 276–288. [Google Scholar]
- Goodrich, L.R.; Hidaka, C.; Robbins, P.D.; Evans, C.H.; Nixon, A.J. Genetic modification of chondrocytes with insulin-like growth factor-1 enhances cartilage healing in an equine model. J. Bone Jt. Surg. Br 2007, 89, 672–685. [Google Scholar]
- Rosselot, G.; Vasilatos-Younken, R.; Leach, R.M. Effect of growth hormone, insulin-like growth factor I, basic fibroblast growth factor, and transforming growth factor beta on cell proliferation and proteoglycan synthesis by avian postembryonic growth plate chondrocytes. J. Bone Miner. Res 1994, 9, 431–439. [Google Scholar]
- Tsukazaki, T.; Usa, T.; Matsumoto, T.; Enomoto, H.; Ohtsuru, A.; Namba, H.; Iwasaki, K.; Yamashita, S. Effect of transforming growth factor-beta on the insulin-like growth factor-I autocrine/paracrine axis in cultured rat articular chondrocytes. Exp. Cell Res 1994, 215, 9–16. [Google Scholar]
- Yaeger, P.C.; Masi, T.L.; de Ortiz, J.L.; Binette, F.; Tubo, R.; McPherson, J.M. Synergistic action of transforming growth factor-beta and insulin-like growth factor-I induces expression of type II collagen and aggrecan genes in adult human articular chondrocytes. Exp. Cell Res 1997, 237, 318–325. [Google Scholar]
- Longobardi, L.; O’Rear, L.; Aakula, S.; Johnstone, B.; Shimer, K.; Chytil, A.; Horton, W.A.; Moses, H.L.; Spagnoli, A. Effect of IGF-I in the chondrogenesis of bone marrow mesenchymal stem cells in the presence or absence of TGF-beta signaling. J. Bone Miner. Res 2006, 21, 626–636. [Google Scholar]
- Worster, A.A.; Brower-Toland, B.D.; Fortier, L.A.; Bent, S.J.; Williams, J.; Nixon, A.J. Chondrocytic differentiation of mesenchymal stem cells sequentially exposed to transforming growth factor-beta1 in monolayer and insulin-like growth factor-I in a three-dimensional matrix. J. Orthop. Res 2001, 19, 738–749. [Google Scholar]
- Fortier, L.A.; Miller, B.J. Signaling through the small G-protein Cdc42 is involved in insulin-like growth factor-I resistance in aging articular chondrocytes. J. Orthop. Res 2006, 24, 1765–1772. [Google Scholar]
- Loeser, R.F.; Carlson, C.S.; del Carlo, M.; Cole, A. Detection of nitrotyrosine in aging and osteoarthritic cartilage: Correlation of oxidative damage with the presence of interleukin-1beta and with chondrocyte resistance to insulin-like growth factor 1. Arthritis Rheum 2002, 46, 2349–2357. [Google Scholar]
- Martin, J.A.; Ellerbroek, S.M.; Buckwalter, J.A. Age-related decline in chondrocyte response to insulin-like growth factor-I: The role of growth factor binding proteins. J. Orthop. Res 1997, 15, 491–498. [Google Scholar]
- Dore, S.; Pelletier, J.P.; DiBattista, J.A.; Tardif, G.; Brazeau, P.; Martel-Pelletier, J. Human osteoarthritic chondrocytes possess an increased number of insulin-like growth factor 1 binding sites but are unresponsive to its stimulation. Possible role of IGF-1-binding proteins. Arthritis Rheum 1994, 37, 253–263. [Google Scholar]
- Loeser, R.F.; Shanker, G.; Carlson, C.S.; Gardin, J.F.; Shelton, B.J.; Sonntag, W.E. Reduction in the chondrocyte response to insulin-like growth factor 1 in aging and osteoarthritis: Studies in a non-human primate model of naturally occurring disease. Arthritis Rheum 2000, 43, 2110–2120. [Google Scholar]
- Morales, T.I. The quantitative and functional relation between insulin-like growth factor-I (IGF) and IGF-binding proteins during human osteoarthritis. J. Orthop. Res 2008, 26, 465–474. [Google Scholar]
- Chubinskaya, S.; Hakimiyan, A.; Pacione, C.; Yanke, A.; Rappoport, L.; Aigner, T.; Rueger, D.C.; Loeser, R.F. Synergistic effect of IGF-1 and OP-1 on matrix formation by normal and OA chondrocytes cultured in alginate beads. Osteoarthr. Cartil 2007, 15, 421–430. [Google Scholar]
- Ellman, M.B.; An, H.S.; Muddasani, P.; Im, H.J. Biological impact of the fibroblast growth factor family on articular cartilage and intervertebral disc homeostasis. Gene 2008, 420, 82–89. [Google Scholar]
- Fortier, L.A.; Barker, J.U.; Strauss, E.J.; McCarrel, T.M.; Cole, B.J. The role of growth factors in cartilage repair. Clin. Orthop. Relat. Res 2011, 469, 2706–2715. [Google Scholar]
- Hiraide, A.; Yokoo, N.; Xin, K.Q.; Okuda, K.; Mizukami, H.; Ozawa, K.; Saito, T. Repair of articular cartilage defect by intraarticular administration of basic fibroblast growth factor gene, using adeno-associated virus vector. Hum. Gene Ther 2005, 16, 1413–1421. [Google Scholar]
- Deng, T.; Huang, S.; Zhou, S.; He, L.; Jin, Y. Cartilage regeneration using a novel gelatin-chondroitin-hyaluronan hybrid scaffold containing bFGF-impregnated microspheres. J. Microencapsul 2007, 24, 163–174. [Google Scholar]
- Inoue, A.; Takahashi, K.A.; Arai, Y.; Tonomura, H.; Sakao, K.; Saito, M.; Fujioka, M.; Fujiwara, H.; Tabata, Y.; Kubo, T. The therapeutic effects of basic fibroblast growth factor contained in gelatin hydrogel microspheres on experimental osteoarthritis in the rabbit knee. Arthritis Rheum 2006, 54, 264–270. [Google Scholar]
- Kaul, G.; Cucchiarini, M.; Arntzen, D.; Zurakowski, D.; Menger, M.D.; Kohn, D.; Trippel, S.B.; Madry, H. Local stimulation of articular cartilage repair by transplantation of encapsulated chondrocytes overexpressing human fibroblast growth factor 2 (FGF-2) in vivo. J. Gene Med. 2006, 8, 100–111. [Google Scholar]
- Stewart, K.; Pabbruwe, M.; Dickinson, S.; Sims, T.; Hollander, A.P.; Chaudhuri, J.B. The effect of growth factor treatment on meniscal chondrocyte proliferation and differentiation on polyglycolic acid scaffolds. Tissue Eng 2007, 13, 271–280. [Google Scholar]
- Loeser, R.F.; Chubinskaya, S.; Pacione, C.; Im, H.J. Basic fibroblast growth factor inhibits the anabolic activity of insulin-like growth factor 1 and osteogenic protein 1 in adult human articular chondrocytes. Arthritis Rheum 2005, 52, 3910–3917. [Google Scholar]
- Im, H.J.; Li, X.; Muddasani, P.; Kim, G.H.; Davis, F.; Rangan, J.; Forsyth, C.B.; Ellman, M.; Thonar, E.J. Basic fibroblast growth factor accelerates matrix degradation via a neuro-endocrine pathway in human adult articular chondrocytes. J. Cell. Physiol 2008, 215, 452–463. [Google Scholar]
- Im, H.J.; Muddasani, P.; Natarajan, V.; Schmid, T.M.; Block, J.A.; Davis, F.; van Wijnen, A.J.; Loeser, R.F. Basic fibroblast growth factor stimulates matrix metalloproteinase-13 via the molecular cross-talk between the mitogen-activated protein kinases and protein kinase Cdelta pathways in human adult articular chondrocytes. J. Biol. Chem 2007, 282, 11110–11121. [Google Scholar]
- Muddasani, P.; Norman, J.C.; Ellman, M.; van Wijnen, A.J.; Im, H.J. Basic fibroblast growth factor activates the MAPK and NFkappaB pathways that converge on Elk-1 to control production of matrix metalloproteinase-13 by human adult articular chondrocytes. J. Biol. Chem 2007, 282, 31409–31421. [Google Scholar]
- Ellsworth, J.L.; Berry, J.; Bukowski, T.; Claus, J.; Feldhaus, A.; Holderman, S.; Holdren, M.S.; Lum, K.D.; Moore, E.E.; Raymond, F.; et al. Fibroblast growth factor-18 is a trophic factor for mature chondrocytes and their progenitors. Osteoarthr. Cartil 2002, 10, 308–320. [Google Scholar]
- Davidson, D.; Blanc, A.; Filion, D.; Wang, H.; Plut, P.; Pfeffer, G.; Buschmann, M.D.; Henderson, J.E. Fibroblast growth factor (FGF) 18 signals through FGF receptor 3 to promote chondrogenesis. J. Biol. Chem 2005, 280, 20509–20515. [Google Scholar]
- Liu, Z.; Xu, J.; Colvin, J.S.; Ornitz, D.M. Coordination of chondrogenesis and osteogenesis by fibroblast growth factor 18. Genes Dev 2002, 16, 859–869. [Google Scholar]
- Moore, E.E.; Bendele, A.M.; Thompson, D.L.; Littau, A.; Waggie, K.S.; Reardon, B.; Ellsworth, J.L. Fibroblast growth factor-18 stimulates chondrogenesis and cartilage repair in a rat model of injury-induced osteoarthritis. Osteoarthr. Cartil 2005, 13, 623–631. [Google Scholar]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar]
- Semenza, G.L. HIF-1 and human disease: One highly involved factor. Genes Dev 2000, 14, 1983–1991. [Google Scholar]
- Schipani, E.; Ryan, H.E.; Didrickson, S.; Kobayashi, T.; Knight, M.; Johnson, R.S. Hypoxia in cartilage: HIF-1alpha is essential for chondrocyte growth arrest and survival. Genes Dev 2001, 15, 2865–2876. [Google Scholar]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell Biol 1996, 16, 4604–4613. [Google Scholar]
- Murata, M.; Yudoh, K.; Masuko, K. The potential role of vascular endothelial growth factor (VEGF) in cartilage: How the angiogenic factor could be involved in the pathogenesis of osteoarthritis? Osteoarthr. Cartil 2008, 16, 279–286. [Google Scholar]
- Duval, E.; Leclercq, S.; Elissalde, J.M.; Demoor, M.; Galera, P.; Boumediene, K. Hypoxia-inducible factor 1alpha inhibits the fibroblast-like markers type I and type III collagen during hypoxia-induced chondrocyte redifferentiation: Hypoxia not only induces type II collagen and aggrecan, but it also inhibits type I and type III collagen in the hypoxia-inducible factor 1alpha-dependent redifferentiation of chondrocytes. Arthritis Rheum 2009, 60, 3038–3048. [Google Scholar]
- Aro, E.; Khatri, R.; Gerard-O’Riley, R.; Mangiavini, L.; Myllyharju, J.; Schipani, E. Hypoxia-inducible factor-1 (HIF-1) but not HIF-2 is essential for hypoxic induction of collagen prolyl 4-hydroxylases in primary newborn mouse epiphyseal growth plate chondrocytes. J. Biol. Chem 2012, 287, 37134–37144. [Google Scholar]
- Mobasheri, A.; Richardson, S.; Mobasheri, R.; Shakibaei, M.; Hoyland, J.A. Hypoxia inducible factor-1 and facilitative glucose transporters GLUT1 and GLUT3: Putative molecular components of the oxygen and glucose sensing apparatus in articular chondrocytes. Histol. Histopathol 2005, 20, 1327–1338. [Google Scholar]
- Pfander, D.; Cramer, T.; Swoboda, B. Hypoxia and HIF-1alpha in osteoarthritis. Int. Orthop 2005, 29, 6–9. [Google Scholar]
- Yudoh, K.; Nakamura, H.; Masuko-Hongo, K.; Kato, T.; Nishioka, K. Catabolic stress induces expression of hypoxia-inducible factor (HIF)-1 alpha in articular chondrocytes: Involvement of HIF-1 alpha in the pathogenesis of osteoarthritis. Arthritis Res. Ther 2005, 7, R904–R914. [Google Scholar]
- Aigner, T.; Zien, A.; Gehrsitz, A.; Gebhard, P.M.; McKenna, L. Anabolic and catabolic gene expression pattern analysis in normal versus osteoarthritic cartilage using complementary DNA-array technology. Arthritis Rheum 2001, 44, 2777–2789. [Google Scholar]
- Coimbra, I.B.; Jimenez, S.A.; Hawkins, D.F.; Piera-Velazquez, S.; Stokes, D.G. Hypoxia inducible factor-1 alpha expression in human normal and osteoarthritic chondrocytes. Osteoarthr. Cartil 2004, 12, 336–345. [Google Scholar]
- Stokes, D.G.; Liu, G.; Coimbra, I.B.; Piera-Velazquez, S.; Crowl, R.M.; Jimenez, S.A. Assessment of the gene expression profile of differentiated and dedifferentiated human fetal chondrocytes by microarray analysis. Arthritis Rheum 2002, 46, 404–419. [Google Scholar]
- Hellwig-Burgel, T.; Rutkowski, K.; Metzen, E.; Fandrey, J.; Jelkmann, W. Interleukin-1beta and tumor necrosis factor-alpha stimulate DNA binding of hypoxia-inducible factor-1. Blood 1999, 94, 1561–1567. [Google Scholar]
- Haddad, J.J.; Land, S.C. A non-hypoxic, ROS-sensitive pathway mediates TNF-alpha-dependent regulation of HIF-1alpha. FEBS Lett 2001, 505, 269–274. [Google Scholar]
- Haddad, J.J. Recombinant human interleukin (IL)-1 beta-mediated regulation of hypoxia-inducible factor-1 alpha (HIF-1 alpha) stabilization, nuclear translocation and activation requires an antioxidant/reactive oxygen species (ROS)-sensitive mechanism. Eur. Cytokine Netw 2002, 13, 250–260. [Google Scholar]
- Jung, Y.J.; Isaacs, J.S.; Lee, S.; Trepel, J.; Neckers, L. IL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J 2003, 17, 2115–2117. [Google Scholar]
- Petersen, W.; Varoga, D.; Zantop, T.; Hassenpflug, J.; Mentlein, R.; Pufe, T. Cyclic strain influences the expression of the vascular endothelial growth factor (VEGF) and the hypoxia inducible factor 1 alpha (HIF-1alpha) in tendon fibroblasts. J. Orthop. Res 2004, 22, 847–853. [Google Scholar]
- Pufe, T.; Lemke, A.; Kurz, B.; Petersen, W.; Tillmann, B.; Grodzinsky, A.J.; Mentlein, R. Mechanical overload induces VEGF in cartilage discs via hypoxia-inducible factor. Am. J. Pathol 2004, 164, 185–192. [Google Scholar]
- Chang, H.; Shyu, K.G.; Wang, B.W.; Kuan, P. Regulation of hypoxia-inducible factor-1alpha by cyclical mechanical stretch in rat vascular smooth muscle cells. Clin. Sci 2003, 105, 447–456. [Google Scholar]
- Ema, M.; Taya, S.; Yokotani, N.; Sogawa, K.; Matsuda, Y.; Fujii-Kuriyama, Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc. Natl. Acad. Sci. USA 1997, 94, 4273–4278. [Google Scholar]
- Patel, S.A.; Simon, M.C. Biology of hypoxia-inducible factor-2alpha in development and disease. Cell Death Differ 2008, 15, 628–634. [Google Scholar]
- Ratcliffe, P.J. HIF-1 and HIF-2: Working alone or together in hypoxia? J. Clin. Investig 2007, 117, 862–865. [Google Scholar]
- Wang, V.; Davis, D.A.; Haque, M.; Huang, L.E.; Yarchoan, R. Differential gene up-regulation by hypoxia-inducible factor-1alpha and hypoxia-inducible factor-2alpha in HEK293T cells. Cancer Res 2005, 65, 3299–3306. [Google Scholar]
- Lafont, J.E.; Talma, S.; Hopfgarten, C.; Murphy, C.L. Hypoxia promotes the differentiated human articular chondrocyte phenotype through SOX9-dependent and -independent pathways. J. Biol. Chem 2008, 283, 4778–4786. [Google Scholar]
- Lafont, J.E.; Talma, S.; Murphy, C.L. Hypoxia-inducible factor 2alpha is essential for hypoxic induction of the human articular chondrocyte phenotype. Arthritis Rheum 2007, 56, 3297–3306. [Google Scholar]
- Thoms, B.L.; Dudek, K.A.; Lafont, J.E.; Murphy, C.L. Hypoxia promotes the production and inhibits the destruction of human articular cartilage. Arthritis Rheum 2013, 65, 1302–1312. [Google Scholar]
- Saito, T.; Fukai, A.; Mabuchi, A.; Ikeda, T.; Yano, F.; Ohba, S.; Nishida, N.; Akune, T.; Yoshimura, N.; Nakagawa, T.; et al. Transcriptional regulation of endochondral ossification by HIF-2alpha during skeletal growth and osteoarthritis development. Nat. Med 2010, 16, 678–686. [Google Scholar]
- Kronenberg, H.M. Developmental regulation of the growth plate. Nature 2003, 423, 332–336. [Google Scholar]
- Mackie, E.J.; Ahmed, Y.A.; Tatarczuch, L.; Chen, K.S.; Mirams, M. Endochondral ossification: How cartilage is converted into bone in the developing skeleton. Int. J. Biochem. Cell Biol 2008, 40, 46–62. [Google Scholar]
- Tamiya, H.; Ikeda, T.; Jeong, J.H.; Saito, T.; Yano, F.; Jung, Y.K.; Ohba, S.; Kawaguchi, H.; Chung, U.I.; Choi, J.Y. Analysis of the Runx2 promoter in osseous and non-osseous cells and identification of HIF2A as a potent transcription activator. Gene 2008, 416, 53–60. [Google Scholar]
- Kawaguchi, H. Endochondral ossification signals in cartilage degradation during osteoarthritis progression in experimental mouse models. Mol. Cells 2008, 25, 1–6. [Google Scholar]
- Yang, S.; Kim, J.; Ryu, J.H.; Oh, H.; Chun, C.H.; Kim, B.J.; Min, B.H.; Chun, J.S. Hypoxia-inducible factor-2alpha is a catabolic regulator of osteoarthritic cartilage destruction. Nat. Med 2010, 16, 687–693. [Google Scholar]
- Mizushima, N.; Klionsky, D.J. Protein turnover via autophagy: Implications for metabolism. Annu. Rev. Nutr 2007, 27, 19–40. [Google Scholar]
- Uchiyama, Y.; Shibata, M.; Koike, M.; Yoshimura, K.; Sasaki, M. Autophagy-physiology and pathophysiology. Histochem. Cell Biol 2008, 129, 407–420. [Google Scholar]
- Mizushima, N. Physiological functions of autophagy. Curr. Top. Microbiol. Immunol 2009, 335, 71–84. [Google Scholar]
- Salminen, A.; Kauppinen, A.; Suuronen, T.; Kaarniranta, K.; Ojala, J. ER stress in Alzheimer’s disease: A novel neuronal trigger for inflammation and Alzheimer’s pathology. J. Neuroinflamm 2009, 6. [Google Scholar] [CrossRef]
- Austin, R.C. The unfolded protein response in health and disease. Antioxid. Redox Signal 2009, 11, 2279–2287. [Google Scholar]
- Bohensky, J.; Terkhorn, S.P.; Freeman, T.A.; Adams, C.S.; Garcia, J.A.; Shapiro, I.M.; Srinivas, V. Regulation of autophagy in human and murine cartilage: Hypoxia-inducible factor 2 suppresses chondrocyte autophagy. Arthritis Rheum 2009, 60, 1406–1415. [Google Scholar]
- Staines, K.A.; Macrae, V.E.; Farquharson, C. Cartilage development and degeneration: A Wnt Wnt situation. Cell Biochem. Funct 2012, 30, 633–642. [Google Scholar]
- Ma, B.; Landman, E.B.; Miclea, R.L.; Wit, J.M.; Robanus-Maandag, E.C.; Post, J.N.; Karperien, M. WNT signaling and cartilage: Of mice and men. Calcif. Tissue Int 2013, 92, 399–411. [Google Scholar]
- Sharma, A.R.; Jagga, S.; Lee, S.S.; Nam, J.S. Interplay between cartilage and subchondral bone contributing to pathogenesis of osteoarthritis. Int. J. Mol. Sci 2013, 14, 19805–19830. [Google Scholar]
- Sassi, N.; Laadhar, L.; Allouche, M.; Achek, A.; Kallel-Sellami, M.; Makni, S.; Sellami, S. WNT signaling and chondrocytes: From cell fate determination to osteoarthritis physiopathology. J. Recept. Signal Transduct. Res 2014, 34, 73–80. [Google Scholar]
- Bhanot, P.; Brink, M.; Samos, C.H.; Hsieh, J.C.; Wang, Y.; Macke, J.P.; Andrew, D.; Nathans, J.; Nusse, R. A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nature 1996, 382, 225–230. [Google Scholar]
- Dann, C.E.; Hsieh, J.C.; Rattner, A.; Sharma, D.; Nathans, J.; Leahy, D.J. Insights into Wnt binding and signalling from the structures of two Frizzled cysteine-rich domains. Nature 2001, 412, 86–90. [Google Scholar]
- Aberle, H.; Bauer, A.; Stappert, J.; Kispert, A.; Kemler, R. beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J 1997, 16, 3797–3804. [Google Scholar]
- Monroe, D.G.; McGee-Lawrence, M.E.; Oursler, M.J.; Westendorf, J.J. Update on Wnt signaling in bone cell biology and bone disease. Gene 2012, 492, 1–18. [Google Scholar]
- Logan, C.Y.; Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol 2004, 20, 781–810. [Google Scholar]
- Nusse, R. Wnt signaling in disease and in development. Cell Res 2005, 15, 28–32. [Google Scholar]
- Chun, J.S.; Oh, H.; Yang, S.; Park, M. Wnt signaling in cartilage development and degeneration. BMB Rep 2008, 41, 485–494. [Google Scholar]
- Nalesso, G.; Sherwood, J.; Bertrand, J.; Pap, T.; Ramachandran, M.; de Bari, C.; Pitzalis, C.; dell’accio, F. WNT-3A modulates articular chondrocyte phenotype by activating both canonical and noncanonical pathways. J. Cell Biol 2011, 193, 551–564. [Google Scholar] [Green Version]
- Yasuhara, R.; Ohta, Y.; Yuasa, T.; Kondo, N.; Hoang, T.; Addya, S.; Fortina, P.; Pacifici, M.; Iwamoto, M.; Enomoto-Iwamoto, M. Roles of beta-catenin signaling in phenotypic expression and proliferation of articular cartilage superficial zone cells. Lab. Investig 2011, 91, 1739–1752. [Google Scholar]
- Golovchenko, S.; Hattori, T.; Hartmann, C.; Gebhardt, M.; Gebhard, S.; Hess, A.; Pausch, F.; Schlund, B.; von der Mark, K. Deletion of beta catenin in hypertrophic growth plate chondrocytes impairs trabecular bone formation. Bone 2013, 55, 102–112. [Google Scholar]
- Dell’Accio, F.; de Bari, C.; El Tawil, N.M.; Barone, F.; Mitsiadis, T.A.; O’Dowd, J.; Pitzalis, C. Activation of WNT and BMP signaling in adult human articular cartilage following mechanical injury. Arthritis Res. Ther 2006, 8, R139. [Google Scholar]
- Dell’accio, F.; de Bari, C.; Eltawil, N.M.; Vanhummelen, P.; Pitzalis, C. Identification of the molecular response of articular cartilage to injury, by microarray screening: Wnt-16 expression and signaling after injury and in osteoarthritis. Arthritis Rheum 2008, 58, 1410–1421. [Google Scholar]
- Eltawil, N.M.; de Bari, C.; Achan, P.; Pitzalis, C.; dell’accio, F. A novel in vivo murine model of cartilage regeneration. Age and strain-dependent outcome after joint surface injury. Osteoarthr. Cartil 2009, 17, 695–704. [Google Scholar]
- Lodewyckx, L.; Cailotto, F.; Thysen, S.; Luyten, F.P.; Lories, R.J. Tight regulation of wingless-type signaling in the articular cartilage—Subchondral bone biomechanical unit: Transcriptomics in Frzb-knockout mice. Arthritis Res. Ther 2012, 14, R16. [Google Scholar]
- Miclea, R.L.; Siebelt, M.; Finos, L.; Goeman, J.J.; Lowik, C.W.; Oostdijk, W.; Weinans, H.; Wit, J.M.; Robanus-Maandag, E.C.; Karperien, M. Inhibition of Gsk3beta in cartilage induces osteoarthritic features through activation of the canonical Wnt signaling pathway. Osteoarthr. Cartil 2011, 19, 1363–1372. [Google Scholar]
- Zhu, M.; Tang, D.; Wu, Q.; Hao, S.; Chen, M.; Xie, C.; Rosier, R.N.; O’Keefe, R.J.; Zuscik, M.; Chen, D. Activation of beta-catenin signaling in articular chondrocytes leads to osteoarthritis-like phenotype in adult beta-catenin conditional activation mice. J. Bone Miner. Res 2009, 24, 12–21. [Google Scholar]
- Loughlin, J.; Dowling, B.; Chapman, K.; Marcelline, L.; Mustafa, Z.; Southam, L.; Ferreira, A.; Ciesielski, C.; Carson, D.A.; Corr, M. Functional variants within the secreted frizzled-related protein 3 gene are associated with hip osteoarthritis in females. Proc. Natl. Acad. Sci. USA 2004, 101, 9757–9762. [Google Scholar]
- Urano, T.; Shiraki, M.; Narusawa, K.; Usui, T.; Sasaki, N.; Hosoi, T.; Ouchi, Y.; Nakamura, T.; Inoue, S. Q89R polymorphism in the LDL receptor-related protein 5 gene is associated with spinal osteoarthritis in postmenopausal Japanese women. Spine 2007, 32, 25–29. [Google Scholar]
- Diarra, D.; Stolina, M.; Polzer, K.; Zwerina, J.; Ominsky, M.S.; Dwyer, D.; Korb, A.; Smolen, J.; Hoffmann, M.; Scheinecker, C.; et al. Dickkopf-1 is a master regulator of joint remodeling. Nat. Med 2007, 13, 156–163. [Google Scholar]
- Lane, N.E.; Nevitt, M.C.; Lui, L.Y.; de Leon, P.; Corr, M. Wnt signaling antagonists are potential prognostic biomarkers for the progression of radiographic hip osteoarthritis in elderly Caucasian women. Arthritis Rheum 2007, 56, 3319–3325. [Google Scholar]
- Honsawek, S.; Tanavalee, A.; Yuktanandana, P.; Ngarmukos, S.; Saetan, N.; Tantavisut, S. Dickkopf-1 (Dkk-1) in plasma and synovial fluid is inversely correlated with radiographic severity of knee osteoarthritis patients. BMC Musculoskelet. Disord 2010, 11, 257. [Google Scholar]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol 2009, 1, a000034. [Google Scholar]
- Marcu, K.B.; Otero, M.; Olivotto, E.; Borzi, R.M.; Goldring, M.B. NF-kappaB signaling: Multiple angles to target OA. Curr. Drug Targets 2010, 11, 599–613. [Google Scholar]
- Rigoglou, S.; Papavassiliou, A.G. The NF-kappaB signalling pathway in osteoarthritis. Int. J. Biochem. Cell Biol 2013, 45, 2580–2584. [Google Scholar]
- Karin, M.; Greten, F.R. NF-kappaB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol 2005, 5, 749–759. [Google Scholar]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar]
- Ge, X.P.; Gan, Y.H.; Zhang, C.G.; Zhou, C.Y.; Ma, K.T.; Meng, J.H.; Ma, X.C. Requirement of the NF-kappaB pathway for induction of Wnt-5A by interleukin-1beta in condylar chondrocytes of the temporomandibular joint: Functional crosstalk between the Wnt-5A and NF-kappaB signaling pathways. Osteoarthr. Cartil 2011, 19, 111–117. [Google Scholar]
- Niederberger, E.; Geisslinger, G. The IKK-NF-kappaB pathway: A source for novel molecular drug targets in pain therapy? FASEB J 2008, 22, 3432–3442. [Google Scholar]
- Pomerantz, J.L.; Baltimore, D. Two pathways to NF-kappaB. Mol. Cell 2002, 10, 693–695. [Google Scholar]
- Bonizzi, G.; Karin, M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol 2004, 25, 280–288. [Google Scholar]
- Karin, M.; Yamamoto, Y.; Wang, Q.M. The IKK NF-kappa B system: A treasure trove for drug development. Nat. Rev. Drug Discov 2004, 3, 17–26. [Google Scholar]
- Yamamoto, Y.; Gaynor, R.B. IkappaB kinases: Key regulators of the NF-kappaB pathway. Trends Biochem. Sci 2004, 29, 72–79. [Google Scholar]
- Yasuda, T. Activation of Akt leading to NF-kappaB up-regulation in chondrocytes stimulated with fibronectin fragment. Biomed. Res 2011, 32, 209–215. [Google Scholar]
- Ulivi, V.; Giannoni, P.; Gentili, C.; Cancedda, R.; Descalzi, F. p38/NF-κB-dependent expression of COX-2 during differentiation and inflammatory response of chondrocytes. J. Cell Biochem 2008, 104, 1393–1406. [Google Scholar]
- DeGroot, J.; Verzijl, N.; Wenting-van Wijk, M.J.; Jacobs, K.M.; van El, B.; van Roermund, P.M.; Bank, R.A.; Bijlsma, J.W.; TeKoppele, J.M.; Lafeber, F.P. Accumulation of advanced glycation end products as a molecular mechanism for aging as a risk factor in osteoarthritis. Arthritis Rheum 2004, 50, 1207–1215. [Google Scholar]
- Steenvoorden, M.M.; Huizinga, T.W.; Verzijl, N.; Bank, R.A.; Ronday, H.K.; Luning, H.A.; Lafeber, F.P.; Toes, R.E.; DeGroot, J. Activation of receptor for advanced glycation end products in osteoarthritis leads to increased stimulation of chondrocytes and synoviocytes. Arthritis Rheum 2006, 54, 253–263. [Google Scholar]
- Goldring, M.B.; Otero, M.; Plumb, D.A.; Dragomir, C.; Favero, M.; El Hachem, K.; Hashimoto, K.; Roach, H.I.; Olivotto, E.; Borzi, R.M.; et al. Roles of inflammatory and anabolic cytokines in cartilage metabolism: Signals and multiple effectors converge upon MMP-13 regulation in osteoarthritis. Eur. Cell Mater 2011, 21, 202–220. [Google Scholar]
- Hirata, M.; Kugimiya, F.; Fukai, A.; Saito, T.; Yano, F.; Ikeda, T.; Mabuchi, A.; Sapkota, B.R.; Akune, T.; Nishida, N.; et al. C/EBPbeta and RUNX2 cooperate to degrade cartilage with MMP-13 as the target and HIF-2alpha as the inducer in chondrocytes. Hum. Mol. Genet 2012, 21, 1111–1123. [Google Scholar]
- Grall, F.; Gu, X.; Tan, L.; Cho, J.Y.; Inan, M.S.; Pettit, A.R.; Thamrongsak, U.; Choy, B.K.; Manning, C.; Akbarali, Y.; et al. Responses to the proinflammatory cytokines interleukin-1 and tumor necrosis factor alpha in cells derived from rheumatoid synovium and other joint tissues involve nuclear factor kappaB-mediated induction of the Ets transcription factor ESE-1. Arthritis Rheum 2003, 48, 1249–1260. [Google Scholar]
- Peng, H.; Tan, L.; Osaki, M.; Zhan, Y.; Ijiri, K.; Tsuchimochi, K.; Otero, M.; Wang, H.; Choy, B.K.; Grall, F.T.; et al. ESE-1 is a potent repressor of type II collagen gene (COL2A1) transcription in human chondrocytes. J. Cell. Physiol 2008, 215, 562–573. [Google Scholar]
- Otero, M.; Plumb, D.A.; Tsuchimochi, K.; Dragomir, C.L.; Hashimoto, K.; Peng, H.; Olivotto, E.; Bevilacqua, M.; Tan, L.; Yang, Z.; et al. E74-like factor 3 (ELF3) impacts on matrix metalloproteinase 13 (MMP13) transcriptional control in articular chondrocytes under proinflammatory stress. J. Biol. Chem 2012, 287, 3559–3572. [Google Scholar]
- Merz, D.; Liu, R.; Johnson, K.; Terkeltaub, R. IL-8/CXCL8 and growth-related oncogene alpha/CXCL1 induce chondrocyte hypertrophic differentiation. J. Immunol 2003, 171, 4406–4415. [Google Scholar]
- Ijiri, K.; Zerbini, L.F.; Peng, H.; Otu, H.H.; Tsuchimochi, K.; Otero, M.; Dragomir, C.; Walsh, N.; Bierbaum, B.E.; Mattingly, D.; et al. Differential expression of GADD45beta in normal and osteoarthritic cartilage: Potential role in homeostasis of articular chondrocytes. Arthritis Rheum 2008, 58, 2075–2087. [Google Scholar]
- Husa, M.; Liu-Bryan, R.; Terkeltaub, R. Shifting HIFs in osteoarthritis. Nat. Med 2010, 16, 641–644. [Google Scholar]
- Scanzello, C.R.; Goldring, S.R. The role of synovitis in osteoarthritis pathogenesis. Bone 2012, 51, 249–257. [Google Scholar]
- Liu, S.C.; Hsu, C.J.; Chen, H.T.; Tsou, H.K.; Chuang, S.M.; Tang, C.H. CTGF increases IL-6 expression in human synovial fibroblasts through integrin-dependent signaling pathway. PLoS One 2012, 7, e51097. [Google Scholar]
- Hou, C.H.; Tang, C.H.; Hsu, C.J.; Hou, S.M.; Liu, J.F. CCN4 induces IL-6 production through alphavbeta5 receptor, PI3K, Akt, and NF-kappaB singling pathway in human synovial fibroblasts. Arthritis Res. Ther 2013, 15, R19. [Google Scholar]
- Krasnokutsky, S.; Attur, M.; Palmer, G.; Samuels, J.; Abramson, S.B. Current concepts in the pathogenesis of osteoarthritis. Osteoarthr. Cartil 2008, 16, S1–S3. [Google Scholar]
- Xing, L.; Carlson, L.; Story, B.; Tai, Z.; Keng, P.; Siebenlist, U.; Boyce, B.F. Expression of either NF-kappaB p50 or p52 in osteoclast precursors is required for IL-1-induced bone resorption. J. Bone Miner. Res 2003, 18, 260–269. [Google Scholar]
- Nakashima, T.; Takayanagi, H. The dynamic interplay between osteoclasts and the immune system. Arch. Biochem. Biophys 2008, 473, 166–171. [Google Scholar]
- Sagar, D.R.; Ashraf, S.; Xu, L.; Burston, J.J.; Menhinick, M.R.; Poulter, C.L.; Bennett, A.J.; Walsh, D.A.; Chapman, V. Osteoprotegerin reduces the development of pain behaviour and joint pathology in a model of osteoarthritis. Ann. Rheum. Dis 2013. [Google Scholar] [CrossRef]
- Castaneda, S.; Roman-Blas, J.A.; Largo, R.; Herrero-Beaumont, G. Subchondral bone as a key target for osteoarthritis treatment. Biochem. Pharmacol 2012, 83, 315–323. [Google Scholar]
- Kwan Tat, S.; Amiable, N.; Pelletier, J.P.; Boileau, C.; Lajeunesse, D.; Duval, N.; Martel-Pelletier, J. Modulation of OPG, RANK and RANKL by human chondrocytes and their implication during osteoarthritis. Rheumatology 2009, 48, 1482–1490. [Google Scholar]
- Trouvin, A.P.; Goeb, V. Receptor activator of nuclear factor-kappaB ligand and osteoprotegerin: Maintaining the balance to prevent bone loss. Clin. Interv. Aging 2010, 5, 345–354. [Google Scholar]
- Martin, T.J. Historically significant events in the discovery of RANK/RANKL/OPG. World J. Orthop 2013, 4, 186–197. [Google Scholar]
- Bobick, B.E.; Kulyk, W.M. Regulation of cartilage formation and maturation by mitogen-activated protein kinase signaling. Birth Defects Res. C Embryo Today 2008, 84, 131–154. [Google Scholar]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar]
- Takebe, K.; Nishiyama, T.; Hayashi, S.; Hashimoto, S.; Fujishiro, T.; Kanzaki, N.; Kawakita, K.; Iwasa, K.; Kuroda, R.; Kurosaka, M. Regulation of p38 MAPK phosphorylation inhibits chondrocyte apoptosis in response to heat stress or mechanical stress. Int. J. Mol. Med 2011, 27, 329–335. [Google Scholar]
- Prasadam, I.; van Gennip, S.; Friis, T.; Shi, W.; Crawford, R.; Xiao, Y. ERK-1/2 and p38 in the regulation of hypertrophic changes of normal articular cartilage chondrocytes induced by osteoarthritic subchondral osteoblasts. Arthritis Rheum 2010, 62, 1349–1360. [Google Scholar] [Green Version]
- Thalhamer, T.; McGrath, M.A.; Harnett, M.M. MAPKs and their relevance to arthritis and inflammation. Rheumatology 2008, 47, 409–414. [Google Scholar]
- Kyriakis, J.M.; Banerjee, P.; Nikolakaki, E.; Dai, T.; Rubie, E.A.; Ahmad, M.F.; Avruch, J.; Woodgett, J.R. The stress-activated protein kinase subfamily of c-Jun kinases. Nature 1994, 369, 156–160. [Google Scholar]
- Raingeaud, J.; Gupta, S.; Rogers, J.S.; Dickens, M.; Han, J.; Ulevitch, R.J.; Davis, R.J. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J. Biol. Chem 1995, 270, 7420–7426. [Google Scholar]
- Freshney, N.W.; Rawlinson, L.; Guesdon, F.; Jones, E.; Cowley, S.; Hsuan, J.; Saklatvala, J. Interleukin-1 activates a novel protein kinase cascade that results in the phosphorylation of Hsp27. Cell 1994, 78, 1039–1049. [Google Scholar]
- Berenbaum, F. Signaling transduction: Target in osteoarthritis. Curr. Opin. Rheumatol 2004, 16, 616–622. [Google Scholar]
- Tetsunaga, T.; Nishida, K.; Furumatsu, T.; Naruse, K.; Hirohata, S.; Yoshida, A.; Saito, T.; Ozaki, T. Regulation of mechanical stress-induced MMP-13 and ADAMTS-5 expression by RUNX-2 transcriptional factor in SW1353 chondrocyte-like cells. Osteoarthr. Cartil 2011, 19, 222–232. [Google Scholar]
- Thirunavukkarasu, K.; Pei, Y.; Moore, T.L.; Wang, H.; Yu, X.P.; Geiser, A.G.; Chandrasekhar, S. Regulation of the human ADAMTS-4 promoter by transcription factors and cytokines. Biochem. Biophys. Res. Commun 2006, 345, 197–204. [Google Scholar]
- Houard, X.; Goldring, M.B.; Berenbaum, F. Homeostatic mechanisms in articular cartilage and role of inflammation in osteoarthritis. Curr. Rheumatol. Rep 2013, 15. [Google Scholar] [CrossRef]
- Stanton, L.A.; Underhill, T.M.; Beier, F. MAP kinases in chondrocyte differentiation. Dev. Biol 2003, 263, 165–175. [Google Scholar]
- Greenblatt, M.B.; Shim, J.H.; Glimcher, L.H. Mitogen-activated protein kinase pathways in osteoblasts. Annu. Rev. Cell Dev. Biol 2013, 29, 63–79. [Google Scholar]
- Prasadam, I.; Friis, T.; Shi, W.; van Gennip, S.; Crawford, R.; Xiao, Y. Osteoarthritic cartilage chondrocytes alter subchondral bone osteoblast differentiation via MAPK signalling pathway involving ERK1/2. Bone 2010, 46, 226–235. [Google Scholar]
- Oh, C.D.; Chang, S.H.; Yoon, Y.M.; Lee, S.J.; Lee, Y.S.; Kang, S.S.; Chun, J.S. Opposing role of mitogen-activated protein kinase subtypes, erk-1/2 and p38, in the regulation of chondrogenesis of mesenchymes. J. Biol. Chem 2000, 275, 5613–5619. [Google Scholar]
- Sondergaard, B.C.; Schultz, N.; Madsen, S.H.; Bay-Jensen, A.C.; Kassem, M.; Karsdal, M.A. MAPKs are essential upstream signaling pathways in proteolytic cartilage degradation—Divergence in pathways leading to aggrecanase and MMP-mediated articular cartilage degradation. Osteoarthr. Cartil 2010, 18, 279–288. [Google Scholar]
- Goldring, M.B.; Otero, M.; Tsuchimochi, K.; Ijiri, K.; Li, Y. Defining the roles of inflammatory and anabolic cytokines in cartilage metabolism. Ann. Rheum. Dis 2008, 67, iii75–iii82. [Google Scholar]
- Wang, X.; Li, F.; Fan, C.; Wang, C.; Ruan, H. Effects and relationship of ERK1 and ERK2 in interleukin-1β-induced alterations in MMP3, MMP13, type II collagen and aggrecan expression in human chondrocytes. Int. J. Mol. Med 2011, 27, 583–589. [Google Scholar]
- Djouad, F.; Rackwitz, L.; Song, Y.; Janjanin, S.; Tuan, R.S. ERK1/2 activation induced by inflammatory cytokines compromises effective host tissue integration of engineered cartilage. Tissue Eng. Part A 2009, 15, 2825–2835. [Google Scholar]
- Chung, U.I.; Schipani, E.; McMahon, A.P.; Kronenberg, H.M. Indian hedgehog couples chondrogenesis to osteogenesis in endochondral bone development. J. Clin. Investig 2001, 107, 295–304. [Google Scholar]
- Mak, K.K.; Kronenberg, H.M.; Chuang, P.T.; Mackem, S.; Yang, Y. Indian hedgehog signals independently of PTHrP to promote chondrocyte hypertrophy. Development 2008, 135, 1947–1956. [Google Scholar]
- Maeda, Y.; Nakamura, E.; Nguyen, M.T.; Suva, L.J.; Swain, F.L.; Razzaque, M.S.; Mackem, S.; Lanske, B. Indian Hedgehog produced by postnatal chondrocytes is essential for maintaining a growth plate and trabecular bone. Proc. Natl. Acad. Sci. USA 2007, 104, 6382–6387. [Google Scholar]
- Kobayashi, T.; Soegiarto, D.W.; Yang, Y.; Lanske, B.; Schipani, E.; McMahon, A.P.; Kronenberg, H.M. Indian hedgehog stimulates periarticular chondrocyte differentiation to regulate growth plate length independently of PTHrP. J. Clin. Investig 2005, 115, 1734–1742. [Google Scholar]
- Vortkamp, A.; Lee, K.; Lanske, B.; Segre, G.V.; Kronenberg, H.M.; Tabin, C.J. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science 1996, 273, 613–622. [Google Scholar]
- Lanske, B.; Karaplis, A.C.; Lee, K.; Luz, A.; Vortkamp, A.; Pirro, A.; Karperien, M.; Defize, L.H.; Ho, C.; Mulligan, R.C.; et al. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science 1996, 273, 663–666. [Google Scholar]
- Ng, T.C.; Chiu, K.W.; Rabie, A.B.; Hagg, U. Repeated mechanical loading enhances the expression of Indian hedgehog in condylar cartilage. Front. Biosci 2006, 11, 943–948. [Google Scholar]
- Wilson, C.W.; Chuang, P.T. Mechanism and evolution of cytosolic Hedgehog signal transduction. Development 2010, 137, 2079–2094. [Google Scholar]
- Corbit, K.C.; Aanstad, P.; Singla, V.; Norman, A.R.; Stainier, D.Y.; Reiter, J.F. Vertebrate Smoothened functions at the primary cilium. Nature 2005, 437, 1018–1021. [Google Scholar]
- Wuelling, M.; Vortkamp, A. Transcriptional networks controlling chondrocyte proliferation and differentiation during endochondral ossification. Pediatr. Nephrol 2010, 25, 625–631. [Google Scholar]
- Lin, A.C.; Seeto, B.L.; Bartoszko, J.M.; Khoury, M.A.; Whetstone, H.; Ho, L.; Hsu, C.; Ali, S.A.; Alman, B.A. Modulating hedgehog signaling can attenuate the severity of osteoarthritis. Nat. Med 2009, 15, 1421–1425. [Google Scholar]
- St-Jacques, B.; Hammerschmidt, M.; McMahon, A.P. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev 1999, 13, 2072–2086. [Google Scholar]
- Chen, X.; Macica, C.M.; Nasiri, A.; Broadus, A.E. Regulation of articular chondrocyte proliferation and differentiation by indian hedgehog and parathyroid hormone-related protein in mice. Arthritis Rheum 2008, 58, 3788–3797. [Google Scholar]
- Wei, F.; Zhou, J.; Wei, X.; Zhang, J.; Fleming, B.C.; Terek, R.; Pei, M.; Chen, Q.; Liu, T.; Wei, L. Activation of Indian hedgehog promotes chondrocyte hypertrophy and upregulation of MMP-13 in human osteoarthritic cartilage. Osteoarthr. Cartil 2012, 20, 755–763. [Google Scholar]
- Steinert, A.F.; Weissenberger, M.; Kunz, M.; Gilbert, F.; Ghivizzani, S.C.; Gobel, S.; Jakob, F.; Noth, U.; Rudert, M. Indian hedgehog gene transfer is a chondrogenic inducer of human mesenchymal stem cells. Arthritis Res. Ther 2012, 14, R168. [Google Scholar]
- Lin, L.; Shen, Q.; Xue, T.; Duan, X.; Fu, X.; Yu, C. Sonic hedgehog improves redifferentiation of dedifferentiated chondrocytes for articular cartilage repair. PLoS One 2014, 9, e88550. [Google Scholar]
- Kafienah, W.; Mistry, S.; Dickinson, S.C.; Sims, T.J.; Learmonth, I.; Hollander, A.P. Three-dimensional cartilage tissue engineering using adult stem cells from osteoarthritis patients. Arthritis Rheum 2007, 56, 177–187. [Google Scholar]
- Wu, L.; Huang, X.; Li, L.; Huang, H.; Xu, R.; Luyten, W. Insights on biology and pathology of HIF-1α/-2α, TGFβ/BMP, Wnt/β-catenin, and NF-κB pathways in osteoarthritis. Curr. Pharm. Des 2012, 18, 3293–3312. [Google Scholar]
- Van den Bosch, M.H.; Blom, A.B.; van Lent, P.L.; van Beuningen, H.M.; Blaney Davidson, E.N.; van der Kraan, P.M.; van den Berg, W.B. Canonical Wnt signaling skews TGF-β signaling in chondrocytes towards signaling via ALK1 and Smad 1/5/8. Cell Signal 2014, 26, 951–958. [Google Scholar]
- Mak, K.K.; Chen, M.H.; Day, T.F.; Chuang, P.T.; Yang, Y. Wnt/beta-catenin signaling interacts differentially with Ihh signaling in controlling endochondral bone and synovial joint formation. Development 2006, 133, 3695–3707. [Google Scholar]
- Dao, D.Y.; Jonason, J.H.; Zhang, Y.; Hsu, W.; Chen, D.; Hilton, M.J.; O’Keefe, R.J. Cartilage-specific β-catenin signaling regulates chondrocyte maturation, generation of ossification centers, and perichondrial bone formation during skeletal development. J. Bone Miner. Res 2012, 27, 1680–1694. [Google Scholar]
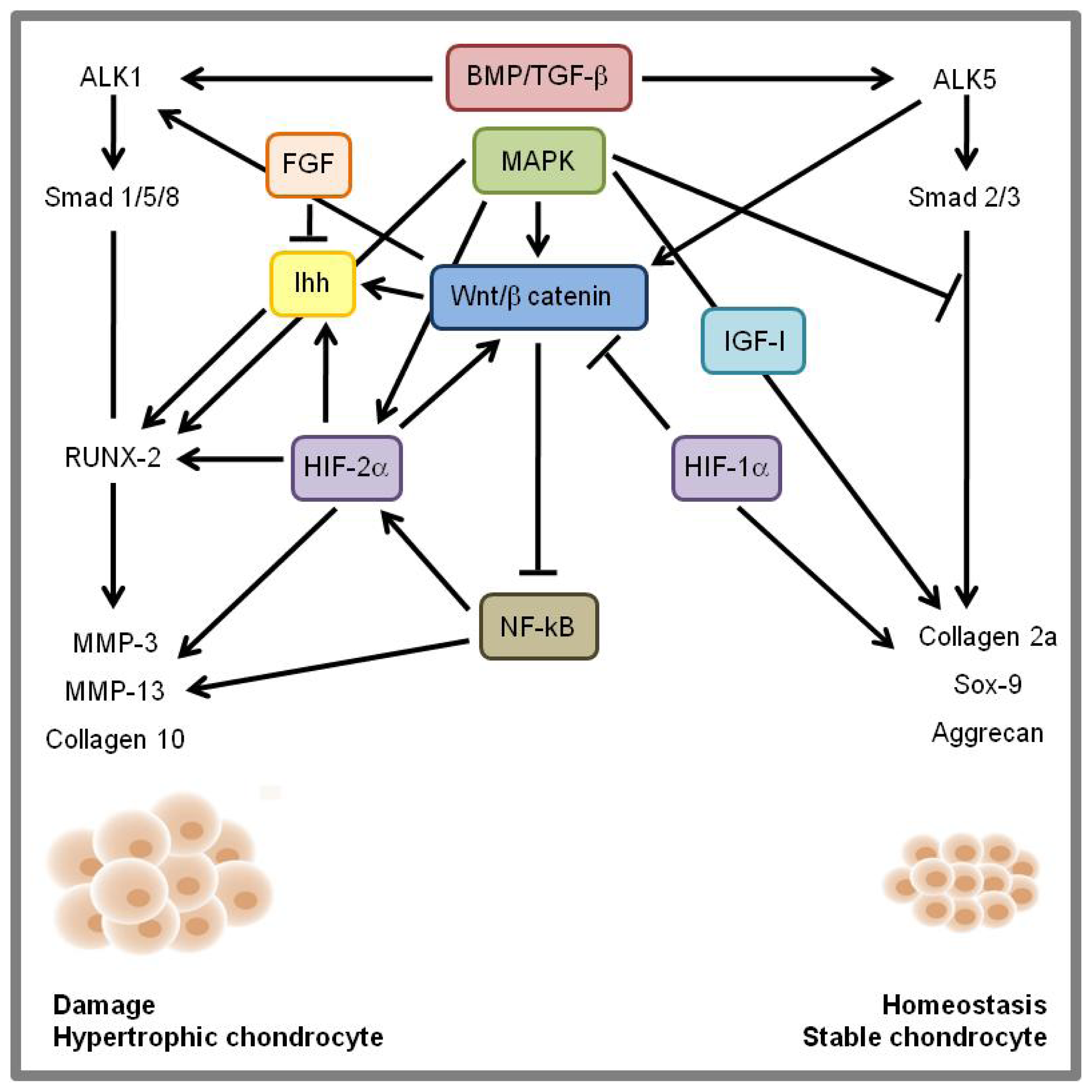

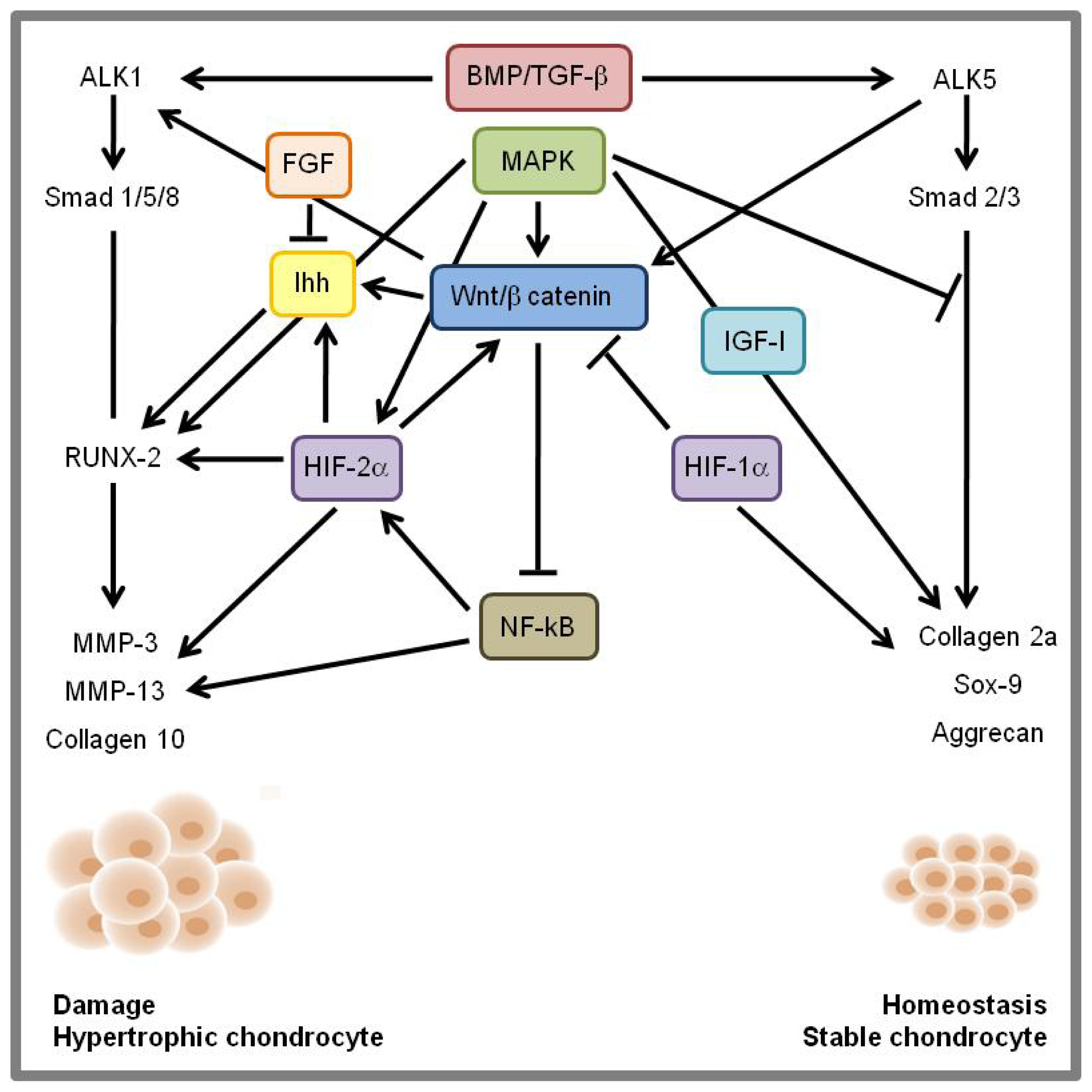{kind=link}
| Promoting Effects | |
| Wnt5a | Expressed in perichondrium surrounding condensations Recruits mesenchymal stem cells Delays chondrocyte differentiation |
| Wnt5b | Expressed in pre-hypertrophic chondrocytes and in the perichondrium Promotes initial steps of chondrogenesis in micromass cultures by stimulating cartilage nodule formation Delays terminal differentiation in vivo |
| β-catenin low level | Expressed in chondrogenic mesenchymal condensations Induces expression of Sox9 and promotes chondrogenic differentiation |
| Dkk1sFRP1 | Promotes early chondrogenesis in human mesenchymal stem cells |
| Wif-1 | Expressed in mesenchyme surrounding cartilage elements and articular cartilage Promotes chondrogenic differentiation by neutralizing Wnt3a mediated inhibition of chondrogenesis |
| Inhibitory Effects | |
| Wnt1 | Inhibits cell condensation and thus cartilage formation |
| Wnt3a | Expressed in early stages of chondrogenesis, decreased when chondrogenic differentiation proceeds Increases self-renewal and decreases apoptosis of MSCs Stabilizes cell-cell adhesion through promoting sustained expression of N-cadherin and β-catenin Blocks collagen II expression and proteoglycan deposition |
| Wnt4 | Expressed in developing joint interzone Inhibits cell condensation and thus chondrogenesis condensation |
| Wnt6 | Inhibits chondrogenesis at an early stage prior to chondrogenic differentiation (up-stream of SOX-9) |
| Wnt7a | Expressed in dorsal ectoderm in developing limb Inhibits chondrogenic differentiation in vitro and in vivo mediated by MAPK and AP-1 signaling Prolongs stabilization of N-cadherin Blocks transition from the condensation state to the cartilaginous nodule formation |
| Wnt9a (14) | Expressed in developing joint interzone Expressed in synovium and joint capsule in the mature joint Arrests or reverses chondrogenic differentiation Blocks transition from the condensation state to the cartilaginous nodule formation |
| Fzl7 | Inhibits mesenchymal condensation at the pre-cartilage aggregate formation by suppressing N-cadherin expression |
| Promoting Effects | |
| Wnt-4 | Expressed in periphery of joint interzone and hypertrophic chondrocytes Accelerates chondrocyte terminal differentiation Induces terminal differentiation of growth plate cartilage |
| Wnt8 | Promotes chondrocyte hypertrophy |
| β-catenin high level | Increases cell hypertrophy through RUNX-2 or IHH signaling activation Induces collagen X expression |
| Inhibitory Effects | |
| Wnt-5aWnt-5b | Inhibits hypertrophic maturation of chondrocytes through NF-κB stimulation and RUNX-2 inhibition |
| FRZB | Expressed in prechondrogenic mesenchymal condensations and in epiphyseal pre-articular chondrocytes Blocks chondrocyte maturation and prevents endochondral ossification in vivo Promotes glycosaminoglycan synthesis and expression of Sox9 and collagen II in vitro |
| DKK-1 | Expressed at sites of programmed cell death in apical ectodermal ridge Expressed at higher level in articular cartilage than growth plate cartilage Promotes glycosaminoglycan synthesis and expression of Sox9 and collagen II in vitro Inhibits chondrocyte hypertrophy |
| Fzl1Fzl7 | Blocks/delays chondrocyte hypertrophy |
| sFRP-1sFRP-3 | Delays terminal differentiation of hypertrophic chondrocytes |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mariani, E.; Pulsatelli, L.; Facchini, A. Signaling Pathways in Cartilage Repair. Int. J. Mol. Sci. 2014, 15, 8667-8698. https://doi.org/10.3390/ijms15058667
Mariani E, Pulsatelli L, Facchini A. Signaling Pathways in Cartilage Repair. International Journal of Molecular Sciences. 2014; 15(5):8667-8698. https://doi.org/10.3390/ijms15058667
Chicago/Turabian StyleMariani, Erminia, Lia Pulsatelli, and Andrea Facchini. 2014. "Signaling Pathways in Cartilage Repair" International Journal of Molecular Sciences 15, no. 5: 8667-8698. https://doi.org/10.3390/ijms15058667
APA StyleMariani, E., Pulsatelli, L., & Facchini, A. (2014). Signaling Pathways in Cartilage Repair. International Journal of Molecular Sciences, 15(5), 8667-8698. https://doi.org/10.3390/ijms15058667