Generation and Analysis of Expressed Sequence Tags (ESTs) from Halophyte Atriplex canescens to Explore Salt-Responsive Related Genes


Abstract
:1. Introduction
2. Results and Discussion
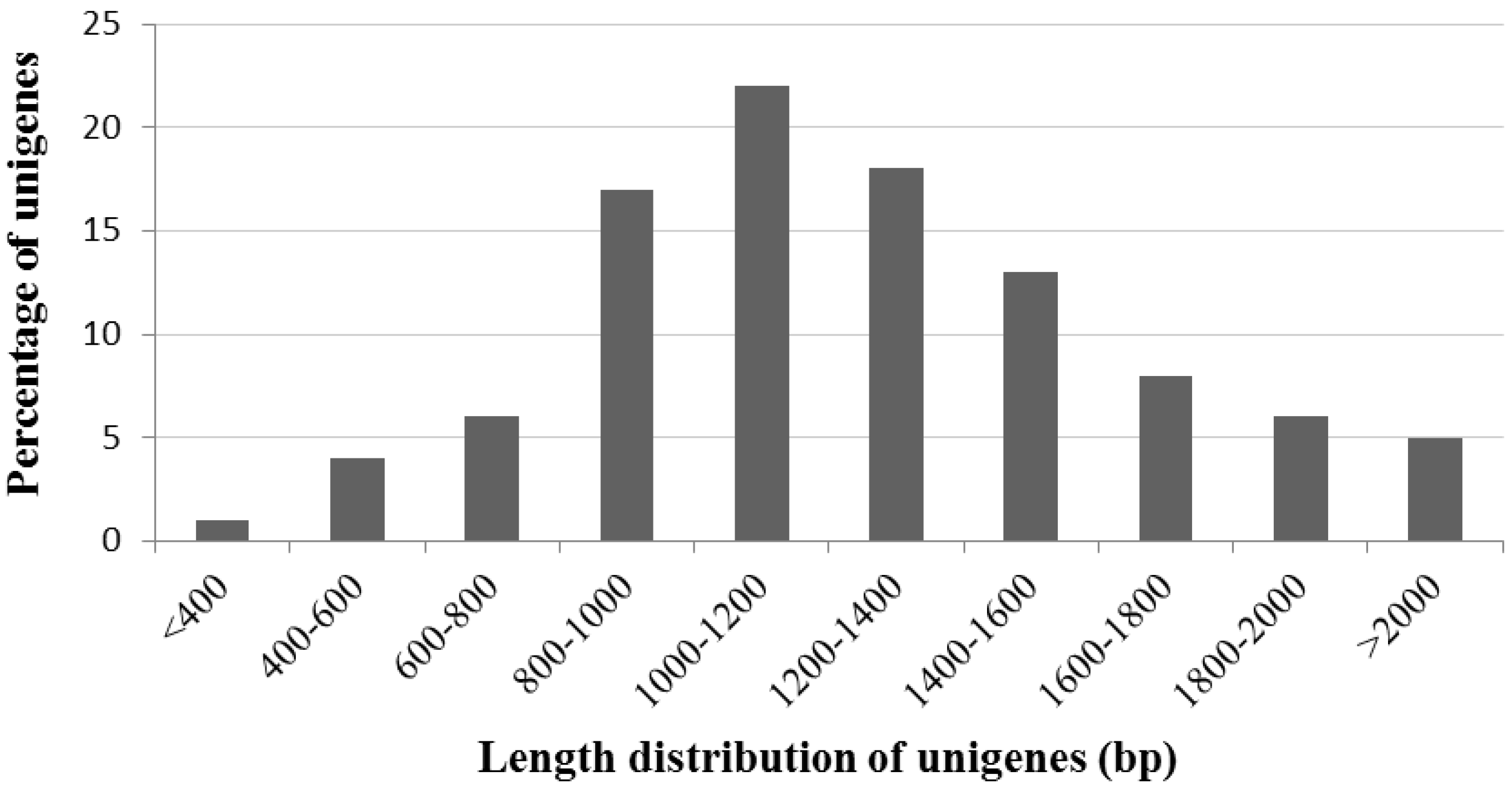
2.1. General Characteristics of the cDNA Library

2.2. General Characteristics of A. canescens ESTs

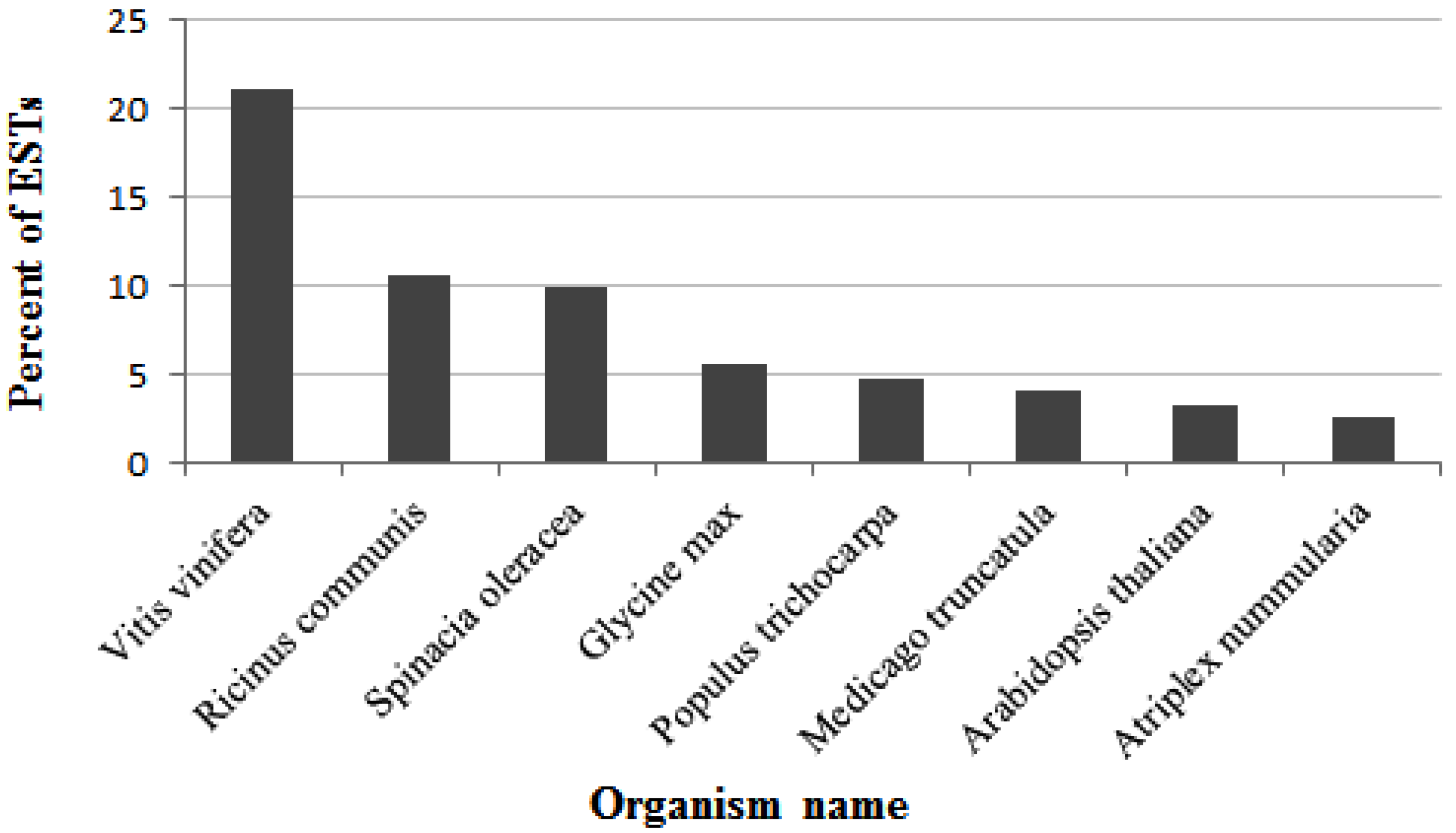
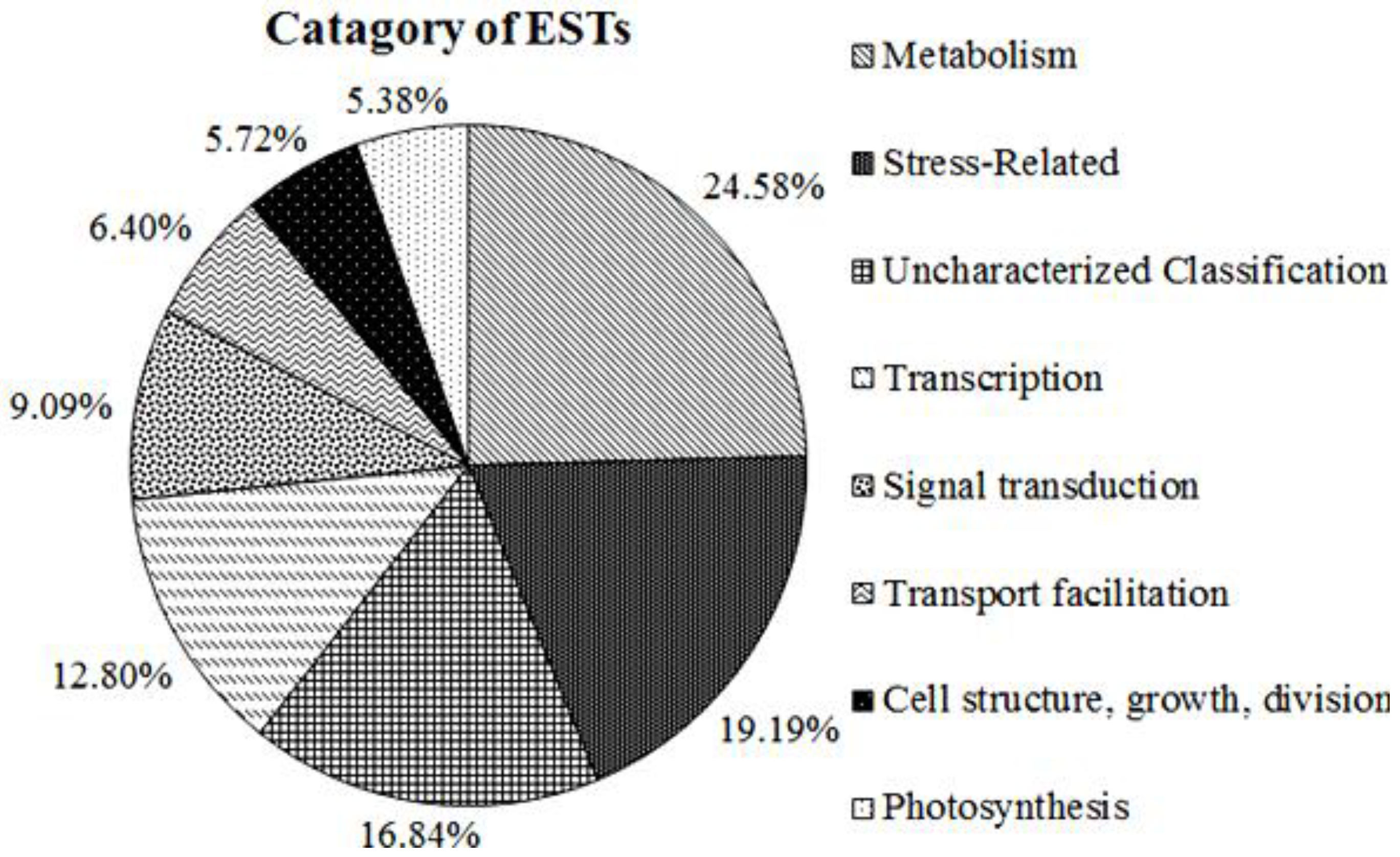
2.3. Functional Annotation and Classification of A. Canescens Unigenes


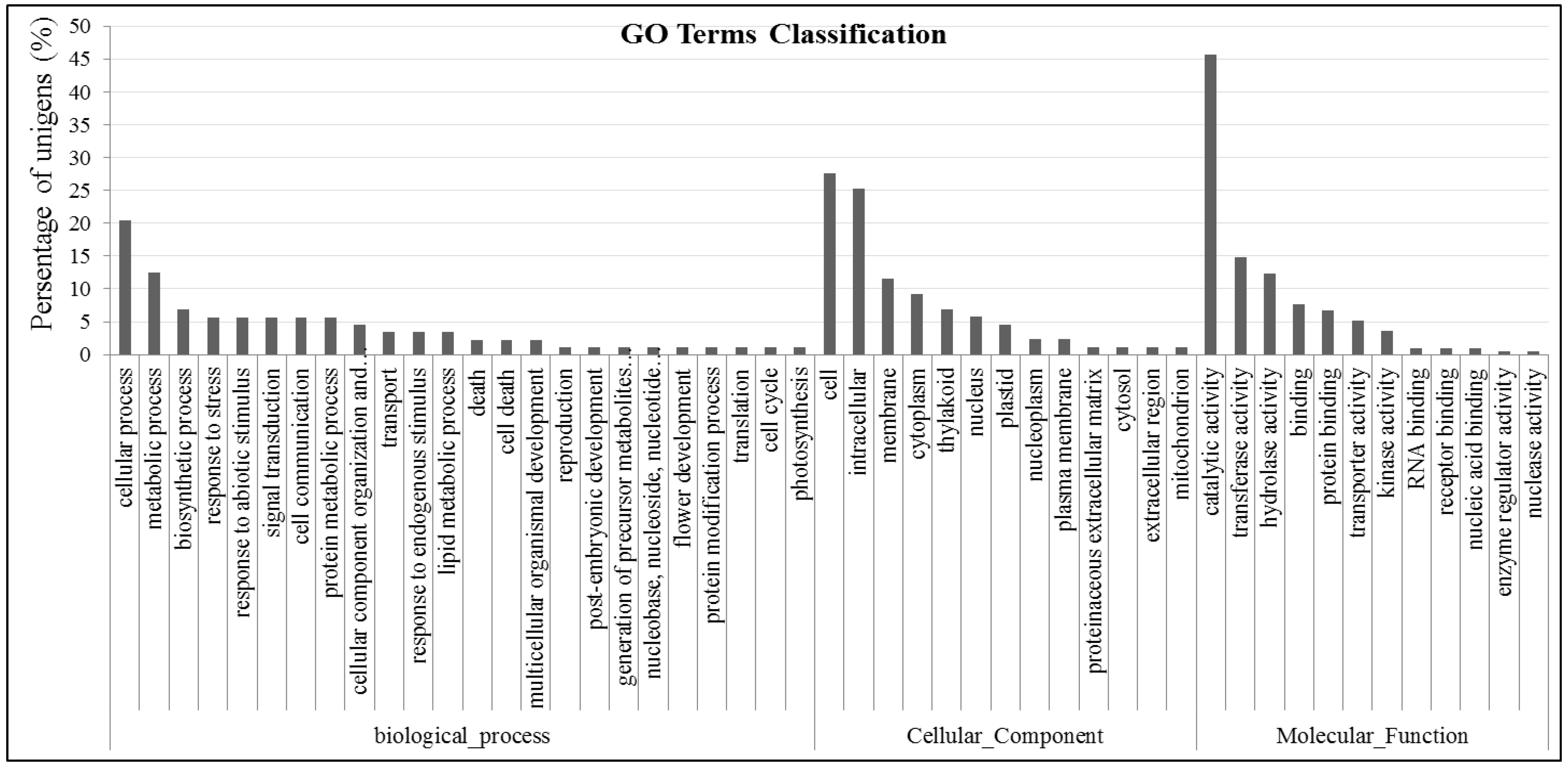
2.4. Gene GO Classifications and Genes Potentially Involved in Abiotic Tolerance


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Accession NO. | Gene Description | Matching Organism | E-Value |
|---|---|---|---|
| response to stress and abiotic stimulus | |||
| JZ535996↑ | Dehydration-responsive element binding protein | Krascheninnikovia arborescens | 3 × 10−98 |
| JZ535839↑ | Stress-induced protein sti1-like protein | Atriplex canescens | 1 × 10−161 |
| JZ536071↑ | Manganese tolerance protein | Beta vulgaris | 2 × 10−112 |
| cellular component organization and biogenesis | |||
| JZ536087↓ | Non-specific lipid-transfer protein-like protein | Vitis vinifera | 5 × 10−37 |
| transport | |||
| JZ535867↑ | Bidirectional sugar transporter SWEET1-like | Glycine max | 1 × 10−110 |
| response to endogenous stimulus | |||
| JZ535960↑ | Ethylene response factor 3 | Malus x domestica | 3 × 10−21 |
| lipid metabolic process | |||
| JZ535825↑ | Abscisic acid stress ripening protein | Salicornia brachiata | 3 × 10−20 |
| JZ535968↑ | Glycine and proline-rich protein | Ipomoea batatas | 0.62 |
| cell death | |||
| JZ535907↑ | Leucine-rich repeat receptor-like protein kinase | Theobroma cacao | 2 × 10−105 |
| thylakoid | |||
| JZ535969↓ | Chlorophyll a/b binding protein | Amaranthus hypochondriacus | 0.0 |
| JZ535848↑ | 23 kDa Precursor protein of the oxygen-evolving complex | Salicornia europaea | 4 × 10−138 |
| protein binding | |||
| JZ536063↑ | General transcription factor IIE subunit 1-like | Vitis vinifera | 7 × 10−31 |
| JZ535986↓ | Ankyrin domain protein | Nicotiana tabacum | 2 × 10−148 |
| JZ535815↑ | Ubiquitin | Medicago truncatula | 1 × 10−161 |
| JZ536095↑ | Dof-type zinc finger domain-containing protein | Arabidopsis lyrata | 1 × 10−30 |
| catalytic activity (partly) | |||
| JZ536113↑ | NADH dehydrogenase | Brachypodium distachyon | 3 × 10−64 |
| JZ536089↓ | S-adenosylmethionine synthase | Atriplex nummularia | 0.0 |
| JZ536067↑ | 3-ketoacyl CoA thiolase | Petunia x hybrida | 4 × 10−120 |
| JZ535984↑ | Short chain alcohol dehydrogenase-like | Arabidopsis thaliana | 6 × 10−65 |
| JZ536011↑ | Chitinase | Chenopodium amaranticolor | 2 × 10−123 |
| transporter activity | |||
| JZ535943↑ | Aquaporin | Knorringia sibirica | 1 × 10−159 |
| JZ535964↓ | Early nodulin 55-2 precursor | Ricinus communis | 2 × 10−33 |
| JZ535896↓ | Sodium-bile acid cotransporter | Ricinus communis | 5 × 10−120 |
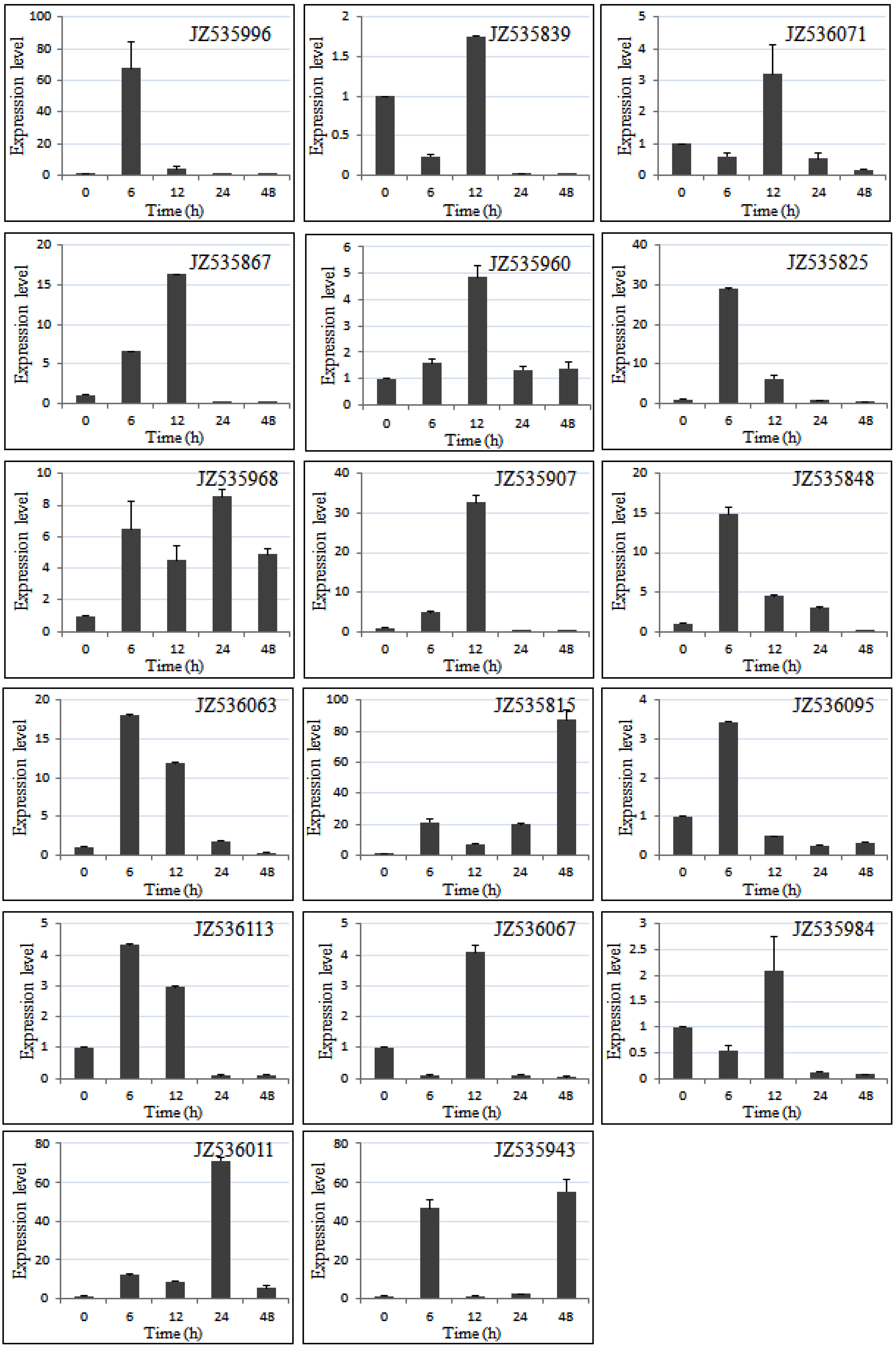
2.5. Expression Level of Salt-Responsive Genes in A. canescens Using Quantitative RT-PCR


2.6. Identification and Characterization of SSRs
| Sequence Accession No. | Repeat Motif | Repeat Numbers | Within ORF | 5'UTR * | 3'UTR * | Motif No. (Total, %) |
|---|---|---|---|---|---|---|
| JZ535808 | CT | 5 | 1 | |||
| JZ535828 | CT | 8 | 1 | |||
| JZ535877 | CT | 5 | 1 | |||
| JZ535901 | CT | 6 | 1 | |||
| JZ536002 | CT | 7 | 1 | |||
| JZ536029 | CT | 5 | 1 | Di- | ||
| JZ536099 | AG | 5 | 1 | (15, 68.2%) | ||
| JZ535947 | TC | 5 | 1 | |||
| JZ536047 | TC | 8 | 1 | |||
| JZ536117 | TC | 7 | 1 | |||
| JZ535983 | GA | 5 | 1 | |||
| JZ535992 | GA | 5 | 1 | |||
| JZ535812 | AT | 7 | 1 | |||
| JZ535835 | AT | 5 | 1 | |||
| JZ536041 | AT | 5 | 1 | |||
| JZ536078 | TCA | 6 | 1 | |||
| JZ536097 | TCA | 5 | 1 | |||
| JZ535851 | AAT | 8 | 1 | Tri- | ||
| JZ536097 | CAA | 9 | 1 | (6, 27.3%) | ||
| JZ536018 | GAT | 5 | 1 | |||
| JZ535928 | GTG | 6 | 1 | |||
| JZ535875 | AAAC | 5 | 1 | Tetra- | ||
| (1, 4.5%) | ||||||
| Total (%) | - | - | 11 (50%) | 6 (27.3%) | 5 (22.7%) | 22 |
3. Experimental
3.1. Plant Growth Conditions, Treatments and cDNA Library Construction
3.2. cDNA Sequencing Strategy
3.3. Sequence Processing and Analyses
3.4. Functional Annotation and Functional Categorization
3.5. Quantitative RT-PCR Validation of Salt-Related Genes
3.6. Frequency and Distribution of EST-SSRs Found in dbEST Sequences
4. Conclusions
Supplementary Files
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef]
- Benzarti, M.; Ben Rejeb, K.; Debez, A.; Abdelly, C. Environmental and economical opportunities for the valorisation of the genus atriplex: New insights. Grop Improv. 2013, 6, 441–457. [Google Scholar]
- Sakamoto, A.; Murata, N. Genetic engineering of glycinebetaine synthesis in plants current status and implications for enhancement of stress tolerance. J. Exp. Bot. 2000, 51, 81–88. [Google Scholar] [CrossRef]
- Mohanty, A.; Kathuria, H.; Ferjani, A.; Sakamoto, A.; Mohanty, P.; Murata, N.; Tyagi, A.K. Transgenics of an elite indica rice variety Pusa Basmati 1 harbouring the codA gene are highly tolerant to salt stress. Theor. Appl. Genet. 2002, 106, 51–57. [Google Scholar]
- Bohnert, H.J.; Ayoubi, P.; Borchert, C.; Bressan, R.A.; Burnap, R.L.; Cushman, J.C.; Cushman, M.A.; Deyholos, M.; Fischer, R.; Galbraith, D.W.; et al. A genomics approach towards salt stress tolerance. Plant Physiol. Biochem. 2001, 39, 295–311. [Google Scholar] [CrossRef]
- Wang, Y.; Chu, Y.; Liu, G.; Wang, M.H.; Jiang, J.; Hou, Y.; Qu, G.; Yang, C. Identification of expressed sequence tags in an alkali grass (Puccinellia tenuiflora) cDNA library. J. Plant Physiol. 2007, 164, 78–89. [Google Scholar] [CrossRef]
- Shavrukov, Y. Salt stress or salt shock: which genes are we studying? J. Exp. Bot. 2013, 64, 119–127. [Google Scholar] [CrossRef]
- Iturriaga, G.; Cushman, M.A.F.; Cushman, J.C. An EST catalogue from the resurrection plant Selaginella lepidophylla reveals abiotic stress-adaptive genes. Plant Sci. 2006, 170, 1173–1184. [Google Scholar] [CrossRef]
- Swarbreck, S.M.; Lindquist, E.A.; Ackerly, D.D.; Andersen, G.L. Analysis of leaf and root transcriptomes of soil-grown Avena barbata plants. Plant Cell Physiol. 2011, 52, 317–332. [Google Scholar]
- Zhang, L.; Ma, X.L.; Zhang, Q.; Ma, C.L.; Wang, P.-P.; Sun, Y.F.; Zhao, Y.-X.; Zhang, H. Expressed sequence tags from a NaCl treated Suaeda Salsa cDNA library. Gene 2001, 267, 193–200. [Google Scholar] [CrossRef]
- Wang, Z.L.; Li, P.H.; Fredricksen, M.; Gong, Z.Z.; Kim, C.S.; Zhang, C.; Bohnert, H.J.; Zhu, J.K.; Bressan, R.A.; Hasegawa, P.M.; et al. Expressed sequence tags from Thellungiella halophila, a new model to study plant salt-tolerance. Plant Sci. 2004, 166, 609–616. [Google Scholar] [CrossRef]
- Kore-eda, S.; Cushman, M.A.; Akselrod, I.; Bufford, D.; Fredrickson, M.; Clark, E.; Cushman, J.C. Transcript profiling of salinity stress responses by large-scale expressed sequence tag analysis in Mesembryanthemum crystallinum. Gene 2004, 341, 83–92. [Google Scholar] [CrossRef]
- Mehta, P.A.; Sivaprakash, K.; Parani, M.; Venkataraman, G.; Parida, A.K. Generation and analysis of expressed sequence tags from the salt-tolerant mangrove species Avicennia marina (Forsk) Vierh. Theor. Appl. Genet. 2004, 110, 416–424. [Google Scholar]
- Wang, Y.; Ma, H.; Liu, G.; Zhang, D.; Ban, Q.; Zhang, G.; Xu, C.; Yang, C. Generation and analysis of expressed sequence tags from a NaHCO3-treated Limonium bicolor cDNA library. Plant Physiol. Biochem. 2008, 46, 977–986. [Google Scholar] [CrossRef]
- Ohta, M.; Hayashi, Y.; Nakashima, A.; Hamada, A.; Tanaka, A.; Nakamura, T.; Hayakawa, T. Introduction of a Na+/H+ antiporter gene from Atriplex gmeliniconfers salt tolerance to rice. FEBS Lett. 2002, 532, 279–282. [Google Scholar] [CrossRef]
- Shen, Y.G.; Du, B.X.; Zhang, W.K.; Zhang, J.S.; Chen, S.Y. AhCMO, regulated by stresses in Atriplex hortensis, can improve drought tolerance in transgenic tobacco. Theor. Appl. Genet. 2002, 105, 815–821. [Google Scholar] [CrossRef]
- Zhang, H.; Dong, H.; Li, W.; Sun, Y.; Chen, S.; Kong, X. Increased glycine betaine synthesis and salinity tolerance in AhCMO transgenic cotton lines. Mol. Breed. 2009, 23, 289–298. [Google Scholar] [CrossRef]
- Jia, G.X.; Zhu, Z.Q.; Chang, F.Q.; Li, Y.X. Transformation of tomato with the BADH gene from Atriplex improves salt tolerance. Plant Cell Rep. 2002, 21, 141–146. [Google Scholar] [CrossRef]
- Fu, X.; Khan, E.U.; Hu, S.; Fan, Q.; Liu, J. Overexpression of the betaine aldehyde dehydrogenase gene from Atriplex hortensis enhances salt tolerance in the transgenic trifoliate orange (Poncirus trifoliata L. Raf.). Environ. Exp. Bot. 2011, 74, 106–113. [Google Scholar] [CrossRef]
- Shen, Y.G.; Zhang, W.K.; Yan, D.Q.; Du, B.X.; Zhang, J.S.; Liu, Q.; Chen, S.Y. Characterization of a DRE-binding transcription factor from a halophyte Atriplex hortensis. Theor. Appl. Genet. 2003, 107, 155–161. [Google Scholar]
- Wei, W.; Qi, X.; Wang, L.; Zhang, Y.; Hua, W.; Li, D.; Lv, H.; Zhang, X. Characterization of the sesame (Sesamum indicum L.) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers. BMC Genomics 2011. [Google Scholar] [CrossRef]
- Garcia, A.; Benchimol, L.; Barbosa, A.; Geraldi, I.; Souza, C.; Souza, A. Comparison of RAPD, RFLP, AFLP and SSR markers for diversity studies in tropical maize inbred lines. Genet. Mol. Biol. 2004, 27, 579–588. [Google Scholar]
- Rungis, D.; Berube, Y.; Zhang, J.; Ralph, S.; Ritland, C.E.; Ellis, B.E.; Douglas, C.; Bohlmann, J.R.; Ritland, K. Robust simple sequence repeat markers for spruce (Picea spp.) from expressed sequence tags. Theor. Appl. Genet. 2004, 109, 1283–1294. [Google Scholar] [CrossRef]
- Sui, S.; Luo, J.; Ma, J.; Zhu, Q.; Lei, X.; Li, M. Generation and analysis of expressed sequence tags from Chimonanthus praecox (Wintersweet) flowers for discovering stress-responsive and floral development-related genes. Comp. Funct. Genomics 2012. [Google Scholar] [CrossRef]
- Kohler, A.; Delaruelle, C.; Martin, D.; Encelot, N.; Martin, F. The poplar root transcriptome: Analysis of 7000 expressed sequence tags. FEBS Lett. 2003, 542, 37–41. [Google Scholar] [CrossRef]
- Fizames, C.; Munos, S.; Cazettes, C. The arabidopsis root transcriptome by serial analysis of gene expression. Gene identification using the genome sequence. Plant Physiol. 2004, 134, 67–80. [Google Scholar] [CrossRef]
- Poroyko, V.; Hejlek, L.; Spollen, W.; Springer, G.; Nguyen, H.; Sharp, R.; Bohnert, H. The maize root transcriptome by serial analysis of gene expression. Plant Physiol. 2005, 138, 1700–1710. [Google Scholar] [CrossRef]
- Andersen, G.R.; Nissen, P.; Nyborg, J. Elongation factors in protein biosynthesis. Trends Biochem. Sci. 2003, 28, 434–441. [Google Scholar] [CrossRef]
- The Gene Ontology. Available online: http://www.geneontology.org/ (accessed on 8 May 2014).
- Park, J.H.; Liu, L.; Kim, I.H.; Kim, J.H.; You, K.R.; Kim, D.G. Identification of the genes involved in enhanced fenretinide-induced apoptosis by parthenolide in human hepatoma cells. Cancer Res. 2005, 65, 2804–2814. [Google Scholar] [CrossRef]
- Chu, W.; Burns, D.K.; Swerlick, R.A.; Presky, D.H. Identification and characterization of a novel cytokine-inducible nuclear protein from human endothelial cells. J. Biol. Chem. 1995, 270, 10236–10245. [Google Scholar]
- Liu, M.; Shi, J.; Lu, C. Identification of stress-responsive genes in Ammopiptanthus mongolicus using ESTs generated from cold- and drought-stressed seedlings. BMC Plant Biol. 2013, 13, 1471–2229. [Google Scholar]
- Jasoni, R.L.; Cothren, J.T.; Morgan, P.W.; Sohan, D.E. Circadian ethylene production in cotton. Plant Growth Regul. 2002, 36, 127–133. [Google Scholar]
- VecScreen: Screen a Sequence for Vector Contamination. Available online: http://www.ncbi.nlm.nih.gov/tools/vecscreen/ (accessed on 8 May 2014).
- Huang, X.; Madan, A. CAP3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. Available online: http://www.ncbi.nlm.nih.gov (accessed on 8 May 2014).
- Sterky, F.; Regan, S.; Karlsson, J.; Hertzberg, M.; Rohde, A.; Holmberg, A.; Amini, B.; Bhalerao, R.; Larsson, M.; Raimundo, V.; et al. Gene discovery in the wood-forming tissues of poplar: Analysis of 5692 expressed sequence tags. Proc. Natl. Acad. Sci. USA 1998, 95, 13330–13335. [Google Scholar] [CrossRef]
- Arabidopsis Thaliana Project. Available online: http://mips.helmholtz-muenchen.de/plant/athal/ (accessed on 8 May 2014).
- Carbon, S.; Ireland, A.; Mungall, C.J.; Shu, S.; Marshall, B.; Lewis, S. AmiGO: Online access to ontology and annotation data. Bioinformatics 2008, 25, 288–289. [Google Scholar]
- CateGOrizer. Available online: http://www.animalgenome.org/bioinfo/tools/countgo/ (accessed on 8 May 2014).
- Wang, C.; Jing, R.; Mao, X.; Chang, X.; Li, A. TaABC1, a member of the activity of bc1 complex protein kinase family from common wheat, confers enhanced tolerance to abiotic stresses in Arabidopsis. J. Exp. Bot. 2010, 62, 1299–1311. [Google Scholar]
- TM4: Microarray Software Suite. Available online: http://www.tm4.org/mev.html (accessed on 8 May 2014).
- SSRIT-Simple Sequence Repeat Identification Tool. Available online: http://www.gramene.org/db/markers/ssrtool (accessed on 8 May 2014).
- ORF Finder (Open Reading Frame Finder). Available online: http://www.ncbi.nlm.nih.gov/gorf/orfig.cgi (accessed on 8 May 2014).
- Flowers, T.J.; Colmer, T.D. Salinity tolerance in halophytes. New Phytol. 2008, 179, 945–963. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, J.; Sun, X.; Yu, G.; Jia, C.; Liu, J.; Pan, H. Generation and Analysis of Expressed Sequence Tags (ESTs) from Halophyte Atriplex canescens to Explore Salt-Responsive Related Genes. Int. J. Mol. Sci. 2014, 15, 11172-11189. https://doi.org/10.3390/ijms150611172
Li J, Sun X, Yu G, Jia C, Liu J, Pan H. Generation and Analysis of Expressed Sequence Tags (ESTs) from Halophyte Atriplex canescens to Explore Salt-Responsive Related Genes. International Journal of Molecular Sciences. 2014; 15(6):11172-11189. https://doi.org/10.3390/ijms150611172
Chicago/Turabian StyleLi, Jingtao, Xinhua Sun, Gang Yu, Chengguo Jia, Jinliang Liu, and Hongyu Pan. 2014. "Generation and Analysis of Expressed Sequence Tags (ESTs) from Halophyte Atriplex canescens to Explore Salt-Responsive Related Genes" International Journal of Molecular Sciences 15, no. 6: 11172-11189. https://doi.org/10.3390/ijms150611172
APA StyleLi, J., Sun, X., Yu, G., Jia, C., Liu, J., & Pan, H. (2014). Generation and Analysis of Expressed Sequence Tags (ESTs) from Halophyte Atriplex canescens to Explore Salt-Responsive Related Genes. International Journal of Molecular Sciences, 15(6), 11172-11189. https://doi.org/10.3390/ijms150611172