p62/Sequestosome 1 Regulates Aggresome Formation of Pathogenic Ataxin-3 with Expanded Polyglutamine


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
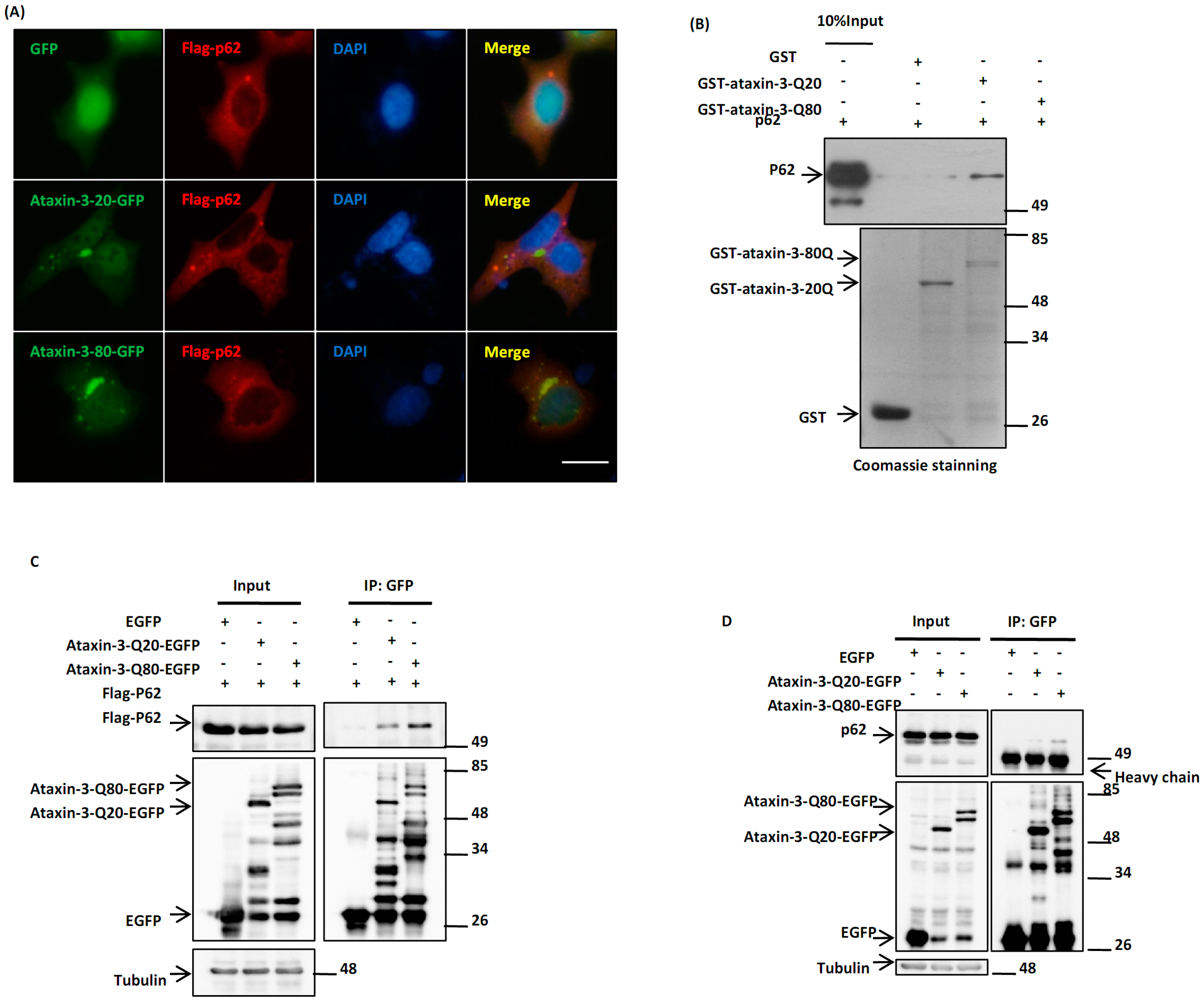
2.1. p62 Directly Interacts with Ataxin-3-Q80

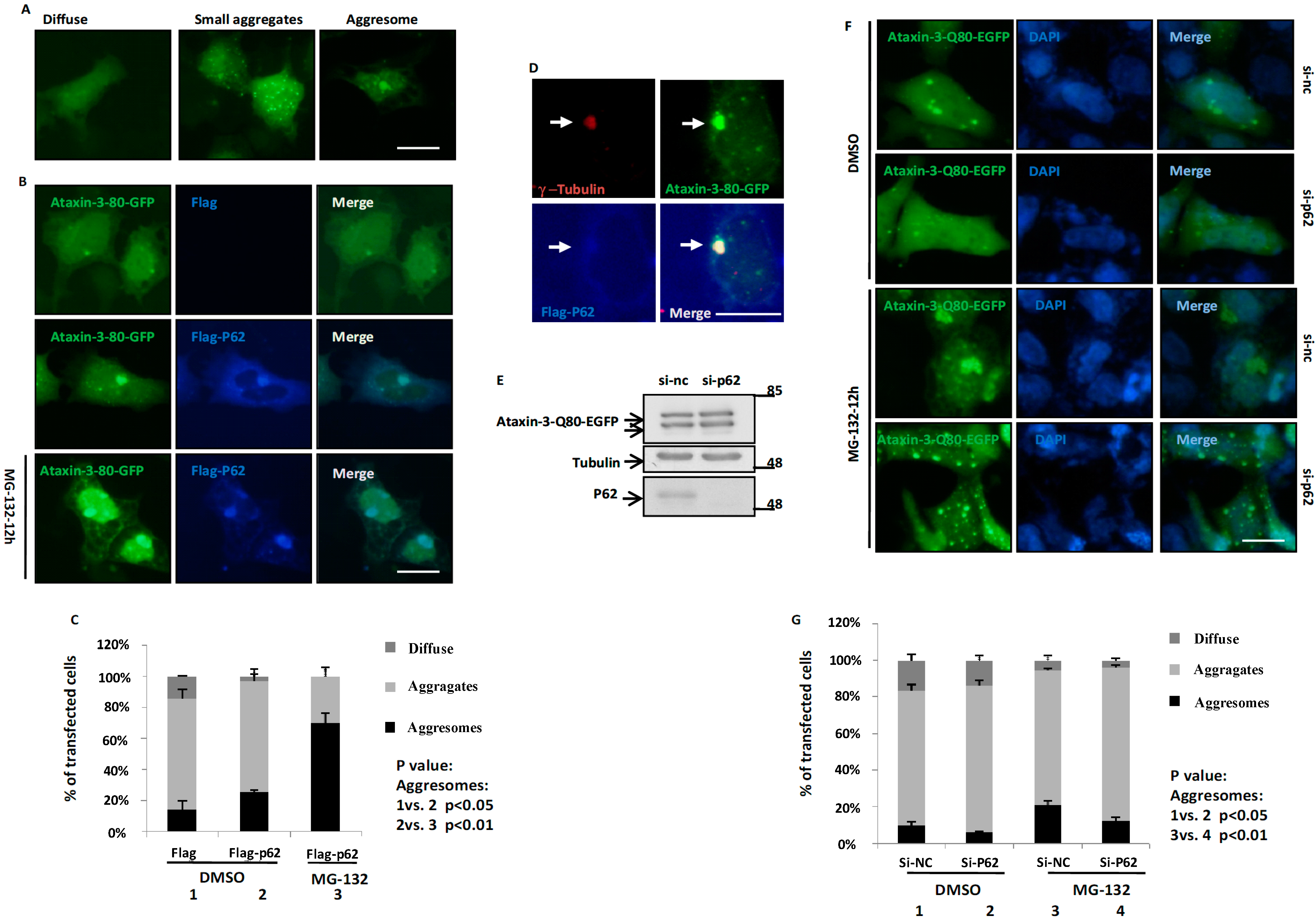
2.2. p62 Promotes the Aggresome Formation of Ataxin-3-Q80
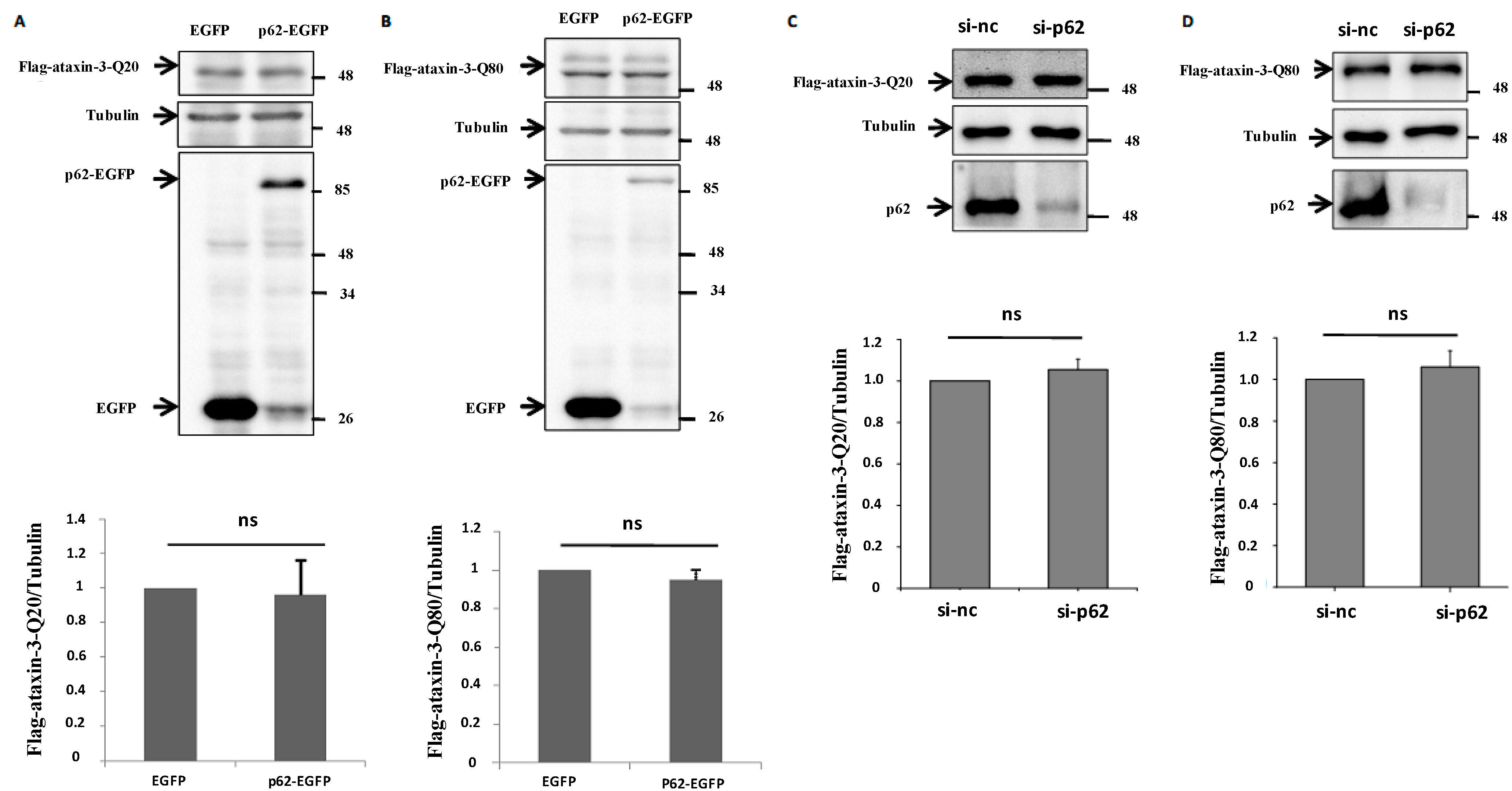
2.3. p62 Has no Effect on the Protein Expression of Ataxin-3
2.4. p62 Promotes the Aggresome Formation of Ataxin-3-80 in a Microtubule-Dependent Manner
2.5. p62 Protects Cells against Ataxin-3-Q80-Induced Cell Death


2.6. Discussion


3. Experimental Section
3.1. Plasmid Construction
3.2. Cell Culture and Transfection
3.3. Fluorescent Microscopy
3.4. Immunoblot and Antibodies
3.5. Immunoprecipitation
3.6. GST Pulldown Assay
3.7. MTT Assay
3.8. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schols, L.; Bauer, P.; Schmidt, T.; Schulte, T.; Riess, O. Autosomal dominant cerebellar ataxias: Clinical features, genetics, and pathogenesis. Lancet Neurol. 2004, 3, 291–304. [Google Scholar] [CrossRef]
- Tang, B.; Liu, C.; Shen, L.; Dai, H.; Pan, Q.; Jing, L.; Ouyang, S.; Xia, J. Frequency of sca1, sca2, sca3/mjd, sca6, sca7, and drpla cag trinucleotide repeat expansion in patients with hereditary spinocerebellar ataxia from chinese kindreds. Arch. Neurol. 2000, 57, 540–544. [Google Scholar] [CrossRef]
- Paulson, H. Dominantly inherited ataxias: Lessons learned from machado-joseph disease/spinocerebellar ataxia type 3. Semin. Neurol. 2007, 27, 133–142. [Google Scholar] [CrossRef]
- Perez, M.K.; Paulson, H.L.; Pendse, S.J.; Saionz, S.J.; Bonini, N.M.; Pittman, R.N. Recruitment and the role of nuclear localization in polyglutamine-mediated aggregation. J. Cell Biol. 1998, 143, 1457–1470. [Google Scholar] [CrossRef]
- Klement, I.A.; Skinner, P.J.; Kaytor, M.D.; Yi, H.; Hersch, S.M.; Clark, H.B.; Zoghbi, H.Y.; Orr, H.T. Ataxin-1 nuclear localization and aggregation: Role in polyglutamine-induced disease in sca1 transgenic mice. Cell 1998, 95, 41–53. [Google Scholar] [CrossRef]
- Bhattacharyya, N.P.; Banerjee, M.; Majumder, P. Huntington’s disease: Roles of huntingtin-interacting protein 1 (hip-1) and its molecular partner hippi in the regulation of apoptosis and transcription. FEBS J. 2008, 275, 4271–4279. [Google Scholar] [CrossRef]
- Williams, A.J.; Paulson, H.L. Polyglutamine neurodegeneration: Protein misfolding revisited. Trends Neurosci. 2008, 31, 521–528. [Google Scholar] [CrossRef]
- Perez, M.K.; Paulsonl, H.L.; Pittman, R.N. Ataxin-3 with an altered conformation that exposes the polyglutamine domain is associated with the nuclear matrix. Hum. Mol. Genet. 1999, 8, 2377–2385. [Google Scholar] [CrossRef]
- Ying, Z.; Wang, H.; Fan, H.; Zhu, X.; Zhou, J.; Fei, E.; Wang, G. Gp78, an er associated e3, promotes sod1 and ataxin-3 degradation. Hum. Mol. Genet. 2009, 18, 4268–4281. [Google Scholar] [CrossRef]
- Seidel, K.; den Dunnen, W.F.; Schultz, C.; Paulson, H.; Frank, S.; de Vos, R.A.; Brunt, E.R.; Deller, T.; Kampinga, H.H.; Rub, U. Axonal inclusions in spinocerebellar ataxia type 3. Acta Neuropathol. 2010, 120, 449–460. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Wyttenbach, A.; Rankin, J. Intracellular inclusions, pathological markers in diseases caused by expanded polyglutamine tracts? J. Med. Genet. 1999, 36, 265–270. [Google Scholar]
- Burnett, B.; Li, F.; Pittman, R.N. The polyglutamine neurodegenerative protein ataxin-3 binds polyubiquitylated proteins and has ubiquitin protease activity. Hum. Mol. Genet. 2003, 12, 3195–3205. [Google Scholar] [CrossRef]
- Johnston, J.A.; Ward, C.L.; Kopito, R.R. Aggresomes: A cellular response to misfolded proteins. J. Cell Biol. 1998, 143, 1883–1898. [Google Scholar] [CrossRef]
- Kopito, R.R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000, 10, 524–530. [Google Scholar] [CrossRef]
- Wang, H.; Ying, Z.; Wang, G. Ataxin-3 regulates aggresome formation of copper-zinc superoxide dismutase (sod1) by editing k63-linked polyubiquitin chains. J. Biol. Chem. 2012, 287, 28576–28585. [Google Scholar] [CrossRef]
- Olanow, C.W.; Perl, D.P.; DeMartino, G.N.; McNaught, K.S. Lewy-body formation is an aggresome-related process: A hypothesis. Lancet Neurol. 2004, 3, 496–503. [Google Scholar] [CrossRef]
- Waelter, S.; Boeddrich, A.; Lurz, R.; Scherzinger, E.; Lueder, G.; Lehrach, H.; Wanker, E.E. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol. Biol. Cell 2001, 12, 1393–1407. [Google Scholar] [CrossRef]
- Tanaka, M.; Kim, Y.M.; Lee, G.; Junn, E.; Iwatsubo, T.; Mouradian, M.M. Aggresomes formed by alpha-synuclein and synphilin-1 are cytoprotective. J. Biol. Chem. 2004, 279, 4625–4631. [Google Scholar]
- Lim, K.L.; Chew, K.C.; Tan, J.M.; Wang, C.; Chung, K.K.; Zhang, Y.; Tanaka, Y.; Smith, W.; Engelender, S.; Ross, C.A.; et al. Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: Implications for lewy body formation. J. Neurosci. 2005, 25, 2002–2009. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Li, L.; Chudaev, M.V.; Chen, J.; Perez, F.A.; Palmiter, R.D.; Chin, L.S. Parkin-mediated k63-linked polyubiquitination targets misfolded dj-1 to aggresomes via binding to hdac6. J. Cell Biol. 2007, 178, 1025–1038. [Google Scholar] [CrossRef]
- Gamerdinger, M.; Kaya, A.M.; Wolfrum, U.; Clement, A.M.; Behl, C. Bag3 mediates chaperone-based aggresome-targeting and selective autophagy of misfolded proteins. EMBO Rep. 2011, 12, 149–156. [Google Scholar] [CrossRef]
- Gal, J.; Strom, A.L.; Kilty, R.; Zhang, F.; Zhu, H. P62 accumulates and enhances aggregate formation in model systems of familial amyotrophic lateral sclerosis. J. Biol. Chem. 2007, 282, 11068–11077. [Google Scholar] [CrossRef]
- Gal, J.; Strom, A.L.; Kwinter, D.M.; Kilty, R.; Zhang, J.; Shi, P.; Fu, W.; Wooten, M.W.; Zhu, H. Sequestosome 1/p62 links familial als mutant sod1 to lc3 via an ubiquitin-independent mechanism. J. Neurochem. 2009, 111, 1062–1073. [Google Scholar] [CrossRef]
- Homma, T.; Ishibashi, D.; Nakagaki, T.; Satoh, K.; Sano, K.; Atarashi, R.; Nishida, N. Increased expression of p62/sqstm1 in prion diseases and its association with pathogenic prion protein. Sci. Rep. 2014, 4, 4504. [Google Scholar] [CrossRef]
- Bjorkoy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. P62/sqstm1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef]
- Fujita, K.; Maeda, D.; Xiao, Q.; Srinivasula, S.M. Nrf2-mediated induction of p62 controls toll-like receptor-4-driven aggresome-like induced structure formation and autophagic degradation. Proc. Natl. Acad. Sci. USA 2011, 108, 1427–1432. [Google Scholar] [CrossRef]
- Watanabe, Y.; Tanaka, M. P62/sqstm1 in autophagic clearance of a non-ubiquitylated substrate. J. Cell Sci. 2011, 124, 2692–2701. [Google Scholar] [CrossRef]
- Babu, J.R.; Geetha, T.; Wooten, M.W. Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J. Neurochem. 2005, 94, 192–203. [Google Scholar] [CrossRef]
- Bevivino, A.E. An expanded glutamine repeat destabilizes native ataxin-3 structure and mediates formation of parallel beta-fibrils. Proc. Natl. Acad. Sci. USA 2001, 98, 11955–11960. [Google Scholar] [CrossRef]
- Apicella, A.; Natalello, A.; Frana, A.M.; Baserga, A.; Casari, C.S.; Bottani, C.E.; Doglia, S.M.; Tortora, P.; Regonesi, M.E. Temperature profoundly affects ataxin-3 fibrillogenesis. Biochimie 2012, 94, 1026–1031. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The deacetylase hdac6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef]
- Ouellet, J.; Barral, Y. Organelle segregation during mitosis: Lessons from asymmetrically dividing cells. J. Cell Biol. 2012, 196, 305–313. [Google Scholar] [CrossRef]
- Burnett, B.G.; Pittman, R.N. The polyglutamine neurodegenerative protein ataxin 3 regulates aggresome formation. Proc. Natl. Acad. Sci. USA 2005, 102, 4330–4335. [Google Scholar] [CrossRef]
- Taylor, J.P.; Tanaka, F.; Robitschek, J.; Sandoval, C.M.; Taye, A.; Markovic-Plese, S.; Fischbeck, K.H. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum. Mol. Genet. 2003, 12, 749–757. [Google Scholar] [CrossRef]
- Smith, W.W.; Liu, Z.; Liang, Y.; Masuda, N.; Swing, D.A.; Jenkins, N.A.; Copeland, N.G.; Troncoso, J.C.; Pletnikov, M.; Dawson, T.M.; et al. Synphilin-1 attenuates neuronal degeneration in the a53t alpha-synuclein transgenic mouse model. Hum. Mol. Genet. 2010, 19, 2087–2098. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. Opinion: What is the role of protein aggregation in neurodegeneration? Nat. Rev. Mol. Cell Biol. 2005, 6, 891–898. [Google Scholar] [CrossRef]
- Zaarur, N.; Meriin, A.B.; Gabai, V.L.; Sherman, M.Y. Triggering aggresome formation. Dissecting aggresome-targeting and aggregation signals in synphilin 1. J. Biol. Chem. 2008, 283, 27575–27584. [Google Scholar] [CrossRef]
- Uchihara, T.; Fujigasaki, H.; Koyano, S.; Nakamura, A.; Yagishita, S.; Iwabuchi, K. Non-expanded polyglutamine proteins in intranuclear inclusions of hereditary ataxias—Triple-labeling immunofluorescence study. Acta Neuropathol. 2001, 102, 149–152. [Google Scholar]
- Wang, Q.; Li, L.; Ye, Y. Regulation of retrotranslocation by p97-associated deubiquitinating enzyme ataxin-3. J. Cell Biol. 2006, 174, 963–971. [Google Scholar] [CrossRef]
- Zhong, X.; Pittman, R.N. Ataxin-3 binds vcp/p97 and regulates retrotranslocation of erad substrates. Hum. Mol. Genet. 2006, 15, 2409–2420. [Google Scholar] [CrossRef]
- Wang, H.; Jia, N.; Fei, E.; Wang, Z.; Liu, C.; Zhang, T.; Fan, J.; Wu, M.; Chen, L.; Nukina, N.; et al. P45, an atpase subunit of the 19s proteasome, targets the polyglutamine disease protein ataxin-3 to the proteasome. J. Neurochem. 2007, 101, 1651–1661. [Google Scholar] [CrossRef]
- Matsumoto, M.; Yada, M.; Hatakeyama, S.; Ishimoto, H.; Tanimura, T.; Tsuji, S.; Kakizuka, A.; Kitagawa, M.; Nakayama, K.I. Molecular clearance of ataxin-3 is regulated by a mammalian e4. EMBO J. 2004, 23, 659–669. [Google Scholar] [CrossRef]
- Boeddrich, A.; Gaumer, S.; Haacke, A.; Tzvetkov, N.; Albrecht, M.; Evert, B.O.; Muller, E.C.; Lurz, R.; Breuer, P.; Schugardt, N.; et al. An arginine/lysine-rich motif is crucial for vcp/p97-mediated modulation of ataxin-3 fibrillogenesis. EMBO J. 2006, 25, 1547–1558. [Google Scholar] [CrossRef]
- Duran, A.; Amanchy, R.; Linares, J.F.; Joshi, J.; Abu-Baker, S.; Porollo, A.; Hansen, M.; Moscat, J.; Diaz-Meco, M.T. P62 is a key regulator of nutrient sensing in the mtorc1 pathway. Mol. Cell 2011, 44, 134–146. [Google Scholar] [CrossRef]
- Chen, L.; Xu, B.; Liu, L.; Luo, Y.; Zhou, H.; Chen, W.; Shen, T.; Han, X.; Kontos, C.D.; Huang, S. Cadmium induction of reactive oxygen species activates the mtor pathway, leading to neuronal cell death. Free Radic. Biol. Med. 2011, 50, 624–632. [Google Scholar] [CrossRef]
- Chen, L.; Xu, B.; Liu, L.; Luo, Y.; Yin, J.; Zhou, H.; Chen, W.; Shen, T.; Han, X.; Huang, S. Hydrogen peroxide inhibits mtor signaling by activation of ampkalpha leading to apoptosis of neuronal cells. Lab. Investig. 2010, 90, 762–773. [Google Scholar] [CrossRef]
- Xu, B.; Chen, S.; Luo, Y.; Chen, Z.; Liu, L.; Zhou, H.; Chen, W.; Shen, T.; Han, X.; Chen, L.; et al. Calcium signaling is involved in cadmium-induced neuronal apoptosis via induction of reactive oxygen species and activation of mapk/mtor network. PLoS One 2011, 6, e19052. [Google Scholar] [CrossRef]
- Ren, H.; Fu, K.; Mu, C.; Li, B.; Wang, D.; Wang, G. Dj-1, a cancer and parkinson’s disease associated protein, regulates autophagy through jnk pathway in cancer cells. Cancer Lett. 2010, 297, 101–108. [Google Scholar] [CrossRef]
- Ying, Z.; Wang, H.; Fan, H.; Wang, G. The endoplasmic reticulum (er)-associated degradation system regulates aggregation and degradation of mutant neuroserpin. J. Biol. Chem. 2011, 286, 20835–20844. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhou, L.; Wang, H.; Chen, D.; Gao, F.; Ying, Z.; Wang, G. p62/Sequestosome 1 Regulates Aggresome Formation of Pathogenic Ataxin-3 with Expanded Polyglutamine. Int. J. Mol. Sci. 2014, 15, 14997-15010. https://doi.org/10.3390/ijms150914997
Zhou L, Wang H, Chen D, Gao F, Ying Z, Wang G. p62/Sequestosome 1 Regulates Aggresome Formation of Pathogenic Ataxin-3 with Expanded Polyglutamine. International Journal of Molecular Sciences. 2014; 15(9):14997-15010. https://doi.org/10.3390/ijms150914997
Chicago/Turabian StyleZhou, Liang, Hongfeng Wang, Dong Chen, Feng Gao, Zheng Ying, and Guanghui Wang. 2014. "p62/Sequestosome 1 Regulates Aggresome Formation of Pathogenic Ataxin-3 with Expanded Polyglutamine" International Journal of Molecular Sciences 15, no. 9: 14997-15010. https://doi.org/10.3390/ijms150914997
APA StyleZhou, L., Wang, H., Chen, D., Gao, F., Ying, Z., & Wang, G. (2014). p62/Sequestosome 1 Regulates Aggresome Formation of Pathogenic Ataxin-3 with Expanded Polyglutamine. International Journal of Molecular Sciences, 15(9), 14997-15010. https://doi.org/10.3390/ijms150914997