1. Introduction
Skeletal muscle, which composes approximately half of total body mass, is an important tissue involved in regulating the metabolism, locomotion and strength of the animal body [
1]. The formation of skeletal muscle requires the mononucleated myoblasts to withdraw from the cell cycle and to fuse with each other to form nascent, multinucleated myotubes. Then, the nascent myotubes undergo further cell fusion and express contractile proteins to form mature myotubes. The fusion of myoblasts is a fundamental step during muscle differentiation, and this step involves several cellular and molecular behaviours, such as cell migration, recognition, adhesion, membrane alignment, signalling transduction and actin cytoskeletal reorganization, leading up to the final membrane fusion [
2]. Many of these cellular and molecular events are conserved in vertebrates [
3]; therefore, studies in flies, zebrafish, mice and other vertebrate model systems have provided a clearer understanding about these events [
3,
4].
Myoblast fusion is a highly regulated process. Recent advances in this field have revealed many molecules and signalling pathways that are involved in this process [
3]. Among these regulatory molecules, transmembrane proteins, which are a type of membrane protein that spans the entirety of the biological membrane, play important roles during myoblast fusion. Many cellular events, such as cell migration, recognition and adhesion, require this type of protein to complete the fusion process. Myoferlin, a transmembrane protein that is expressed at apposed membranes sites undergoing fusion, can bind to phospholipids in a calcium-sensitive manner [
5]. A mutation in myoferlin C2A can disrupt this binding and decrease the fusion efficiency of large myotubes [
5]. As a type I transmembrane protein, the mannose receptor is also required for the fusion of myoblasts due to its role in directing myoblast migration [
6]. In
Drosophila, direct evidence suggests that the fusion of myoblasts are dependent on transmembrane proteins of the immunoglobulin superfamily, which include Kin of IrreC/Dumbfounded (Kirre/Duf) [
7], Roughest/Irregular-optic chiasma C (Rst/Irre-C) [
8], Hibris (Hbs) and Sticks-and-stones (Sns) [
9,
10]. However, none of the above proteins is muscle-specific, and many of them have redundant roles during myoblast fusion. Therefore, a muscle-specific transmembrane protein with a direct and essential role in myoblast fusion remains an attractive target for discovery.
Recently, the muscle-specific transmembrane protein transmembrane protein 8c (Tmem8c), also called Myomaker, was found to be necessary for myoblast fusion [
11]. During myogenesis and muscle regeneration, Myomaker is expressed transiently and promotes myoblast fusion efficiently [
11,
12].
Myomaker genetic disruption in mice not only completely abolishes muscle regeneration by adult satellite cells but also causes perinatal death of embryos due to a complete block of myoblast fusion. The protein sequence of Myomaker is highly conserved across vertebrate organisms [
11], and its function in myogenesis is conserved between mice and zebrafish [
13]. However, the expression pattern and function of Myomaker in avian myogenesis have not been explored, and the cellular mechanism of its function and the regulatory mechanism of its expression during myogenesis remain to be determined. MYOG and MYOD are critical transcription factors in myogenesis and can regulate the transcription of most of the muscle-specific genes [
14,
15,
16,
17]. Both of them play an important role in the regulation of myoblast differentiation.
MYOD act as a myogenic determination gene [
15], whereas
MYOG is essential for the terminal differentiation of committed myoblasts [
17]. Here, we found the regulatory role of MYOG and MYOD in the transcription of
Myomaker, and report the expression pattern of these genes during chicken embryonic skeletal muscle development and the differentiation of primary myoblast. Myomaker function in chicken myoblast fusion was explored by overexpression and loss-of-function assays. In addition, we analysed the mRNA expression patterns of
MYOD,
MYOG and
Myomaker and found that MYOD and MYOG can bind directly to the promoter of
Myomaker and induce its transcription during myoblast fusion. Finally, to understand the post-transcriptional regulation of
Myomaker expression, we analysed the 3ʹ UTR of
Myomaker and found that miR-140-3p can inhibit
Myomaker expression by binding to the
Myomaker 3ʹ UTR
in vitro. miR-140-3p overexpression inhibited the late stage of myoblast differentiation but promoted myoblast proliferation. Collectively, our results confirmed the important roles of Myomaker in avian myoblast fusion and found that MYOD, MYOG and miR-140-3p could regulate
Myomaker expression.
3. Discussion
In mice and zebrafish, Myomaker is a muscle-specific transmembrane protein with important roles in promoting myoblast fusion [
11,
13]. However, its roles in avians have not yet been elucidated. In this study, we first confirmed the important roles of
Myomaker in chicken myoblast fusion and identified some of the regulatory mechanisms of its expression during myoblast fusion. Importantly, this study is the first to demonstrate that miR-140-3p can target inhibit
Myomaker expression during myoblast differentiation. These findings not only provide evidence for the function and regulation of
Myomaker during chicken myoblast fusion but also provide insight regarding the regulators and biological roles of
Myomaker, which is essential for muscle formation and regeneration.
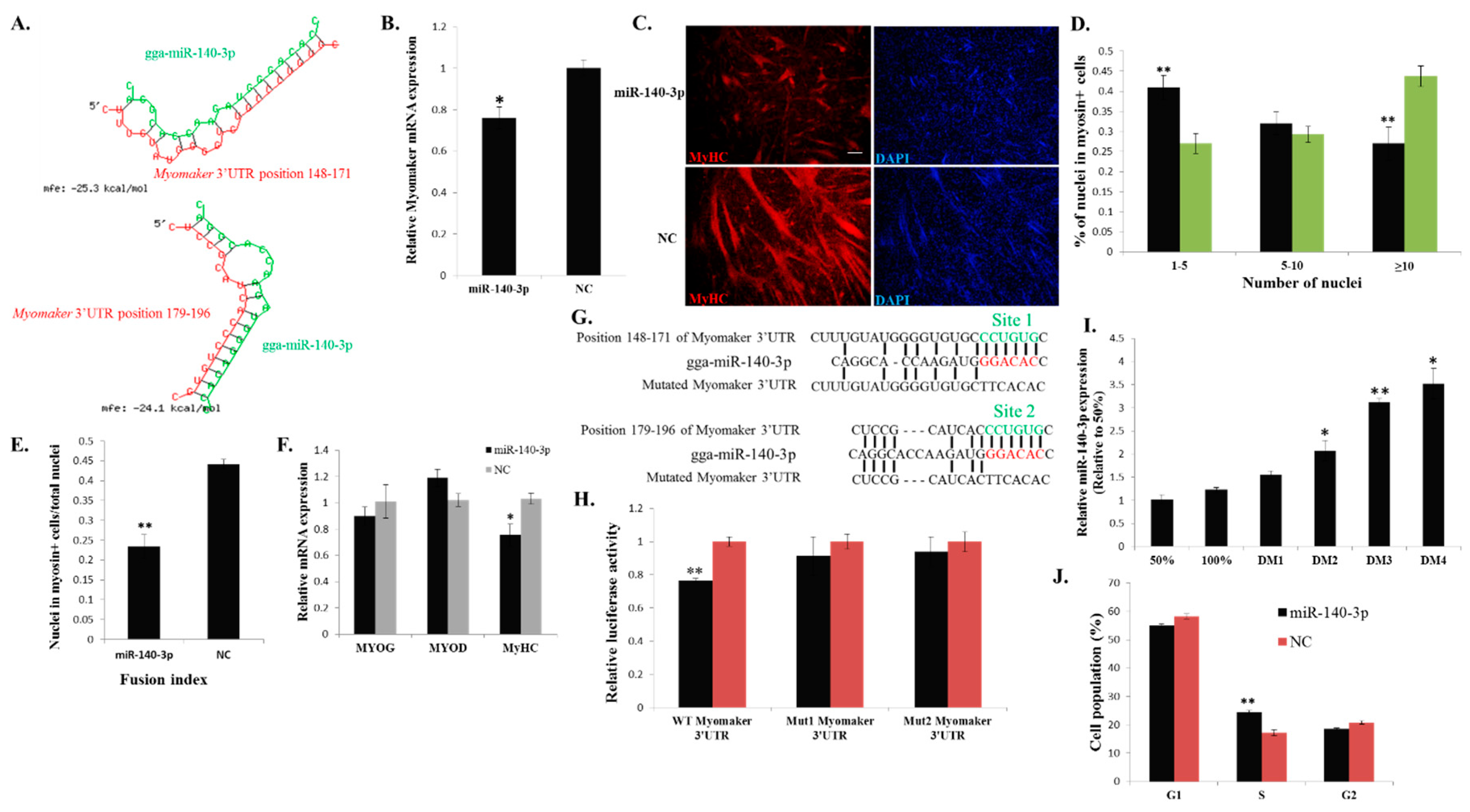
Figure 6.
miR-140-3p directly binds to the 3ʹ UTR of Myomaker and inhibits Myomaker expression and myoblast fusion. (A) Schematic representation of the duplexes of miR-140-3p and the Myomaker 3ʹ UTR target region; (B) Myomaker mRNA expression after miR-140-3p mimic or NC duplexes transfection into DM2 myoblasts; (C) MyHC immunostaining of primary myoblasts transduced with miR-140-3p mimic or NC duplexes and differentiated for 48 h. Bar, 100 µm; (D) The distribution of myonuclear content in the cells with transfection of miR-140-3p mimic or NC duplexes; (E) The fusion index of the cells transfected with miR-140-3p mimic or NC duplexes; (F) The mRNA expression of MYOG, MYOD and MyHC after transfection of miR-140-3p mimic or NC duplexes in DM2 myoblasts; (G) Schematic representation of the predicted binding sites (green) and mutated sites of miR-140-3p in the 3ʹ UTR of Myomaker; (H) Myomaker 3ʹ UTR wild-type or mutant luciferase reporters were transfected into DF-1 cells overexpressing miR-140-3p mimic or NC duplexes. Luciferase activity was determined at 36 h after transfection; (I) miR-140-3p expression during myoblast differentiation; (J) Cell cycle analysis of myoblasts at 36 h after miR-140-3p mimic or NC duplexes transfection. The results are shown as the mean ±S.E.M. of at least three cultures (n = 3 cultures in D, E and J; n = 4 cultures in B, F, H and I). One-sample t test was used to assess the change from each data point to the control (50% in I). * p < 0.05; ** p < 0.01.
Figure 6.
miR-140-3p directly binds to the 3ʹ UTR of Myomaker and inhibits Myomaker expression and myoblast fusion. (A) Schematic representation of the duplexes of miR-140-3p and the Myomaker 3ʹ UTR target region; (B) Myomaker mRNA expression after miR-140-3p mimic or NC duplexes transfection into DM2 myoblasts; (C) MyHC immunostaining of primary myoblasts transduced with miR-140-3p mimic or NC duplexes and differentiated for 48 h. Bar, 100 µm; (D) The distribution of myonuclear content in the cells with transfection of miR-140-3p mimic or NC duplexes; (E) The fusion index of the cells transfected with miR-140-3p mimic or NC duplexes; (F) The mRNA expression of MYOG, MYOD and MyHC after transfection of miR-140-3p mimic or NC duplexes in DM2 myoblasts; (G) Schematic representation of the predicted binding sites (green) and mutated sites of miR-140-3p in the 3ʹ UTR of Myomaker; (H) Myomaker 3ʹ UTR wild-type or mutant luciferase reporters were transfected into DF-1 cells overexpressing miR-140-3p mimic or NC duplexes. Luciferase activity was determined at 36 h after transfection; (I) miR-140-3p expression during myoblast differentiation; (J) Cell cycle analysis of myoblasts at 36 h after miR-140-3p mimic or NC duplexes transfection. The results are shown as the mean ±S.E.M. of at least three cultures (n = 3 cultures in D, E and J; n = 4 cultures in B, F, H and I). One-sample t test was used to assess the change from each data point to the control (50% in I). * p < 0.05; ** p < 0.01.
![Ijms 16 25946 g006]()
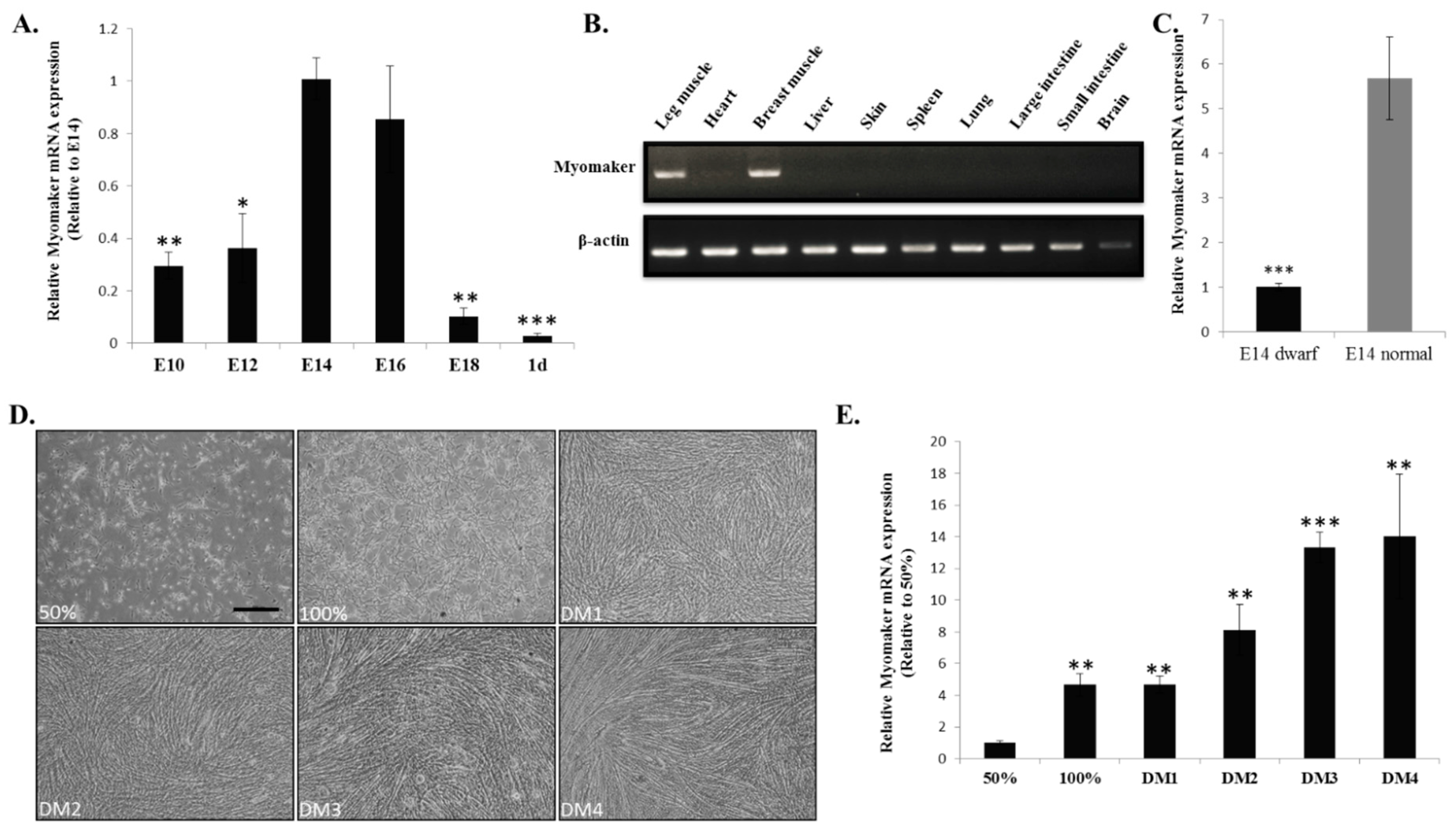
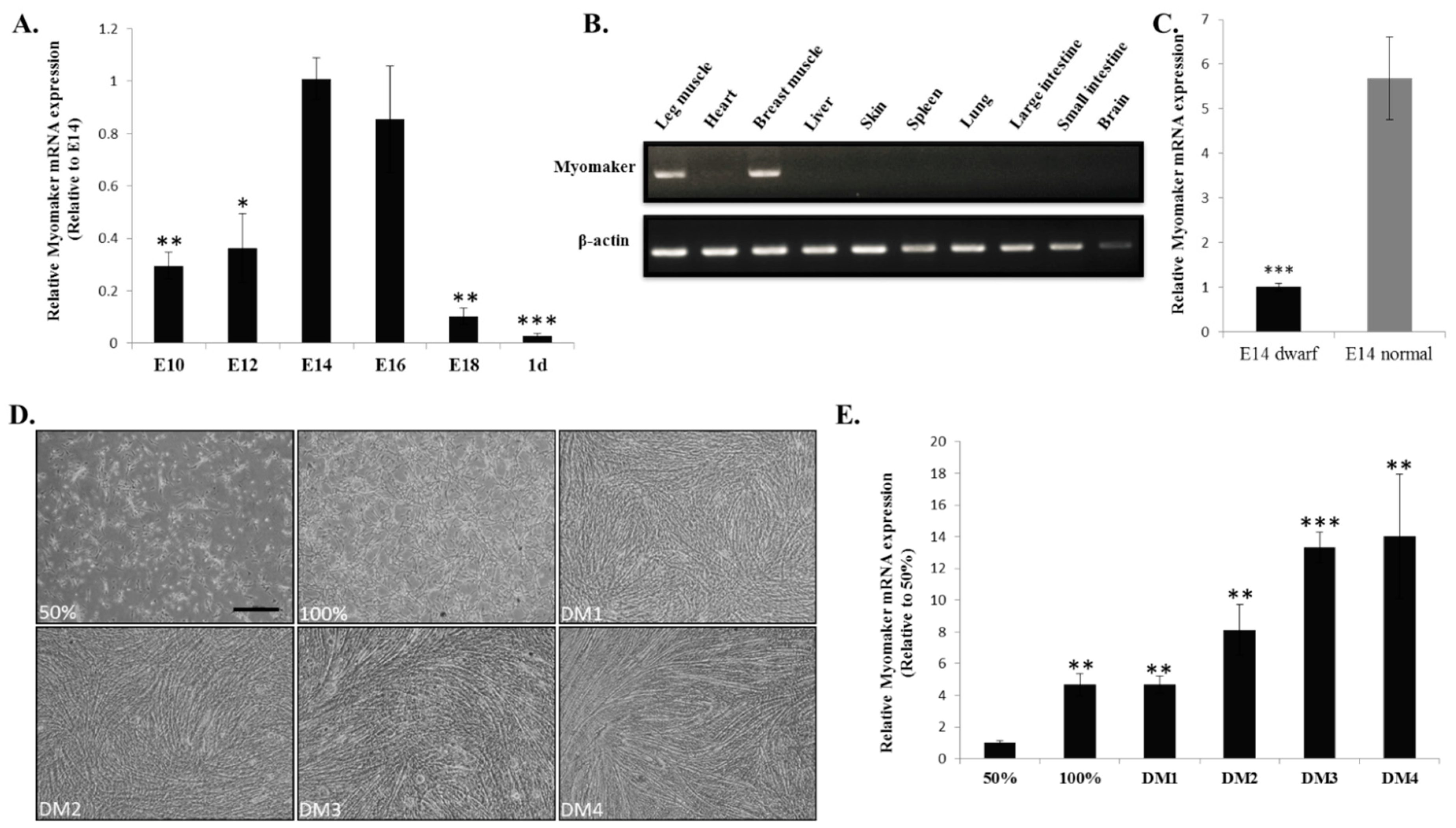
Our previous work demonstrated that E10-E16 is an important period of chicken muscle fibre formation [
23]. In this study, we found that
Myomaker mRNA expression is up-regulated during this period. The up-regulated expression of
Myomaker may contribute to the promotion of myoblast fusion and muscle fibre formation. Studies show that the phenotype of sex-linked dwarf chicken is a result from a mutation of the
GHR gene [
24], and the mutation can lead to a decrease in muscle fibers number and fiber diameter [
18]. To understand whether
Myomaker is involve in the regulatory network of the
GHR mutation induced muscle development difference, we tested its expression between the muscle of E14 dwarf and normal chickens. The results of reduced
Myomaker mRNA expression in dwarf chickens suggesting that
Myomaker may communicate with
GHR gene by direct or indirect signalling pathway. Additionally, lower
Myomaker expression in dwarf chickens may also result in the decrease of myoblast fusion during chicken muscle development. Therefore,
Myomaker expression is important for chicken skeletal muscle development, and the
Myomaker gene can be considered a candidate gene for molecular breeding in broilers.
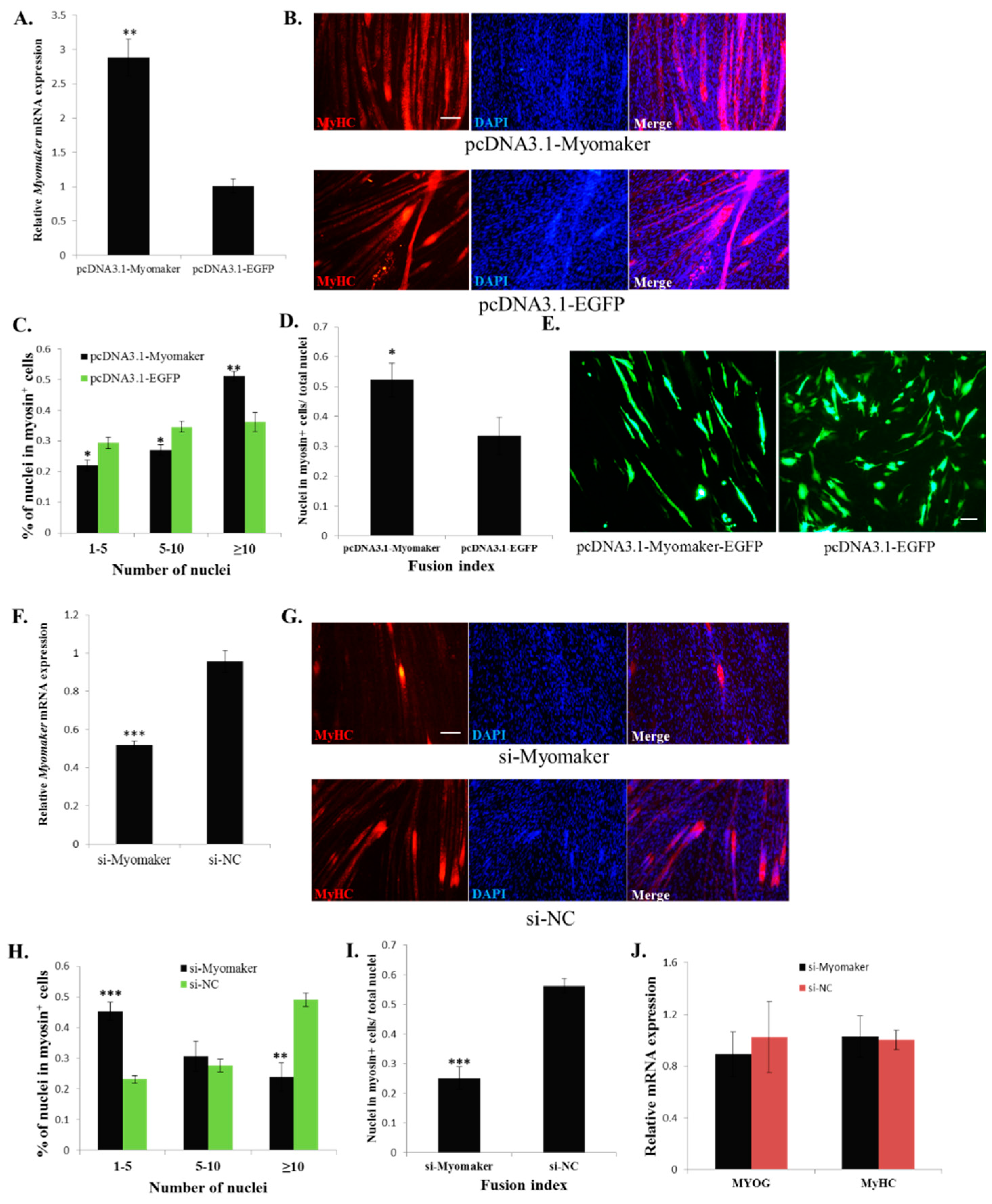
Myomaker is a transmembrane protein. Previous studies have demonstrated that some transmembrane proteins can regulate myoblast fusion by influencing cell migration [
6], recognition and adhesion [
5,
9,
10], which are important processes during myoblast fusion [
3]. Therefore, we examined whether Myomaker could regulate myoblast migration (
Supplementary File 3). Results from Classic scratch and Transwell migration assays indicated that
Myomaker had no significant effect on cell migration. Additionally, in
Myomaker knockout mice, myoblasts are present in embryonic limbs [
11], suggesting that
Myomaker null myoblasts can migrate from somites to the limbs. Therefore, the above results indicate that Myomaker may be not a regulator of myoblast migration. Recently, a cell surface protein BAI3 was found to interact with ELMO and promote myoblast fusion by the ELMO-Dock1-Rac1 pathway in chick embryos [
21]. This BAI3-ELMO-Dock1-Rac1 pathway is able to regulate the downstream actin cytoskeleton network, which plays an essential role during myoblast fusion [
3]. As the actin cytoskeleton is also critical for Myomaker-induced myoblast fusion [
11], it is possible that Myomaker can crosstalk with the pathway of BAI3-ELMO-Dock1-Rac1 to regulate myoblast fusion [
21]. However, no related investigations have been performed to illustrate this interaction. The precise mechanism by which Myomaker regulates myoblast fusion still requires further exploration.
Although the function of Myomaker in myoblast fusion has been clearly demonstrated, the regulatory mechanism of
Myomaker expression remains unknown. Myogenic regulatory factors (MRFs), which include MYOD, MYF5, MRF4 and MYOG, are able to activate many downstream genes to initiate muscle cell differentiation [
14,
15]. However, the specific roles of these factors are different.
MYF5 and
MYOD act as myogenic determination genes, whereas
MYOG is essential for the terminal differentiation of committed myoblasts [
15,
16,
17].
MRF4 seems to have both of these roles [
25]. In this study, we tested the function of
MYOG and
MYOD, which are transcriptional factors that play essential roles in muscle-specific gene transcription [
20,
26], in the regulation of
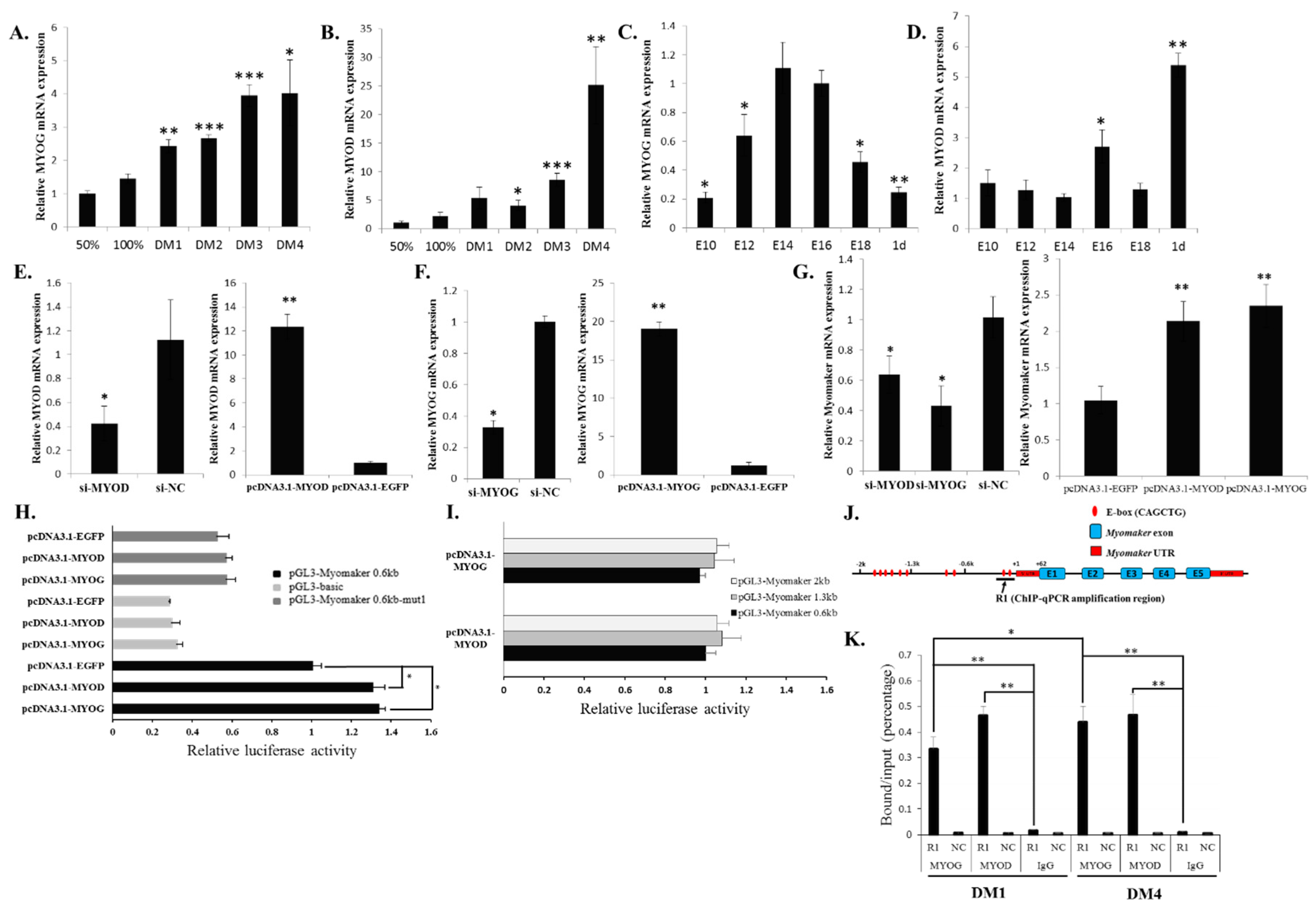
Myomaker transcription. Our results confirmed that both MYOG and MYOD can directly bind and activate
Myomaker expression. However, in the luciferase assay, the luciferase activity of the 0.6-kb promoter only increased about 1.4 fold when overexpressing
MYOD and
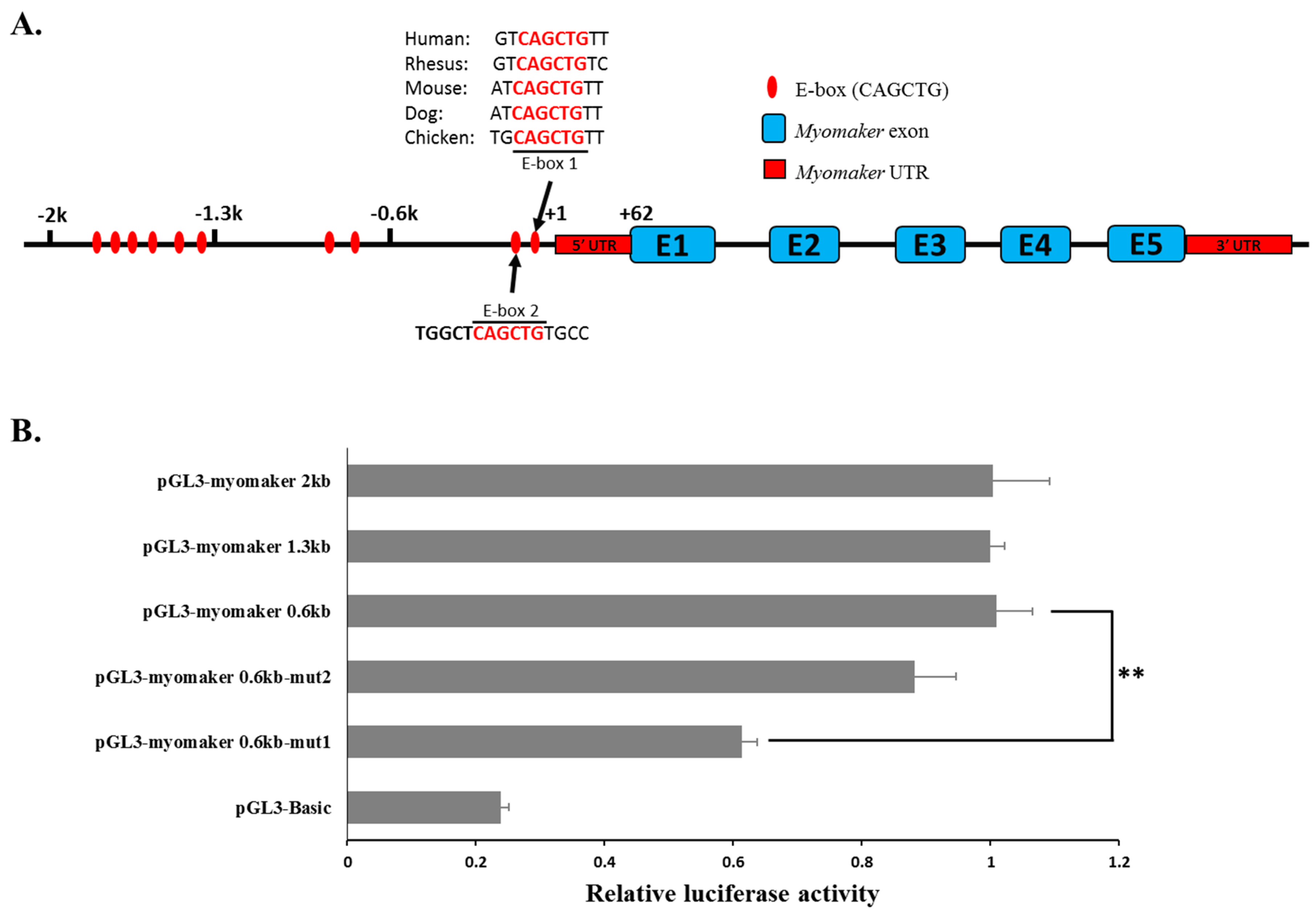
MYOG. This may be due to the impact of endogenous MYOD and MYOG levels. As the deletion of E-box 1 can lower the promoter activity of pGL3-Myomaker 0.6kb (
Figure 3B), its deletion can also reduce the promoter activity of pGL3-Myomaker 0.6kb when only pcDNA3.1-EGFP was transfected (
Figure 4H). Moreover, other distal elements outside of the 2-kb promoter may be needed for a cooperative activation effect of the promoter, because many distal enhancers have been found to be involved in the activation of gene transcription.
Recently, Millay
et al. found that two E-boxes located proximal to the
Myomaker TSS were essential to
Myomaker transcription in mice [
12]. By analysing available ChIP-sequence data sets from the ENCODE Project, these authors revealed the significant binding of both MYOG and MYOD at these two E-boxes during C2C12 differentiation. Therefore, MYOG and MYOD can regulate
Myomaker transcription directly in both mice and chicken.
A previous study showed that MYOD and MYOG play different roles in the regulation of a similar target genes set [
20]. MYOD can initiate early gene expression and regional histone modification, whereas MYOG enhances the MYOD-initiated expression of a subset of genes. In our study, we found that MYOD was bound abundantly to the
Myomaker promoter during both early and late differentiation. MYOG was bound less during early differentiation but bound more during late differentiation. Therefore, during early chicken myoblast differentiation, MYOD may first bind to the promoter of
Myomaker and initiate
Myomaker expression and regional histone modification. During late differentiation, MYOG binds to the promoter more efficiently with MYOD and then enhances the expression of
Myomaker.
Many miRNAs have been found to be involved in skeletal muscle differentiation [
27,
28,
29]. Here, we found that the miRNA miR-140-3p is another candidate involved in myoblast fusion
in vitro. miR-140-3p can play roles in chondrogenic differentiation [
30], testis differentiation [
31] and spinal chordoma prognosis [
32]. However, few studies have examined miR-140-3p involvement in muscle development. A previous study showed that miR-140-3p was immediately down-regulated in skeletal muscle within one hour of refeeding after fasting for one week [
33], and target prediction and expressional analyses suggested that miR-140-3p might bind and inhibit the expression of the
myostatin gene, which is a well-known negative regulator of muscle growth [
34,
35,
36]. Another study in human airway smooth muscle cells showed that miR-140-3p attenuates the activation of NF-κB and p38 MAPK by indirect mechanisms [
37]. Both NF-κB and p38 MAPK signalling are involved in skeletal muscle differentiation [
38,
39,
40]; however, their specific roles are quite different. p38 MAPK signalling is a positive regulator in muscle development [
38,
39], whereas the data from knockout mice support that the NF-κB pathway functions as an inhibitor of myogenesis [
41]. Therefore, the function of miR-140-3p in muscle remains to be explored.
In our results, miR-140-3p overexpression inhibited
Myomaker and
MyHC expression. The
Myomaker gene is a direct target of miR-140-3p. However, the mechanism of miR-140-3p inhibited
MyHC expression remains unclear. It is possible that there are other target genes of miR-140-3p that can regulate
MyHC expression, because
Myomaker loss-of-function has no impact on
MyHC expression. Additionally, miR-140-3p expression during myoblast differentiation is consistent with
Myomaker, suggesting that this miRNA may have another function in this process. Cell cycle arrest is an important event for myoblast differentiation, and our results showed that miR-140-3p promotes myoblast proliferation, suggesting the negative role of miR-140-3p in myoblast differentiation. However, the roles and expression pattern of miR-140-3p are similar to those of miR-133a, which is an important muscle-specific miRNA during muscle development [
42,
43,
44]. miR-133a has sharply increased expression during muscle differentiation and functions not only in the inhibition of muscle differentiation but also in the promotion of myoblast proliferation [
42,
43]. Therefore, miR-140-3p may be a positive regulator during muscle development similar to miR-133a. However, the regulatory mechanism of miR-140-3p during myoblast differentiation remains unclear, and its regulatory role in
Myomaker is limited to the
in vitro system. The specific function and mechanism of miR-140-3p in myoblast differentiation and proliferation remains to be further explored.
4. Experimental Section
4.1. Animals and Cells
The chicken embryos used in this study were as previously described [
23]. The primary myoblasts of chicken were isolated from the leg muscles of E10 chicks and maintained in cell culture using growth medium as previously characterized [
23]. Myoblast differentiation was induced by culture in differentiation medium, which consisting of DMEM medium (Gibco, Grand Island, NY, USA), 2% horse serum (Hyclone, Logan, UT, USA) and 0.2% penicillin/streptomycin (Invitrogen, Carlsbad, CA, USA). DF-1 cells were cultured in DMEM with 10% fetal bovine serum and 0.2% penicillin/streptomycin.
4.2. cDNA Synthesis and Quantitative Real-Time PCR (qPCR)
Total RNA from tissues or cells was extracted using RNAiso reagent (Takara, Otsu, Japan). cDNA synthesis for mRNA was using PrimeScript
TM RT reagent Kit (Perfect Real Time) (Takara, Otsu, Japan). qPCR program was performed in a Bio-rad CFX96 system (Bio-Rad, Hercules, CA, USA) using KAPA SYBR
® FAST qPCR Kit (KAPA Biosystems, Woburn, MA, USA). The relative expression level was calculated using the method as described before [
45]. qPCR primers sequences for all genes are listed in
Supplementary File 4.
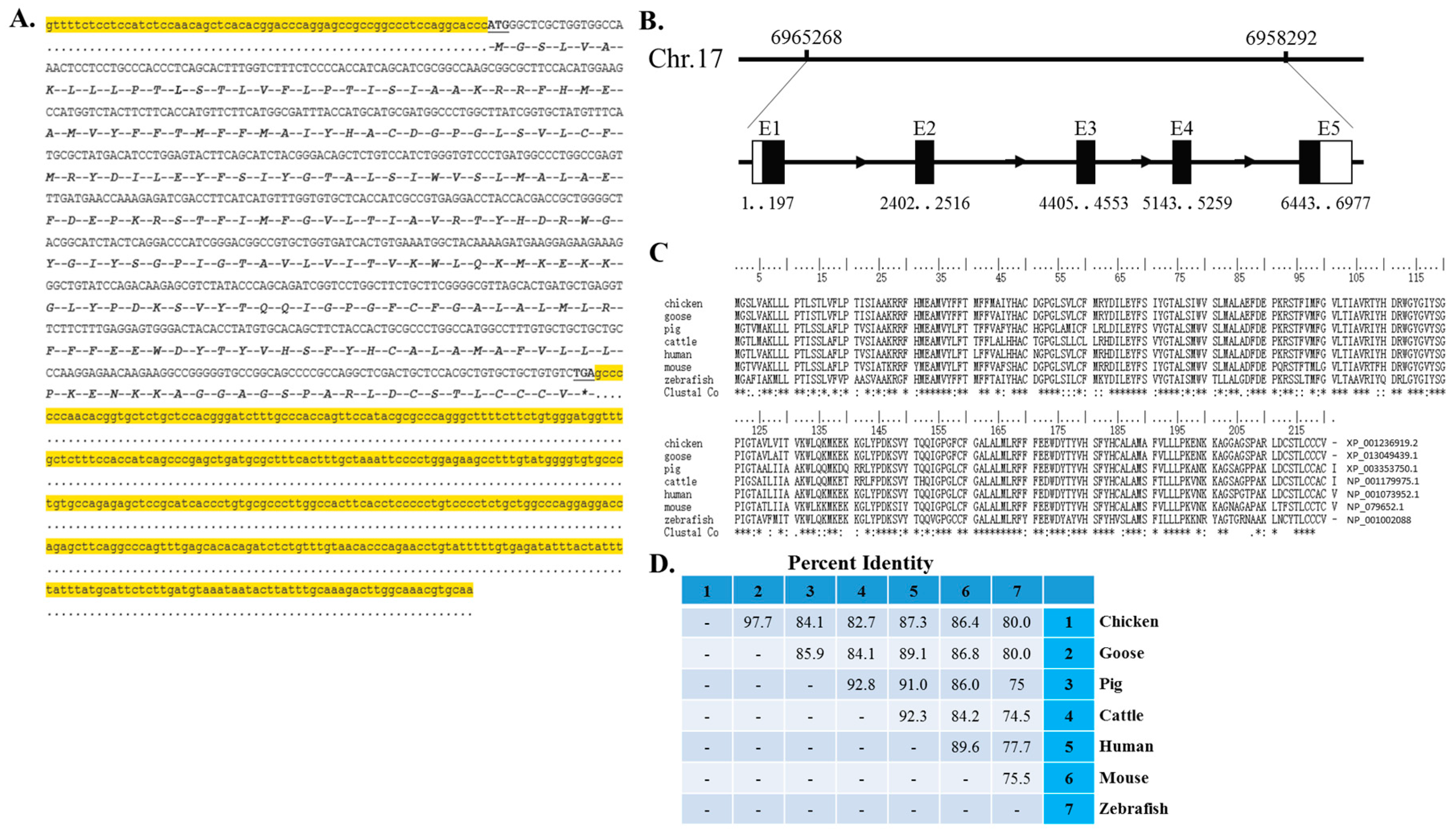
4.3. The 5ʹ and 3ʹ Rapid Amplification of cDNA Ends (RACE)
For 5ʹ RACE, total RNA isolated from chicken embryo skeletal muscle was reversely transcribed using 5ʹ RACE RT-adapter primer and PrimeScript
TM II Reverse Transcriptase (Takara, Otsu, Japan). The obtained first-strand cDNA was subsequently digested by RNase H (Takara, Otsu, Japan) and tailed with terminal deoxynucleotidyl transferase (Beyotime, Shanghai, China) and dCTP. Two rounds of PCR were performed to amplify reverse transcribed products. First round with 5ʹ-RACE outer primer corresponding to RT-adapter and a
Myomaker specific outer primer, and a second round PCR with 5ʹ-RACE inner primer and a nested
Myomaker specific inner primer. For 3ʹ RACE, the synthesis of first-strand cDNA was carried out using the Oligo(dT)-anchor primer and PrimeScript
TM II Reverse Transcriptase (Takara, Otsu, Japan). The following PCR amplification was performed using Myomaker specific outer primer and the 3ʹ-adaptor outer primer, and further nested with
Myomaker specific inner primer and 3ʹ-adaptor inner primer. The above PCR products were then gel-purified, ligated into pGEM-T Easy vector (Promega, Madison, WI, USA) and sequenced. All of the primers used in RACE were summarized in
Supplementary File 4.
4.4. Immunofluorescence
Primary myoblasts seeded in 24-well plates were cultured to 100% confluence and then transfected. Forty-eight hours after transfection with miRNA, siRNA or overexpression vector, the cells were fixed and stained for MyHC and DAPI (Beyotime, Shanghai, China) as previously described [
23]. Images were captured using Nikon Eclipse Ti-U fluorescent microscope.
4.5. ChIP Assays
ChIP assays were carried out using ChIP Assay Kit (Beyotime, Shanghai, China) according to the manufacturer’s instructions. One μg of MYOD (BD Biosciences, San Jose, CA, USA), MYOG (Biorbyt, Cambridge, UK) or control IgG antibody were used in immunoprecipitation. ChIP products were subjected to quantitative PCR using a KAPA SYBR
® FAST qPCR Kit (KAPA Biosystems, Woburn, MA, USA). The primer sequences for ChIP-qPCR analysis are listed in
Supplementary File 4.
4.6. Transfections
Transfection was carried out using Lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA, USA). Cells were transfected with 50 nM miRNA mimics (RiboBio, Guangzhou, China) or 100 nM siRNA (GenePharma, Suzhou, China). Lipofectamine 3000 and nucleic acids were diluted in OPTI-MEM I Reduced Serum Medium (Gibco, Grand Island, NY, USA). The procedure of transfection was performed according to the manufacturer’s direction.
4.7. Plasmid Construction
4.7.1. pcDNA-3.1 Gene Overexpression Vectors
The coding sequences of chicken
Myomaker,
MYOG and
MYOD were amplified using gene-specific clone primers (
Supplementary File 4) and then cloned into the vector of pcDNA-3.1 (Invitrogen, Carlsbad, CA, USA) or pcDNA-3.1-EGFP.
4.7.2. pmirGLO Dual-Luciferase Reporters
Myomaker 3ʹ UTRs were amplified from chicken embryonic leg muscle cDNA and ligated into the pmirGLO vector (Promega, Madison, WI, USA). The mutant Myomaker-3ʹ UTR reporters were generated by changing the miR-140-3p binding site from CCTGTG to TTCACA, and mutagenesis was carried out by PCR amplification and DpnI digestion to remove the parental DNA.
4.7.3. Myomaker Promoter Reporter Plasmid
A 2-kb fragment of the
Myomaker promoter was isolated by PCR using the primers listed in
Supplementary File 4. After the PCR product was digested with
KpnI and
SmaI, the insertion was ligated into the pGL3-basic vector (Promega, Madison, WI, USA) to create the expression vector pGL3-Myomaker-2K. After pGL3-Myomaker-2K was sequenced, this construct was used as a template, and pGL3-Myomaker-1.3K or pGL3-Myomaker-0.6K was isolated by PCR. Site-directed mutagenesis of E-box 1 and E-box 2 were carried out by PCR amplification and
DpnI digestion to remove the parental DNA.
4.8. Target Prediction
RNAhybrid algorithm (
http://bibiserv2.cebitec.uni-bielefeld.de/rnahybrid) was used to predict miRNAs potential target sites for
Myomaker mRNA 3ʹ UTR. The default settings was used to run the algorithm with the extra constraints of perfect base pairing in the seed sequence of miRNA (nucleotides 2 to 7) and with the binding minimum free energy (mfe) lower than −20 kcal/mol.
4.9. Dual Luciferase Reporter Assay
For Myomaker promoter assays, myoblasts were transfected with reporter plasmid or co-transfected with overexpression vectors for MYOG and MYOD, and the TK-Renilla reporter (Promega, Madison, WI, USA) was co-transfected to each sample as an internal control. For miRNA target validation assays, wild-type or mutant Myomaker 3ʹ UTR dual-luciferase reporter (100 ng) and miR-140-3p mimic or NC duplexes (50 nM) were co-transfected into DF-1 cells using the Lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA, USA) in 96-well plates. After the cells were transfected for 36 h, luciferase activities were measured according to the manufacturer’s instructions (Dual-luciferase reporter assay system; Promega, Madison, WI, USA). Synergy 2 Multi-mode Microplate Reader (Biotek, Winooski, VT, USA) was used to quantify the luminescent signal and analysed using Gene5 software (Biotek, Winooski, VT, USA).
4.10. Cell-Cycle Analysis
After 36 h transfection, cells culture in growth medium were collected, fixed, permeabilized and stained with propidium iodide (Sigma, St. Louis, MO, USA) containing 10 µg/mL RNase A (Takara, Otsu, Japan) for flow cytometric cell cycle analysis using a BD Accuri C6 flow cytometer (BD Biosciences, San Jose, CA, USA). Data were analysed using FlowJo 7.6 software (Verity Software House, Tosham, ME, USA).
4.11. Statistical Analysis
Unless otherwise stated, all results are showed as mean ± S.E.M. At least three independent experiments were performed for each treatment. Statistical significance between groups was analyzed by one-sample t tests or ANOVA.
4.12. Ethics Standards
All animal experiments were carried out with the permission of the Animal Care Committee of South China Agricultural University (approval number: SCAU#0014). The experiment was performed in accordance with the regulations and guidelines established by this committee.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}