
DNA Damage: A Sensible Mediator of the Differentiation Decision in Hematopoietic Stem Cells and in Leukemia

Abstract
:1. Introduction
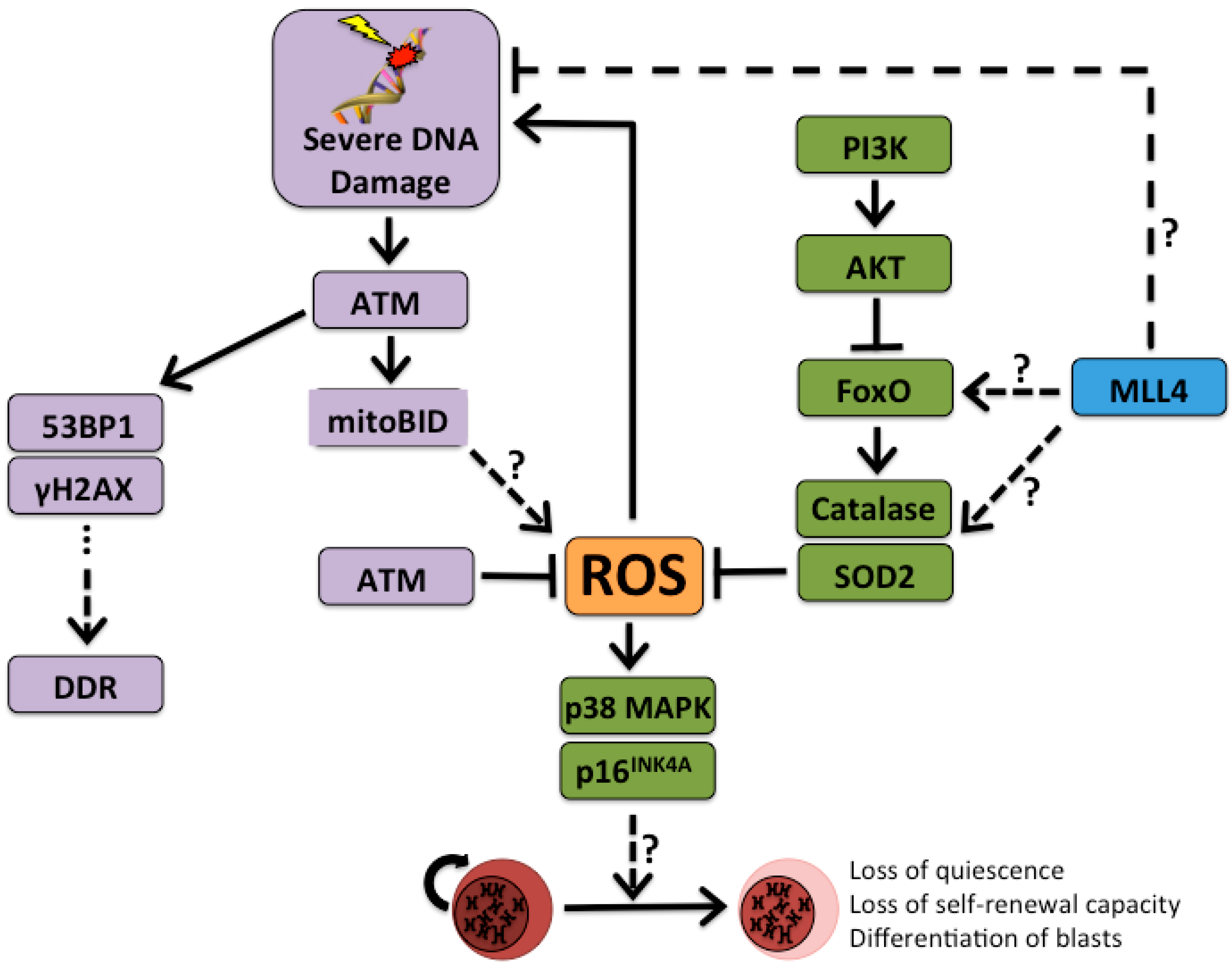
2. The DNA Damage Response as a Potent Oncogenic Driver

3. Sensing Stress and Quitting Quiescence
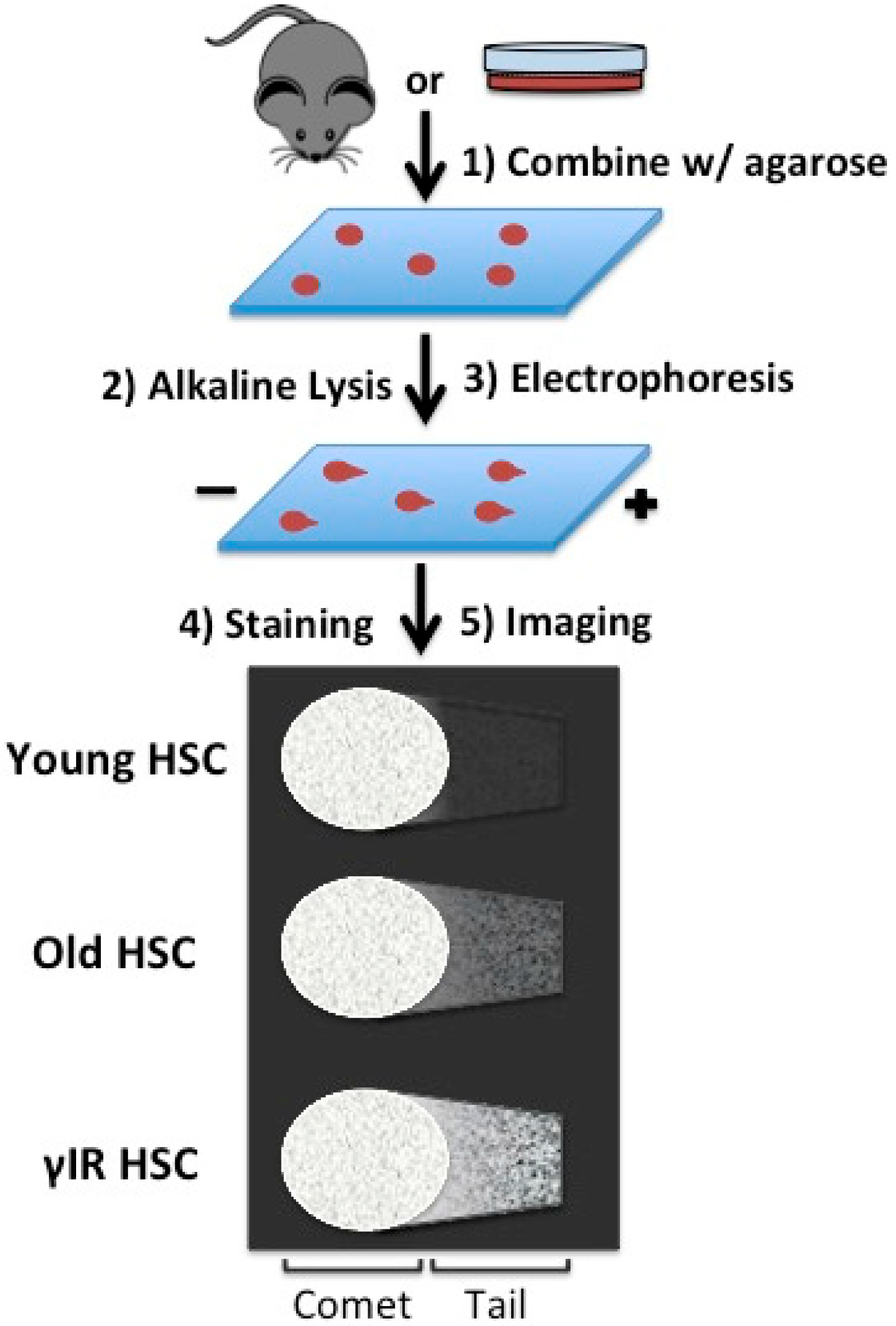
4. Replication: A Source for Rejuvenation or Stress in Aged Hematopoietic Stem Cells (HSCs)


{kind=link}
{kind=link}
| Antigen | Abbreviation | Indication | Notes | References | |
|---|---|---|---|---|---|
| Gamma-H2AX | γH2AX | DSB single stranded DNA | Target of ATM/ATR may serve as repressive epigenetic mark in quiescent aged HSCs | [29,75,76,77,78,79] | |
| Tumor suppressor p53-binding protein 1 | TP53BP1 | DSB | Target of ATM/ATR | [80,81,82] | |
| Phosphorylated checkpoint kinase 1 | pCHK1 | DNA damage repair | Target of ATR mediates cell cycle arrest | [83,84,85,86,87] | |
| Phosphorylated ataxia-telangiectasia mutated | pATM | DNA damage DSBs | Mediates DDR mediates redox homeostasis in HSCs | [18,22,30,88,89,90,91] | |
| Poly (ADP-ribose) | PAR | Single stranded DNA break | Signals for single strand break repair synthesized by PARP | [92,93] | |
| Replication protein A | RPA | Binds single stranded DNA | Prevents formation of secondary structures during replication | [94,95] | |
| ATR interacting protein | ATRIP | Binds RPA coated single stranded DNA | Associates with ATR, leading to its accumulation at intranuclear DNA damage foci | [96,97,98] | |
| Fibrillarin | FBL | Ribosome biosynthesis | Fibrillarin component of SnRNPs | [99,100,101] | |
| Upstream binding factor | UBF | Ribosome biosynthesis | Upstream binding factor transcription factor of rRNAs | [102] | |
| Nucleolin | NCL | Ribosome biosynthesis | Nucleolin invlolved in ribosome synthesis | [103,104] | |
| Nuclear serine/threonine protein phosphatase 4 catalytic subunit | nPP4c | γH2AX phosphatase | [105,106] | ||
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Weissman, I.L.; Anderson, D.J.; Gage, F. Stem and progenitor cells: Origins, phenotypes, lineage commitments, and transdifferentiations. Annu. Rev. Cell Dev. Biol. 2001, 17, 387–403. [Google Scholar] [CrossRef] [PubMed]
- Seita, J.; Weissman, I.L. Hematopoietic stem cell: Self-renewal versus differentiation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 640–653. [Google Scholar] [CrossRef] [PubMed]
- Osawa, M.; Hanada, K.; Hamada, H.; Nakauchi, H. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science 1996, 273, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Kiel, M.J.; Yilmaz, O.H.; Iwashita, T.; Yilmaz, O.H.; Terhorst, C.; Morrison, S.J. Slam family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 2005, 121, 1109–1121. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, O.H.; Kiel, M.J.; Morrison, S.J. Slam family markers are conserved among hematopoietic stem cells from old and reconstituted mice and markedly increase their purity. Blood 2006, 107, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Orkin, S.H.; Zon, L.I. Hematopoiesis: An evolving paradigm for stem cell biology. Cell 2008, 132, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.F.; Fuller, M. Stem cells and cancer: Two faces of eve. Cell 2006, 124, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, C.H.; Ailles, L.E.; Dylla, S.J.; Muijtjens, M.; Jones, C.; Zehnder, J.L.; Gotlib, J.; Li, K.; Manz, M.G.; Keating, A.; et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in BLAST-crisis CML. N. Engl. J. Med. 2004, 351, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Blanpain, C.; Rossi, D.J. DNA damage response in adult stem cells: Pathways and consequences. Nat. Rev. Mol. Cell Biol. 2011, 12, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Naka, K.; Hirao, A. Maintenance of genomic integrity in hematopoietic stem cells. Int. J. Hematol. 2011, 93, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Cheshier, S.H.; Morrison, S.J.; Liao, X.; Weissman, I.L. In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 1999, 96, 3120–3125. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.; Laurenti, E.; Oser, G.; van der Wath, R.C.; Blanco-Bose, W.; Jaworski, M.; Offner, S.; Dunant, C.F.; Eshkind, L.; Bockamp, E.; et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 2008, 135, 1118–1129. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.A.; Ferraro, F.; Roussakis, E.; Klein, A.; Wu, J.; Runnels, J.M.; Zaher, W.; Mortensen, L.J.; Alt, C.; Turcotte, R.; et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014, 508, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Nombela-Arrieta, C.; Pivarnik, G.; Winkel, B.; Canty, K.J.; Harley, B.; Mahoney, J.E.; Park, S.Y.; Lu, J.; Protopopov, A.; Silberstein, L.E. Quantitative imaging of haematopoietic stem and progenitor cell localization and hypoxic status in the bone marrow microenvironment. Nat. Cell Biol. 2013, 15, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Takubo, K.; Semenza, G.L. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 2011, 9, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Hirao, A.; Arai, F.; Matsuoka, S.; Takubo, K.; Hamaguchi, I.; Nomiyama, K.; Hosokawa, K.; Sakurada, K.; Nakagata, N.; et al. Regulation of oxidative stress by atm is required for self-renewal of haematopoietic stem cells. Nature 2004, 431, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Hirao, A.; Arai, F.; Takubo, K.; Matsuoka, S.; Miyamoto, K.; Ohmura, M.; Naka, K.; Hosokawa, K.; Ikeda, Y.; et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 2006, 12, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Suda, T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol. 2014, 15, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Maryanovich, M.; Gross, A. A ROS rheostat for cell fate regulation. Trends Cell Biol. 2013, 23, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Maryanovich, M.; Oberkovitz, G.; Niv, H.; Vorobiyov, L.; Zaltsman, Y.; Brenner, O.; Lapidot, T.; Jung, S.; Gross, A. The Atm-bid pathway regulates quiescence and survival of haematopoietic stem cells. Nat. Cell Biol. 2012, 14, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, K.; Araki, K.Y.; Naka, K.; Arai, F.; Takubo, K.; Yamazaki, S.; Matsuoka, S.; Miyamoto, T.; Ito, K.; Ohmura, M.; et al. FoxO3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell 2007, 1, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Tothova, Z.; Gilliland, D.G. FoxO transcription factors and stem cell homeostasis: Insights from the hematopoietic system. Cell Stem Cell 2007, 1, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Tothova, Z.; Kollipara, R.; Huntly, B.J.; Lee, B.H.; Castrillon, D.H.; Cullen, D.E.; McDowell, E.P.; Lazo-Kallanian, S.; Williams, I.R.; Sears, C.; et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 2007, 128, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Weiss, C.N.; Ito, K. DNA damage response, redox status and hematopoiesis. Blood Cells Mol. Dis. 2014, 52, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.A.; Faryabi, R.B.; Ergen, A.V.; Day, A.M.; Malhowski, A.; Canela, A.; Onozawa, M.; Lee, J.E.; Callen, E.; Gutierrez-Martinez, P.; et al. DNA-damage-induced differentiation of leukaemic cells as an anti-cancer barrier. Nature 2014, 514, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Beerman, I.; Seita, J.; Inlay, M.A.; Weissman, I.L.; Rossi, D.J. Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell 2014, 15, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Flach, J.; Bakker, S.T.; Mohrin, M.; Conroy, P.C.; Pietras, E.M.; Reynaud, D.; Alvarez, S.; Diolaiti, M.E.; Ugarte, F.; Forsberg, E.C.; et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 2014, 512, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Bakkenist, C.J.; Rajpert-De Meyts, E.; Skakkebaek, N.E.; Sehested, M.; Lukas, J.; Kastan, M.B.; Bartek, J. Atm activation in normal human tissues and testicular cancer. Cell Cycle 2005, 4, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Kops, G.J.; Dansen, T.B.; Polderman, P.E.; Saarloos, I.; Wirtz, K.W.; Coffer, P.J.; Huang, T.T.; Bos, J.L.; Medema, R.H.; Burgering, B.M. Forkhead transcription factor FoxO3a protects quiescent cells from oxidative stress. Nature 2002, 419, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Corral, J.; Lavenir, I.; Impey, H.; Warren, A.J.; Forster, A.; Larson, T.A.; Bell, S.; McKenzie, A.N.; King, G.; Rabbitts, T.H. An MLL-AF9 fusion gene made by homologous recombination causes acute leukemia in chimeric mice: A method to create fusion oncogenes. Cell 1996, 85, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Krivtsov, A.V.; Twomey, D.; Feng, Z.; Stubbs, M.C.; Wang, Y.; Faber, J.; Levine, J.E.; Wang, J.; Hahn, W.C.; Gilliland, D.G.; et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 2006, 442, 818–822. [Google Scholar] [CrossRef]
- Somervaille, T.C.; Cleary, M.L. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell 2006, 10, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Cozzio, A.; Passegue, E.; Ayton, P.M.; Karsunky, H.; Cleary, M.L.; Weissman, I.L. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev. 2003, 17, 3029–3035. [Google Scholar] [CrossRef] [PubMed]
- Ugale, A.; Norddahl, G.L.; Wahlestedt, M.; Sawen, P.; Jaako, P.; Pronk, C.J.; Soneji, S.; Cammenga, J.; Bryder, D. Hematopoietic stem cells are intrinsically protected against MLL-ENL-mediated transformation. Cell Rep. 2014, 9, 1246–1255. [Google Scholar] [CrossRef] [PubMed]
- Krivtsov, A.V.; Figueroa, M.E.; Sinha, A.U.; Stubbs, M.C.; Feng, Z.; Valk, P.J.; Delwel, R.; Dohner, K.; Bullinger, L.; Kung, A.L.; et al. Cell of origin determines clinically relevant subtypes of MLL-rearranged AML. Leukemia 2013, 27, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, Q.; Morita, Y.; Jiang, H.; Gross, A.; Lechel, A.; Hildner, K.; Guachalla, L.M.; Gompf, A.; Hartmann, D.; et al. A differentiation checkpoint limits hematopoietic stem cell self-renewal in response to DNA damage. Cell 2012, 148, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Inomata, K.; Aoto, T.; Binh, N.T.; Okamoto, N.; Tanimura, S.; Wakayama, T.; Iseki, S.; Hara, E.; Masunaga, T.; Shimizu, H.; et al. Genotoxic stress abrogates renewal of melanocyte stem cells by triggering their differentiation. Cell 2009, 137, 1088–1099. [Google Scholar] [CrossRef] [PubMed]
- Schoppy, D.W.; Ragland, R.L.; Gilad, O.; Shastri, N.; Peters, A.A.; Murga, M.; Fernandez-Capetillo, O.; Diehl, J.A.; Brown, E.J. Oncogenic stress sensitizes murine cancers to hypomorphic suppression of ATR. J. Clin. Investig. 2012, 122, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C.; Borutaite, V. There is no evidence that mitochondria are the main source of reactive oxygen species in mammalian cells. Mitochondrion 2012, 12, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Giorgi, C.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; de Marchi, E.; Missiroli, S.; Patergnani, S.; Poletti, F.; et al. Mitochondria-ROS crosstalk in the control of cell death and aging. J. Signal Transduct. 2012, 2012, 329635. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W.; Elledge, S.J. The DNA damage response: Ten years after. Mol. Cell 2007, 28, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Kozlov, S.; Lavin, M.F.; Person, M.D.; Paull, T.T. Atm activation by oxidative stress. Science 2010, 330, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Willis, J.; Patel, Y.; Lentz, B.L.; Yan, S. APE2 is required for ATR-CHK1 checkpoint activation in response to oxidative stress. Proc. Natl. Acad. Sci. USA 2013, 110, 10592–10597. [Google Scholar] [CrossRef] [PubMed]
- Takao, N.; Li, Y.; Yamamoto, K. Protective roles for atm in cellular response to oxidative stress. FEBS Lett. 2000, 472, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Kamsler, A.; Daily, D.; Hochman, A.; Stern, N.; Shiloh, Y.; Rotman, G.; Barzilai, A. Increased oxidative stress in ataxia telangiectasia evidenced by alterations in redox state of brains from atm-deficient mice. Cancer Res. 2001, 61, 1849–1854. [Google Scholar] [PubMed]
- Schubert, R.; Reichenbach, J.; Royer, N.; Pichler, M.; Zielen, S. Spontaneous and oxidative stress-induced programmed cell death in lymphocytes from patients with ataxia telangiectasia (AT). Clin. Exp. Immunol. 2000, 119, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Takao, N.; Kato, H.; Mori, R.; Morrison, C.; Sonada, E.; Sun, X.; Shimizu, H.; Yoshioka, K.; Takeda, S.; Yamamoto, K. Disruption of atm in p53-Null cells causes multiple functional abnormalities in cellular response to ionizing radiation. Oncogene 1999, 18, 7002–7009. [Google Scholar] [CrossRef] [PubMed]
- Oguro, H.; Iwama, A.; Morita, Y.; Kamijo, T.; van Lohuizen, M.; Nakauchi, H. Differential impact of INK4A and ARF on hematopoietic stem cells and their bone marrow microenvironment in BMI1-deficient mice. J. Exp. Med. 2006, 203, 2247–2253. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Iwama, A.; Takayanagi, S.; Morita, Y.; Eto, K.; Ema, H.; Nakauchi, H. Cytokine signals modulated via lipid rafts mimic niche signals and induce hibernation in hematopoietic stem cells. EMBO J. 2006, 25, 3515–3523. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.J.; Jamieson, C.H.M.; Weissman, I.L. Stems cells and the pathways to aging and cancer. Cell 2008, 132, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.J.; Bryder, D.; Seita, J.; Nussenzweig, A.; Hoeijmakers, J.; Weissman, I.L. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 2007, 447, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Rube, C.E.; Fricke, A.; Widmann, T.A.; Furst, T.; Madry, H.; Pfreundschuh, M.; Rube, C. Accumulation of DNA damage in hematopoietic stem and progenitor cells during human aging. PLoS One 2011, 6, e17487. [Google Scholar] [CrossRef] [PubMed]
- Nijnik, A.; Woodbine, L.; Marchetti, C.; Dawson, S.; Lambe, T.; Liu, C.; Rodrigues, N.P.; Crockford, T.L.; Cabuy, E.; Vindigni, A.; et al. DNA repair is limiting for haematopoietic stem cells during ageing. Nature 2007, 447, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Olive, P.L.; Banath, J.P. The comet assay: A method to measure DNA damage in individual cells. Nat. Protoc. 2006, 1, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Ostling, O.; Johanson, K.J. Microelectrophoretic study of radiation-induced DNA damages in individual mammalian cells. Biochem. Biophys. Res. Commun. 1984, 123, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.P.; McCoy, M.T.; Tice, R.R.; Schneider, E.L. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp. Cell Res. 1988, 175, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Foudi, A.; Hochedlinger, K.; Van Buren, D.; Schindler, J.W.; Jaenisch, R.; Carey, V.; Hock, H. Analysis of histone 2B-GFP retention reveals slowly cycling hematopoietic stem cells. Nat. Biotechnol. 2009, 27, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Passegue, E.; Wagers, A.J.; Giuriato, S.; Anderson, W.C.; Weissman, I.L. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J. Exp. Med. 2005, 202, 1599–1611. [Google Scholar] [CrossRef] [PubMed]
- Bowie, M.B.; McKnight, K.D.; Kent, D.G.; McCaffrey, L.; Hoodless, P.A.; Eaves, C.J. Hematopoietic stem cells proliferate until after birth and show a reversible phase-specific engraftment defect. J. Clin. Investig. 2006, 116, 2808–2816. [Google Scholar] [CrossRef] [PubMed]
- Mohrin, M.; Bourke, E.; Alexander, D.; Warr, M.R.; Barry-Holson, K.; Le Beau, M.M.; Morrison, C.G.; Passegue, E. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell 2010, 7, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Lee, Y.D.; Wagers, A.J. Stem cell aging: Mechanisms, regulators and therapeutic opportunities. Nat. Med. 2014, 20, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Jan, M.; Snyder, T.M.; Corces-Zimmerman, M.R.; Vyas, P.; Weissman, I.L.; Quake, S.R.; Majeti, R. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci. Transl. Med. 2012, 4, 149ra118. [Google Scholar] [CrossRef] [PubMed]
- Corces-Zimmerman, M.R.; Majeti, R. Pre-leukemic evolution of hematopoietic stem cells: The importance of early mutations in leukemogenesis. Leukemia 2014, 28, 2276–2282. [Google Scholar] [CrossRef] [PubMed]
- Jan, M.; Chao, M.P.; Cha, A.C.; Alizadeh, A.A.; Gentles, A.J.; Weissman, I.L.; Majeti, R. Prospective separation of normal and leukemic stem cells based on differential expression of tim3, a human acute myeloid leukemia stem cell marker. Proc. Natl. Acad. Sci. USA 2011, 108, 5009–5014. [Google Scholar] [CrossRef] [PubMed]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D., Jr.; van Rooijen, N.; Weissman, I.L. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 2010, 11, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Burhans, W.C.; Weinberger, M. DNA replication stress, genome instability and aging. Nucleic Acids Res. 2007, 35, 7545–7556. [Google Scholar] [CrossRef] [PubMed]
- Milyavsky, M.; Gan, O.I.; Trottier, M.; Komosa, M.; Tabach, O.; Notta, F.; Lechman, E.; Hermans, K.G.; Eppert, K.; Konovalova, Z.; et al. A distinctive DNA damage response in human hematopoietic stem cells reveals an apoptosis-independent role for p53 in self-renewal. Cell Stem Cell 2010, 7, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Nieves-Neira, W.; Boon, C.; Pommier, Y.; Bonner, W.M. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J. Biol. Chem. 2000, 275, 9390–9395. [Google Scholar] [CrossRef] [PubMed]
- Burma, S.; Chen, B.P.; Murphy, M.; Kurimasa, A.; Chen, D.J. Atm phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001, 276, 42462–42467. [Google Scholar] [CrossRef] [PubMed]
- Katsube, T.; Mori, M.; Tsuji, H.; Shiomi, T.; Wang, B.; Liu, Q.; Nenoi, M.; Onoda, M. Most hydrogen peroxide-induced histone H2AX phosphorylation is mediated by ATR and is not dependent on DNA double-strand breaks. J. Biochem. 2014, 156, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Ward, I.M.; Chen, J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 2001, 276, 47759–47762. [Google Scholar] [CrossRef] [PubMed]
- Schultz, L.B.; Chehab, N.H.; Malikzay, A.; Halazonetis, T.D. P53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J. Cell Biol. 2000, 151, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Capetillo, O.; Chen, H.T.; Celeste, A.; Ward, I.; Romanienko, P.J.; Morales, J.C.; Naka, K.; Xia, Z.; Camerini-Otero, R.D.; Motoyama, N.; et al. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat. Cell Biol. 2002, 4, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.; Henderson, C.; Adachi, Y. Phosphorylation and rapid relocalization of 53BP1 to nuclear foci upon DNA damage. Mol. Cell. Biol. 2001, 21, 1719–1729. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Gatei, M.; O'Connell, M.J.; Khanna, K.K.; Bugg, S.J.; Hogg, A.; Scott, S.P.; Hobson, K.; Lavin, M.F. Chk1 complements the G2/M checkpoint defect and radiosensitivity of ataxia-telangiectasia cells. Oncogene 1999, 18, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Walworth, N.C.; Bernards, R. Rad-dependent response of the CHK1-encoded protein kinase at the DNA damage checkpoint. Science 1996, 271, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Flaggs, G.; Plug, A.W.; Dunks, K.M.; Mundt, K.E.; Ford, J.C.; Quiggle, M.R.; Taylor, E.M.; Westphal, C.H.; Ashley, T.; Hoekstra, M.F.; et al. ATM-dependent interactions of a mammalian CHK1 homolog with meiotic chromosomes. Curr. Biol. 1997, 7, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, T.H.; Walworth, N.C. Association of CHK1 with 14–3-3 proteins is stimulated by DNA damage. Genes Dev. 1999, 13, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Gatei, M.; Sloper, K.; Sorensen, C.; Syljuasen, R.; Falck, J.; Hobson, K.; Savage, K.; Lukas, J.; Zhou, B.B.; Bartek, J.; et al. Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent phosphorylation of CHK1 on Ser-317 in response to ionizing radiation. J. Biol. Chem. 2003, 278, 14806–14811. [Google Scholar] [CrossRef] [PubMed]
- Savitsky, K.; Bar-Shira, A.; Gilad, S.; Rotman, G.; Ziv, Y.; Vanagaite, L.; Tagle, D.A.; Smith, S.; Uziel, T.; Sfez, S.; et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995, 268, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Kamer, I.; Sarig, R.; Zaltsman, Y.; Niv, H.; Oberkovitz, G.; Regev, L.; Haimovich, G.; Lerenthal, Y.; Marcellus, R.C.; Gross, A. Proapoptotic bid is an ATM effector in the DNA-damage response. Cell 2005, 122, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Takubo, K.; Arai, F.; Satoh, H.; Matsuoka, S.; Ohmura, M.; Naka, K.; Azuma, M.; Miyamoto, K.; Hosokawa, K.; et al. Regulation of reactive oxygen species by atm is essential for proper response to DNA double-strand breaks in lymphocytes. J. Immunol. 2007, 178, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Malanga, M.; Pleschke, J.M.; Kleczkowska, H.E.; Althaus, F.R. Poly(adp-ribose) binds to specific domains of p53 and alters its DNA binding functions. J. Biol. Chem. 1998, 273, 11839–11843. [Google Scholar] [CrossRef] [PubMed]
- Kawamitsu, H.; Hoshino, H.; Okada, H.; Miwa, M.; Momoi, H.; Sugimura, T. Monoclonal antibodies to poly(adenosine diphosphate ribose) recognize different structures. Biochemistry 1984, 23, 3771–3777. [Google Scholar] [CrossRef] [PubMed]
- Heyer, W.D.; Rao, M.R.; Erdile, L.F.; Kelly, T.J.; Kolodner, R.D. An essential saccharomyces cerevisiae single-stranded DNA binding protein is homologous to the large subunit of human RP-A. EMBO J. 1990, 9, 2321–2329. [Google Scholar] [PubMed]
- Wold, M.S.; Kelly, T. Purification and characterization of replication protein A, a cellular protein required for in vitro replication of simian virus 40 DNA. Proc. Natl. Acad. Sci. USA 1988, 85, 2523–2527. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Elledge, S.J. Sensing DNA damage through atrip recognition of RPA-SSDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Unsal-Kacmaz, K.; Sancar, A. Quaternary structure of atr and effects of atrip and replication protein a on its DNA binding and kinase activities. Mol. Cell. Biol. 2004, 24, 1292–1300. [Google Scholar] [CrossRef] [PubMed]
- Bomgarden, R.D.; Yean, D.; Yee, M.C.; Cimprich, K.A. A novel protein activity mediates DNA binding of an ATR-ATRIP complex. J. Biol. Chem. 2004, 279, 13346–13353. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.P.; Hurt, E.C.; Kern, H.; Lehtonen, H.; Carmo-Fonseca, M.; Lapeyre, B.; Tollervey, D. Evolutionary conservation of the human nucleolar protein fibrillarin and its functional expression in yeast. J. Cell Biol. 1991, 113, 715–729. [Google Scholar] [CrossRef] [PubMed]
- Tollervey, D.; Lehtonen, H.; Carmo-Fonseca, M.; Hurt, E.C. The small nucleolar RNP protein NOP1 (fibrillarin) is required for pre-rrna processing in yeast. EMBO J. 1991, 10, 573–583. [Google Scholar] [PubMed]
- Politz, J.C.; Lewandowski, L.B.; Pederson, T. Signal recognition particle rna localization within the nucleolus differs from the classical sites of ribosome synthesis. J. Cell Biol. 2002, 159, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Learned, R.M.; Learned, T.K.; Haltiner, M.M.; Tjian, R.T. Human rRNA transcription is modulated by the coordinate binding of two factors to an upstream control element. Cell 1986, 45, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Bugler, B.; Caizergues-Ferrer, M.; Bouche, G.; Bourbon, H.; Amalric, F. Detection and localization of a class of proteins immunologically related to a 100-kDa nucleolar protein. FEBS J. 1982, 128, 475–480. [Google Scholar] [CrossRef]
- Borer, R.A.; Lehner, C.F.; Eppenberger, H.M.; Nigg, E.A. Major nucleolar proteins shuttle between nucleus and cytoplasm. Cell 1989, 56, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, D.; Keogh, M.C.; Ishii, H.; Peterson, C.L.; Buratowski, S.; Lieberman, J. γ-H2AX dephosphorylation by protein phosphatase 2a facilitates DNA double-strand break repair. Mol. Cell 2005, 20, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Nakada, S.; Chen, G.I.; Gingras, A.C.; Durocher, D. PP4 is a γH2AX phosphatase required for recovery from the DNA damage checkpoint. EMBO Rep. 2008, 9, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, T.; Guillou, E.; Coloma, J.; Montoya, G.; Mendez, J. The human gins complex associates with CDC45 and MCM and is essential for DNA replication. Nucleic Acids Res. 2009, 37, 2087–2095. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, A.; Schwob, E.; Mendez, J. Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc. Natl. Acad. Sci. USA 2008, 105, 8956–8961. [Google Scholar] [CrossRef] [PubMed]
- Durkin, S.G.; Glover, T.W. Chromosome fragile sites. Ann. Rev. Genet. 2007, 41, 169–192. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiss, C.N.; Ito, K. DNA Damage: A Sensible Mediator of the Differentiation Decision in Hematopoietic Stem Cells and in Leukemia. Int. J. Mol. Sci. 2015, 16, 6183-6201. https://doi.org/10.3390/ijms16036183
Weiss CN, Ito K. DNA Damage: A Sensible Mediator of the Differentiation Decision in Hematopoietic Stem Cells and in Leukemia. International Journal of Molecular Sciences. 2015; 16(3):6183-6201. https://doi.org/10.3390/ijms16036183
Chicago/Turabian StyleWeiss, Cary N., and Keisuke Ito. 2015. "DNA Damage: A Sensible Mediator of the Differentiation Decision in Hematopoietic Stem Cells and in Leukemia" International Journal of Molecular Sciences 16, no. 3: 6183-6201. https://doi.org/10.3390/ijms16036183