
Wilson’s Disease: A Comprehensive Review of the Molecular Mechanisms

Abstract
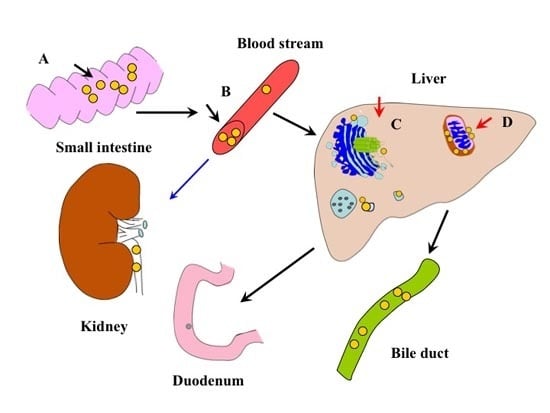
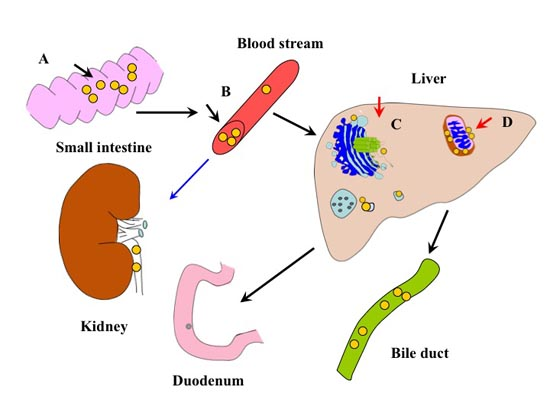
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
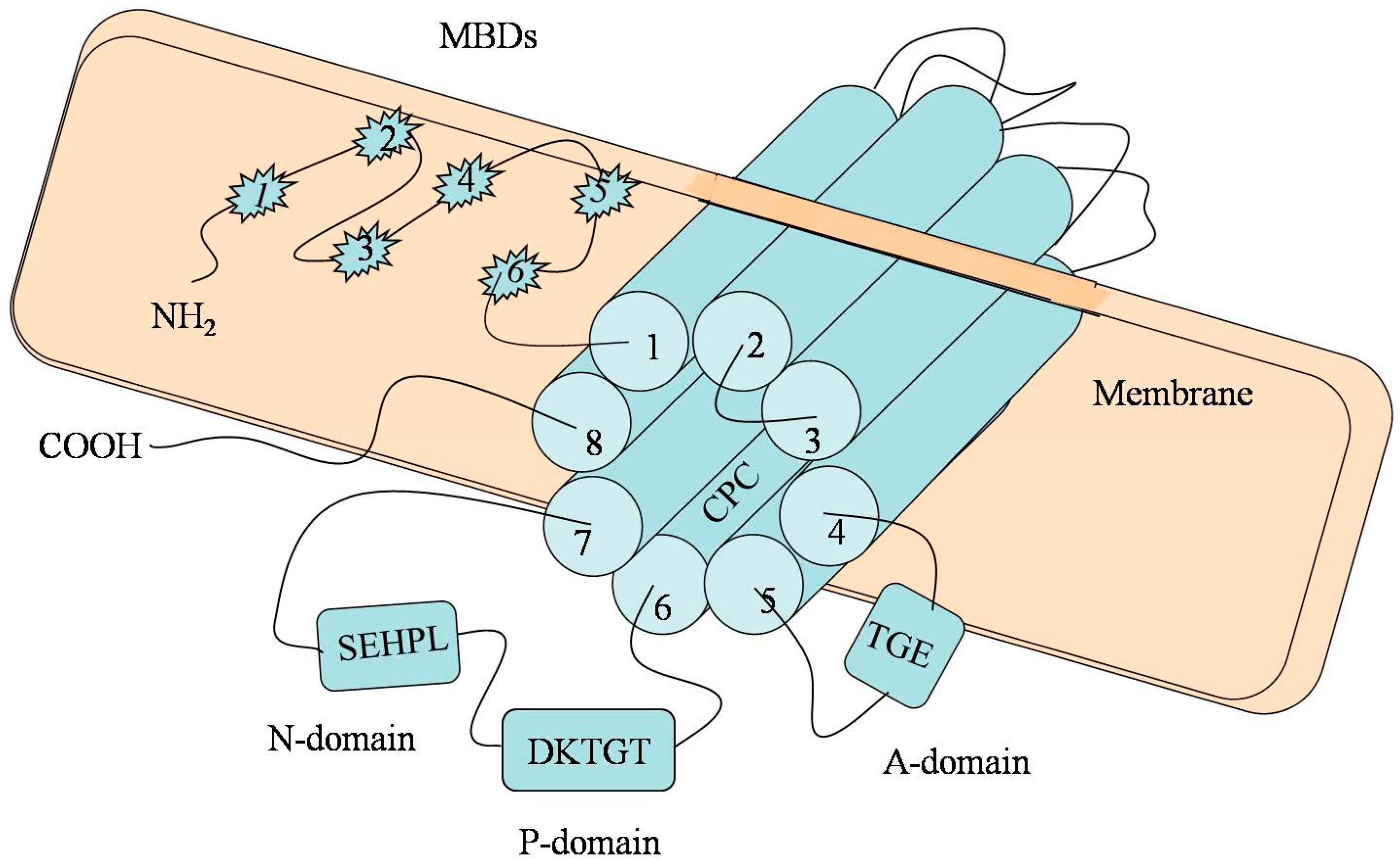
2. The Molecular Architecture of P-Type ATPase (ATP7B)
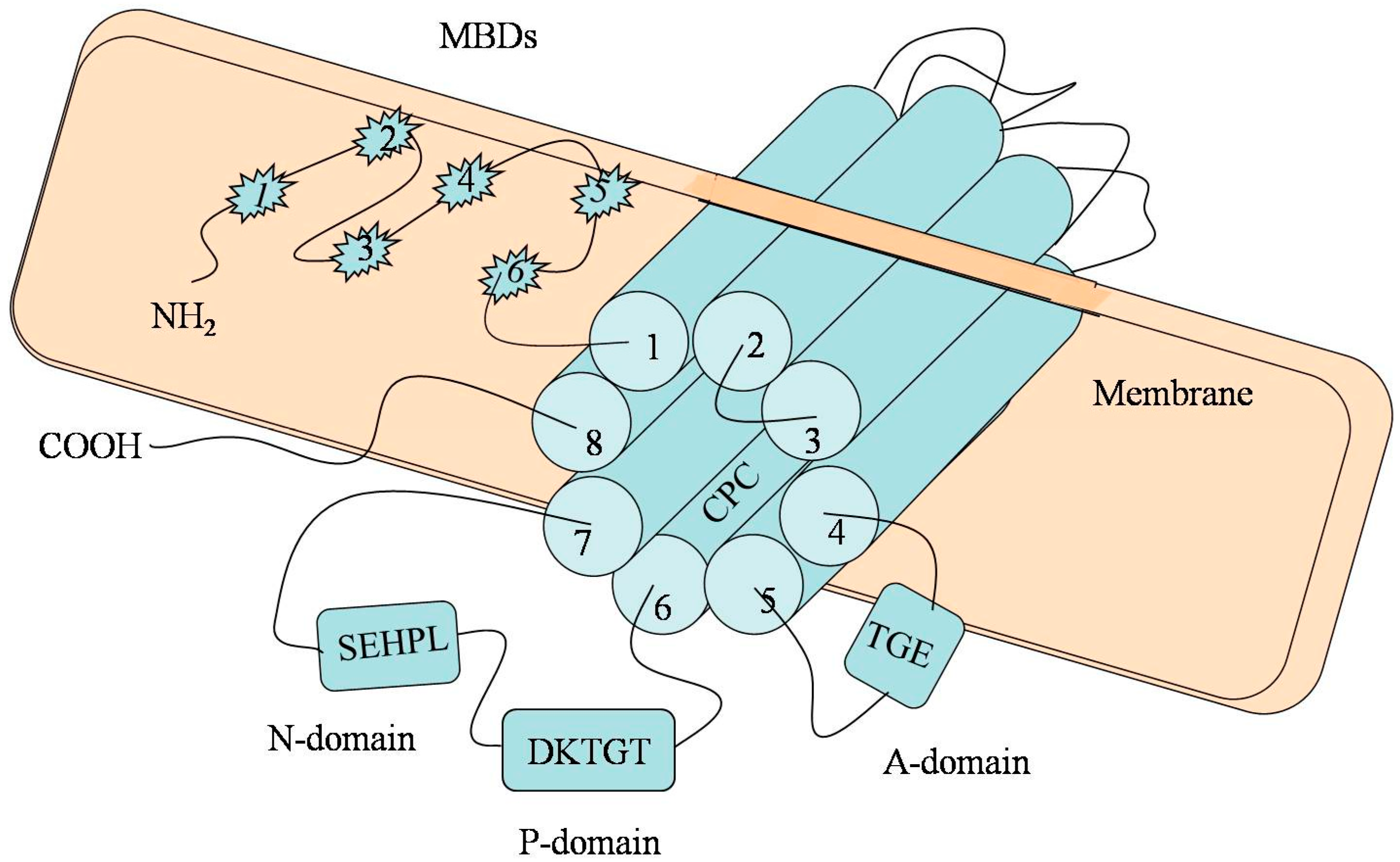
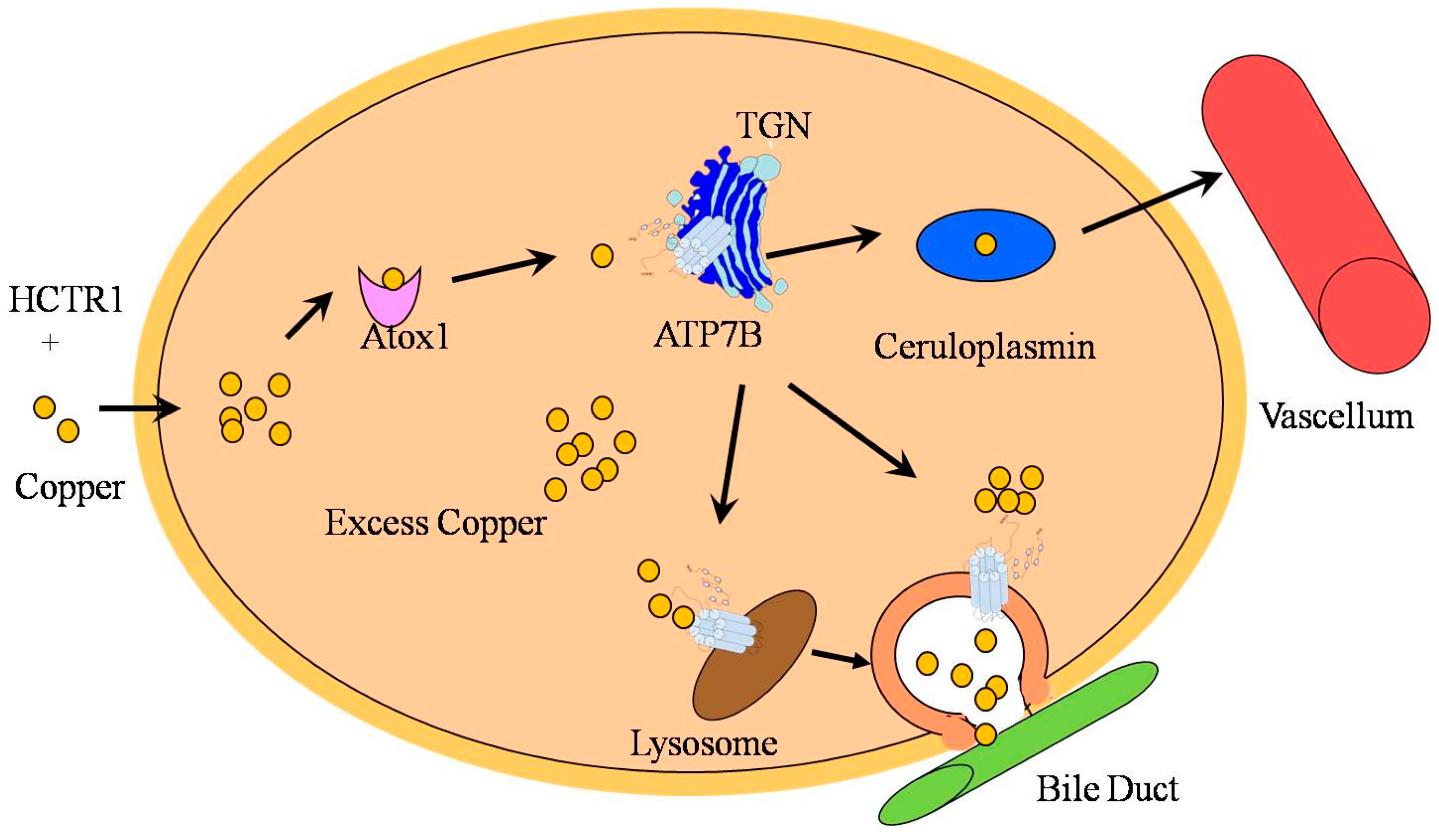
3. Function and Regulation of ATP7B in Copper Transportation
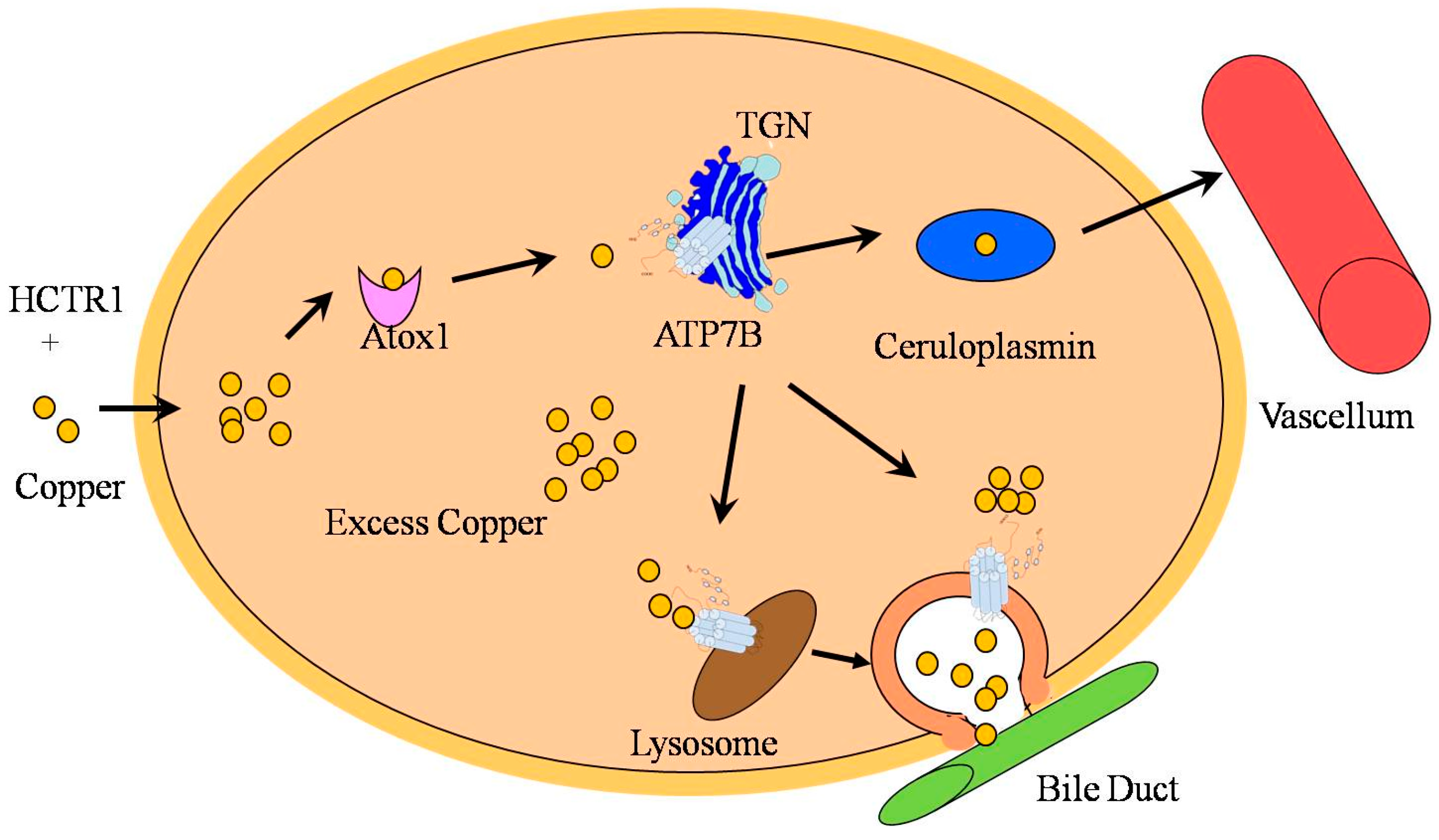
4. Mutation Hotspots of ATP7B
5. Mechanism in Copper-Induced Liver Injury
6. Modifying Factors and Phenotypic Diversity in WD
7. Conclusions
Acknowledgments
Author Contributions
Abbreviations
| ATP7B | P-type adenosine triphosphatase |
| CL | Cardiolipin |
| DNMTs | DNA methyltransferase |
| MBDs | Metal-binding domains |
| PA | Phosphatidic acid |
| SAH | S-adenosylhomocysteine |
| SAM | S-adenosylmethionine |
| TGN | Trans-Golgi network |
| WD | Wilson disease |
Conflicts of Interest
References
- Wilson, S.A.K. Progressive lenticular degeneration: A familial nervous disease associated with cirrhosis of liver. Brain 1912, 34, 295–507. [Google Scholar] [CrossRef]
- Beinhardt, S.; Leiss, W.; Stattermayer, A.F.; Graziadei, I.; Zoller, H.; Stauber, R.; Maieron, A.; Datz, C.; Steindl-Munda, P.; Hofer, H.; et al. Long-term outcomes of patients with Wilson disease in a large Austrian cohort. Clin. Gastroenterol. Hapatol. 2014, 12, 683–689. [Google Scholar] [CrossRef]
- Seniow, J.; Mroziak, B.; Czlonkowska, A.; Jedryka-Goral, A. Self-rated emotional functioning of patients with neurological or asymptomatic form of Wilson’s disease. J. Clin. Neuropsychol. 2004, 17, 367–373. [Google Scholar] [CrossRef]
- Zimbrean, P.C.; Schilsky, M.L. Psychiatric aspects of Wilson disease: A review. Gen. Hosp. Psychiatry 2014, 36, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.A.; Schilsky, M.L. A practice guideline on Wilson disease. Hepatology 2003, 37, 1475–1492. [Google Scholar] [CrossRef] [PubMed]
- Gouider-Khouja, N. Wilson’s disease. Parkinsonism Relat. Disord. 2009, 15, 126–129. [Google Scholar] [CrossRef]
- Dong, Q.Y.; Wu, Z.Y. Advance in the pathogenesis and treatment of Wilson disease. Transl. Neurodegener. 2012. [Google Scholar] [CrossRef]
- Mehta, R.; Templeton, D.; O’Brien, P.J. Mitochondrial involvement in genetically determined transition metal toxicity. II. Copper toxicity. Chem. Biol. Interact. 2006, 163, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.A.; Robinson, B.H.; Yang, S. Mitochondrial structure and function in the untreated Jackson toxic milk (tx-j) mouse, a model for Wilson disease. Mol. Genet. Metab. 2008, 93, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Ala, A.; Walker, A.P.; Ashkan, K.; Dooley, J.S.; Schilsky, M.L. Wilson’s disease. Lancet 2007, 369, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.A. Wilson’s disease. Medicine 2011, 39, 602–604. [Google Scholar] [CrossRef]
- Tanzi, R.E.; Petrukhin, K.; Chernov, I.; Pellequer, J.L.; Wasco, W.; Ross, B.; Romano, D.M.; Pavone, L.; Brzustowicz, L.M. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. J. Nat. Genet. 1993, 5, 344–350. [Google Scholar] [CrossRef]
- Walker, J.M.; Tsivkovskii, R.; Lutsenko, S. Metallochaperone Atox1 transfers copper to the NH2-terminal domain of the Wilson’s disease protein and regulates its catalytic activity. J. Biol. Chem. 2002, 277, 27953–27959. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, N.; Sarkar, B. Molecular mechanism of copper transport in Wilson disease. Environ. Health Perspect. 2002, 110, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.M.; Huster, D.; Ralle, M.; Morgan, C.T.; Blackburn, N.J.; Lutsenko, S. The N-terminal metal-binding site 2 of the Wilson’s disease protein plays a key role in the transfer of copper from Atox1. J. Biol. Chem. 2004, 279, 15376–15384. [Google Scholar] [CrossRef] [PubMed]
- Cater, M.A.; La Fontaine, S.; Cox, D.; Mercer, J.F. Intracellular trafficking of the human Wilson protein: The role of the six N-terminal metal-binding sites. J. Biochem. 2004, 380, 805–813. [Google Scholar] [CrossRef]
- Cater, M.A.; La Fontaine, S.; Mercer, J.F.B. Copper binding to the N-terminal metal-binding sites or the CPC motif is not essential for copper-induced trafficking of the human Wilson protein (ATP7B). Biochem. J. 2007, 401, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Huster, D.; Lusenko, S. The distinct roles of the N-terminal copper-binding sites in regulation of catalytic activity of the Wilson’s disease protein. J. Biol. Chem. 2003, 278, 32212–32218. [Google Scholar] [CrossRef] [PubMed]
- Lenartowicz, M.; Krzeptowski, W. Structure and function of ATP7A and ATP7B proteins—Cu-transporting ATPase. Postepy Biochem. 2010, 56, 317–327. [Google Scholar] [PubMed]
- Lutsenko, S.; Efremov, R.G.; Tsivkovski, R.; Walker, J.M. Human copper-transporting ATPase ATP7B (the Wilson’s disease protein): Biochemical properties and regulation. J. Bioenerg. Biomembr. 2002, 34, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Myari, A.; Hadjiliadis, N.; Fatemi, N.; Sarkar, B. Copper (I) interaction with model peptides of WD6 and TM6 domains of Wilson ATPase: Regulatory and mechanistic implications. J. Inorg. Biochem. 2004, 98, 1483–1494. [Google Scholar] [CrossRef] [PubMed]
- Tsivkovskii, R.; MacArthur, B.C.; Lutsenko, S. The Lys1010-Lys1325 fragment of the Wilson’s disease protein binds nucleotides and interacts with the N-terminal domain of this protein in a copper-dependent manner. J. Biol. Chem. 2001, 19, 2334–2342. [Google Scholar]
- Dmitriev, O.; Tsivkoskii, R.; Abildgaard, F.; Morgan, C.T.; Markley, H.L.; Lutsenko, S. Solution structure of the N-domain of Wilson disease protein: Distinct nucleotide-binding environment and effects of disease mutations. Proc. Natl. Acad. Sci. USA 2006, 103, 5302–5307. [Google Scholar] [CrossRef] [PubMed]
- Sazinsky, M.H.; Mandal, A.K.; Arguello, J.M.; Rosenzweig, A.C. Structure of the ATP bind domain from the Archaeoglobus fulgidus Cu+-ATPase. J. Biol. Chem. 2006, 281, 11161–11166. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.V.; Juul, B.; Maire, M. Structural organization, ion transport, and energy transduction of P-type ATPases. Biochim. Biophys. Acta 1996, 1286, 1–51. [Google Scholar] [CrossRef] [PubMed]
- Petris, M.J.; Voskoboinik, I.; Cater, M.; Smith, K.; Kim, B.E.; Llanos, R.M.; Strausak, D.; Camakaris, J.; Mercer, J.F.B. Copper-regulated trafficking of the Menkes disease copper ATPase is associated with formation of a phosphorylated catalytic intermediate. J. Biol. Chem. 2002, 277, 46736–46742. [Google Scholar] [CrossRef] [PubMed]
- Moller, L.B.; Ott, P.; Lund, C.; Horn, N. Homozygosity for a gross partial gene deletion of the C-terminal end of ATP7B in a Wilson patient with hepatic and no neurological manifestations. Am. J. Med. Genet. 2005, 138, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Huster, D.; Hoppert, M.; Lutsenko, S.; Zinke, J.; Lehmann, C.; Mossner, J.; Berr, F.; Caca, K. Defective cellular localization of mutant ATP7B in Wilson’s disease patients and hepatoma cell lines. Gastroenterology 2003, 124, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Lutsenko, S.; Tsivkovskii, R.; Walker, J.M. Functional properties of the human copper-transporting ATPase ATP7B (the Wilson’s disease protein) and regulation by metallochaperone Atox1. Ann. N. Y. Acad. Sci. 2003, 986, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Hung, I.H.; Suzuki, M.; Yamaguchi, Y.; Yuan, D.S.; Klausner, R.D.; Gitlin, J.D. Biochemical characterization of the Wilson disease protein and function expression in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 1997, 272, 21461–21466. [Google Scholar] [CrossRef] [PubMed]
- Harada, M.; Kawaguchi, T.; Kumemura, H.; Terada, K.; Ninomiya, H.; Taniquchi, E.; Hanada, S.; Maeyama, M.; Koqa, H.; Ueno, T.; et al. The Wilson disease protein ATP7B resides in the late endosomes with Rab7 and the Niemann-Pick C1 protein. Am. J. Pathol. 2005, 166, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Gitlin, J.D. Copper homeostasis: Specialized functions of the late secretory pathway. Dev. Cell 2014, 29, 631–632. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.R.; Yang, H.C.; Hsieh, D.J.; Liu, Z.; Tsai, K.J. Zebrafish sod1 and sp1 expression are modulated by the copper ATPase gene atp7a in response to intracellular copper status. Chem. Biol. Interact. 2011, 189, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Gitschier, J. hCTR1: A human gene for copper uptake identified by complementation in yeast. Proc. Natl. Acad. Sci. USA 1997, 94, 7481–7486. [Google Scholar] [CrossRef] [PubMed]
- Hellman, N.E.; Kono, S.; Mancini, G.M.; Hoogeboom, A.J.; de Jong, G.J.; Gitlin, J.D. Mechanisms of copper incorporation into human ceruloplasmin. J. Biol. Chem. 2002, 277, 46632–46638. [Google Scholar] [CrossRef] [PubMed]
- Polishchuk, E.V.; Concilli, M.; Iacobacci, S.; Chesi, G.; Pastore, N.; Piccolo, P.; Paladino, S.; Baldantoni, D.; van IJzendoorn, S.C.; Chan, J.; et al. Wilson disease protein ATP7B utilized lysosomal exocytosis to maintain copper homeostasis. Dev. Cell 2014, 29, 686–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cater, M.; la Fontaine, S.; Shield, K.; Deal, Y.; Mercer, F.B.J. ATP7B mediates vesicular sequestration of copper; Insight into biliary copper excretion. Gastroenterology 2006, 130, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Kucinskas, L.; Jeroch, J.; Vitkauskiene, A.; Sakalauskas, R.; Petrenkiene, V.; Kucinskas, V.; Naginiene, R.; Schmidt, H.; Kupcinskas, L. High frequency of the c.3207C>A (p.H1069Q) mutation in ATP7B gene of Lithuanian patients with hepatic presentation of Wilson’s disease. World J. Gastroenterol. 2008, 14, 5876–5879. [Google Scholar] [CrossRef] [PubMed]
- Gromadzka, G.; Schmidt, H.H.; Genschel, J.; Bochow, B.; Rodo, M.; Tarnacka, B.; Litwin, T.; Chabik, G.; Czonkowska, A. Frameshift and nonsense mutations in the gene for ATPase are associated with sever impairment of copper metabolism and with an early clinical manifestation of Wilson’s disease. Clin. Genet. 2005, 68, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.W.; Fraser, F.C.; Sass-Kortsak, A. A genetic study of Wilson’s disease: Evidence for heterogeneity. Am. J. Hum. Genet. 1972, 24, 646–666. [Google Scholar] [PubMed]
- Lepori, M.B.; Zappu, A.; Incollu, S.; Dessi, V.; Mameli, E.; Demelia, L.; Nurchi, A.M.; Gheorghe, L.; Maggiore, G.; Sciveres, M.; et al. Mutation analysis of the ATP7B gene in a new group of Wilson’s disease patients: Contribution to diagnosis. Mol. Cell. Probes 2012, 26, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.A.; Schilky, M.L. Diagnosis and treatment of Wilson disease: An update. Hepatology 2008, 47, 2089–2111. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Chattopadhyay, I.; Dey, S.; Nasipuri, P.; Das, S.K.; Gangopadhyay, P.K.; Ray, K. Molecular pathogenesis of Wilson disease among Indians: A perspective on mutation spectrum in ATP7B gene, prevalent defects, clinical heterogeneity and implication toward diagnosis. Cell. Mol. Neurobiol. 2007, 27, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Usta, J.; Daya, H.A.; Halawi, H.; Al-Shareef, I.; EI-Rifai, O.; Malli, A.H.; Sharara, A.I.; Habib, R.H.; Barada, K. Homozygosity for non-H1069Q missense mutation in ATP7B gene and early severe liver disease: Report of two families and a meta-analysis. JIMD Rep. 2012, 4, 129–137. [Google Scholar] [PubMed]
- Sokol, R.J.; Twedt, D.; Mckim, J.M.; Devereaux, M.W.; Karrer, F.M.; Kam, I.; Steiqman, G.; Narkewicz, M.R.; Bacon, B.P.; Britton, R.S. Oxidant injury to hepatic mitochondria in patients with Wilson’s disease and bedlington terriers with copper toxicosis. Gastroenterology 1994, 107, 1788–1798. [Google Scholar] [PubMed]
- Rossi, L.; Lombardo, M.F.; Ciriolo, M.R.; Rotillo, G. Mitochondral dysfunction in neurodegenerative disease associated with copper imbalance. Neurochem. Res. 2004, 29, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Sauer, S.W.; Merle, E.; Opp, S.; Haas, D.; Hoffmann, G.F.; Stremmel, W.; Okun, J.G. Severe dysfunction of respiratory chain and cholesterol metabolism in Atp7b−/− mice as a model for Wilson disease. Biochim. Biophys. Acta 2011, 1812, 1607–1615. [Google Scholar] [CrossRef] [PubMed]
- Arnarez, C.; Mazat, J.P.; Elezgaray, J.; Marrink, S.J.; Periole, X. Evidence for cardiolipin binding sites on the membrane-exposed surface of the cytochrome bc1. J. Am. Chem. Soc. 2013, 135, 3112–3120. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, G.; Ruggiero, F.M.; Paradies, G. Role of reactive oxygen species and cardiolipin in the release of cytochrome c from mitochondria. FAEB J. 2003, 17, 2202–2208. [Google Scholar]
- Montero, J.; Mari, M.; Collell, A.; Morales, A.; Basanez, G.; Garcia-Ruiz, C.; Femandez-Checa, J.C. Cholesterol and peroxidized cardiolipin in mitochondrial membrane properties, permeabilization and cell death. Biochim. Biophys. Acta 2010, 1797, 1217–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shadyro, O.I.; Yurkova, I.L.; Kisel, M.A.; Brede, O.; Arnhold, J. Formation of phosphatidic acid,ceramide and diglyceride on radiolysis of lipids: Identification by MALDI-TOF mass spectrometry. Free Radic. Biol. Med. 2004, 36, 1612–1624. [Google Scholar] [CrossRef] [PubMed]
- Yurkova, I.L.; Stuckert, F.; Kisel, M.A.; Shadyro, O.I.; Arnhold, J.; Huster, D. Formation of phosphatidic acid in stressed mitochondria. Arch. Biochem. Biophys. 2008, 480, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Yurkova, I.L.; Arnhold, J.; Fitzl, G.; Huster, D. Fragmentation of mitochondrial cardiolipin by copper ions in the Atp7b−/− mouse model of Wilson’s disease. Chem. Phys. Lipids 2011, 164, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Su, Y.; Wang, X.M. Phosphatidic acid-mediated signaling. Adv. Exp. Med. Biol. 2013, 991, 159–176. [Google Scholar] [PubMed]
- Kroemer, G.; Reed, J.C. Mitochondrial control of cell death. Nat. Med. 2000, 6, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Zischka, H.; Lichtmannegger, J.; Schmitt, S.; Jagemamm, N.; Schulz, S.; Wartini, D.; Jennen, L.; Rust, C.; Larochette, N.; Galluzzi, L.; et al. Liver mitochondrial membrane crosslinking and destruction in a rat model of Wilson disease. J. Clin. Investig. 2011, 121, 1508–1518. [Google Scholar] [CrossRef] [PubMed]
- Huster, D. Structural and metabolic changes in Atp7b−/− mouse liver and potential for new interventions in Wilson’s disease. Ann. N. Y. Acad. Sci. 2014, 1315, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Czlonkowska, A.; Gromadzka, G.; Chabik, G. Monozygotic female twins discordant for phenotype of Wilson’s disease. Mov. Disord. 2009, 24, 1066–1069. [Google Scholar] [CrossRef] [PubMed]
- Ferenci, P. Polymorphism of methylenetetrahydrofolate reductase as disease modifier—A deja-vu in Wilson disease? J. Hepatol. 2011, 55, 753–755. [Google Scholar] [CrossRef] [PubMed]
- Litwin, T.; Gromadzka, G.; Czlonkowska, A. Apolipoprotein E gene (APOE) genotype in Wilson’s disease: Impact on clinical presentation. Parkinsonism Relat. Disord. 2012, 18, 367–369. [Google Scholar] [CrossRef] [PubMed]
- Cocos, R.; Sendroiu, A.; Schipor, S.; Bohitea, L.C.; Sendroiu, I. Genotype-phenotype correlations in a mountain population community with high prevalence of Wilson’s disease: Genetic and clinical homogeneity. PLoS One 2014, 9, e98520. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Shiono, Y.; Hayashi, H.; Satoh, H.; Sawada, T.; Suzuki, A.; Takeda, Y.; Yano, M.; Michitaka, K.; Onji, M.; et al. Mutational analysis of ATP7B and genotype-phenotype correlation in Japanese with Wilson’s disease. Hum. Mutat. 2000, 15, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Q.; Zhang, Y.F.; Liu, T.T.; Hsiao, K.J.; Zhang, J.M.; Gu, X.F.; Bao, K.R.; Yu, L.H.; Wang, M.X. Correlation of ATP7B genotype with phenotype in Chinese patients with Wilson disease. World J. Gastroenterol. 2004, 15, 590–593. [Google Scholar]
- Lee, B.H.; Kin, J.H.; Lee, S.Y.; Jin, H.Y.; Kim, K.J. Distinct clinical courses according to presenting phenotypes and their correlations to ATP7B mutations in a large Wilson’s disease cohort. Liver Int. 2011, 31, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Krijt, J.; Duta, A.; Kozich, V. Determination of S-adenosylmethionine and S-adenosylhomocysteine by LC-MS/MS and evaluation of their stability in mice tissues. J. Chromatogr. B 2009, 877, 2061–2966. [Google Scholar] [CrossRef]
- Le, A.; Shibata, N.M.; French, S.W.; Kim, K.; Kharbanba, K.K.; Islam, M.S.; LaSalle, J.M.; Halsted, C.H.; Keen, C.L.; Medici, V. Characterization of timed changes in hepatic copper concentrations, methionine metabolism, gene expression, and global DNA methylation in the Jackson toxic milk mouse model of Wilson disease. Int. J. Mol. Sci. 2014, 15, 8004–8023. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Yang, M.; Chen, Z.; Huang, Y.; Zhao, Q.; Xu, J.; Ren, H.; Zhao, H.; Chen, Z.; Ren, Q.; et al. Decreased DNA methyltransferase 3A and 3B mRNA expression in peripheral blood mononuclear cells and increased plasma SAH concentration in adult patients with idiopathic thrombocytopenic purpura. J. Clin. Immunol. 2008, 28, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Medici, V.; Shibata, N.M.; Kharbanda, K.K.; LaSalle, J.M.; Woods, R.; Liu, S.; Engelberg, J.A.; Devaraj, S.; Torok, N.J.; Jiang, J.X.; et al. Wilson’s disease: Changes in methionine metabolism and inflammation affect global DNA methylation in early liver disease. Hepatology 2013, 57, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Oh, B.K.; Kim, H.; Park, H.J.; Shim, Y.H.; Choi, J.; Park, C.; Park, Y.N. DNA methyltransferase expression and DNA methylation in human hepatocellular carcinoma and their clinicopathological correlation. Int. J. Mol. Med. 2007, 20, 65–73. [Google Scholar] [PubMed]
- Levy, E.; Brunet, S.; Alvarez, F.; Seidman, E.; Bouchard, G.; Escobar, E.; Martin, S. Abnormal hepatobiliary and circulating lipid metabolism in the Long-Evans Cinnamon rat model of Wilson’s disease. Life Sci. 2007, 16, 1472–1483. [Google Scholar] [CrossRef]
- Huster, D.; Purnat, T.D.; Burkhead, J.L.; Ralle, M.; Fiehn, O.; Stuckert, F.; Olson, N.E.; Teupser, D.; Lutsenko, S. High copper selectively alters lipid metabolism and cell cycle machinery in the mouse model of Wilson disease. J. Biol. Chem. 2007, 282, 8343–8355. [Google Scholar] [CrossRef] [PubMed]
- European association for the study of the liver. EASL Clinical Practice Guidelines: Wilson’s disease. J. Hepatol. 2012, 56, 671–685. [Google Scholar]
- Burkhead, J.L.; Gray, L.W.; Lutsenko, S. Systems biology approach to Wilson’s disease. Biometals 2011, 24, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.; Hahn, S.H. Clinical molecular diagnosis of Wilson disease. Semin. Liver Dis. 2011, 31, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.T.; Schwarz, K.B.; Swanson, P.D.; Hahn, S.H. An exceptional family with three consecutive generations affected by Wilson disease. JIMD Rep. 2013, 10, 1–4. [Google Scholar] [PubMed]
- Brewer, G.J. Diagnosis of Wilson’s disease. Neth. J. Med. 2009, 67, 195. [Google Scholar] [PubMed]
- Purchase, R. The treatment of Wilson’s disease, a rare genetic disorder of copper metabolism. Sci. Prog. 2013, 96, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Askari, F.K.; Greenson; Dick, R.D.; Johnson, V.D.; Brewre, G.J. Treatment of Wilson’s disease with zinc.XVIII. Initial treatment of the hepatic decompensation presentation with trientine and zinc. J. Lab. Clin. Med. 2003, 142, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Ala, A.; Borjigin, J.; Rochwarger, A.; Schilsky, M. Wlison disease in septuagenarian siblings: Raising the bar for diagnosis. Hepatology 2005, 41, 668–670. [Google Scholar] [CrossRef] [PubMed]
- Marin, C.; Robles, R.; Parrilla, G.; Ramirez, P.; Bueno, F.S.; Parrilla, P. Liver transplantation in Wilson’s disease: Are its indications established? Transplant. Proc. 2007, 39, 2300–2301. [Google Scholar] [CrossRef] [PubMed]
- Schilsky, M.L. Wilson disease: Current status and the future. Biochimie 2009, 91, 1278–1281. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S.T.; Corbineau, S.; Hannan, N.; Marciniak, S.J.; Miranda, E.; Alexander, G.; Huang-Doran, I.; Griffin, J.; Ahrlund-Richter, L.; Skepper, J.; et al. Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. J. Clin. Investig. 2010, 120, 3127–3136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Q.; Chen, S.; Li, W.; Guo, X.P.; Zhao, P.; Xu, J.Y.; Chen, Y.; Pan, Q.; Liu, X.R.; Zychlinski, D.; et al. Rescue of ATP7B function in hepatocyte-like cells from Wilson’s disease induced pluripotent stem cells using gene therapy or the chaperone drug curcumin. Hum. Mol. Genet. 2011, 20, 3176–3187. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S. Cell therapy to remove excess copper in Wilson’s disease. Ann. N. Y. Acad. Sci. 2014, 1315, 70–80. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, F.; Wang, J.; Pu, C.; Qiao, L.; Jiang, C. Wilson’s Disease: A Comprehensive Review of the Molecular Mechanisms. Int. J. Mol. Sci. 2015, 16, 6419-6431. https://doi.org/10.3390/ijms16036419
Wu F, Wang J, Pu C, Qiao L, Jiang C. Wilson’s Disease: A Comprehensive Review of the Molecular Mechanisms. International Journal of Molecular Sciences. 2015; 16(3):6419-6431. https://doi.org/10.3390/ijms16036419
Chicago/Turabian StyleWu, Fei, Jing Wang, Chunwen Pu, Liang Qiao, and Chunmeng Jiang. 2015. "Wilson’s Disease: A Comprehensive Review of the Molecular Mechanisms" International Journal of Molecular Sciences 16, no. 3: 6419-6431. https://doi.org/10.3390/ijms16036419
APA StyleWu, F., Wang, J., Pu, C., Qiao, L., & Jiang, C. (2015). Wilson’s Disease: A Comprehensive Review of the Molecular Mechanisms. International Journal of Molecular Sciences, 16(3), 6419-6431. https://doi.org/10.3390/ijms16036419