2.2. Structures and Vibrational Frequencies
In the present section, we will attempt to localize and identify the structural characteristics and energetic of the C–Br and O–Br bonding interactions.
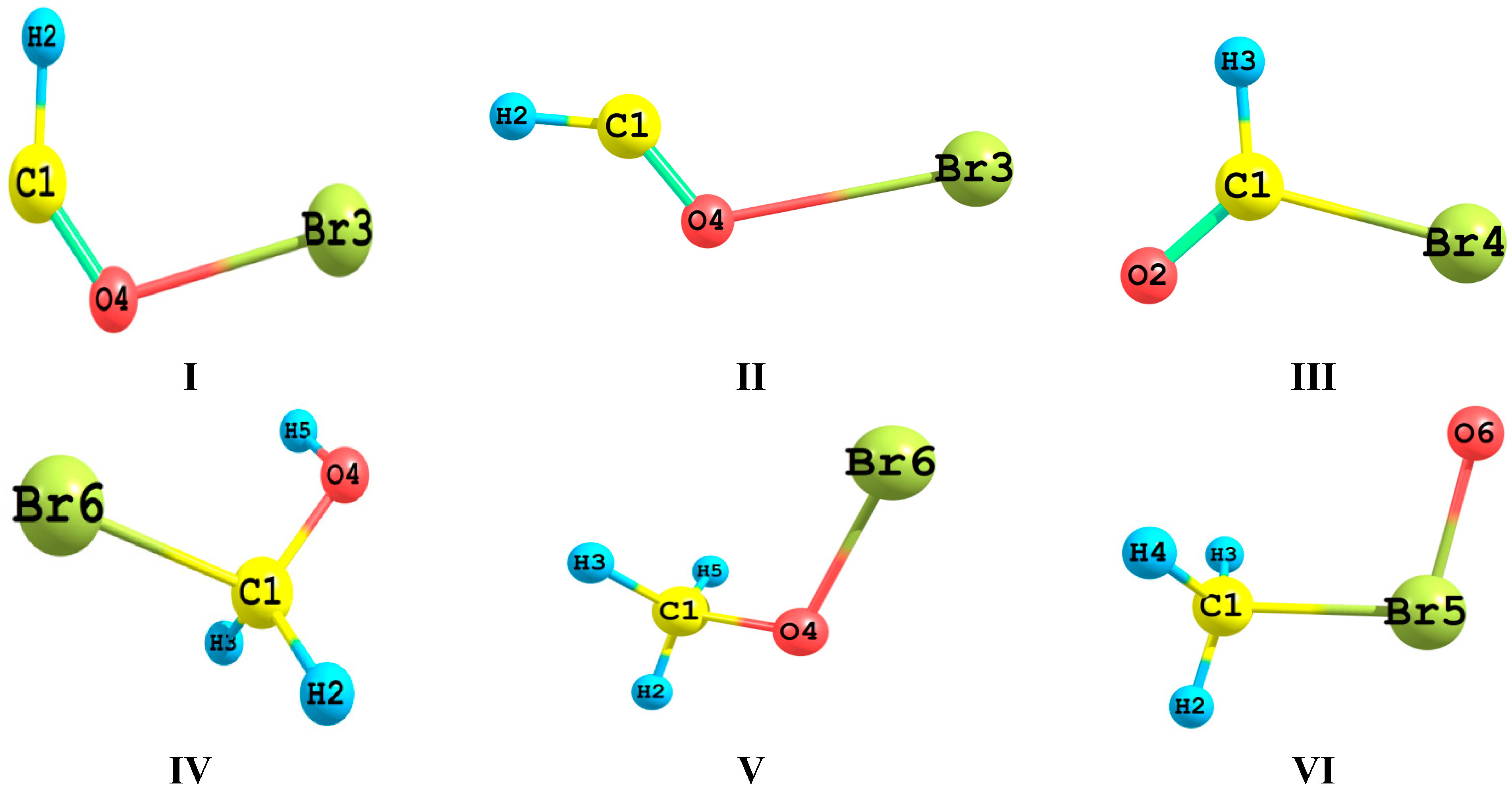
Figure 1 presented the optimized geometries of the bromine-containing species which will be the subject of the present study.
Let us start by studying the geometric features of the isomeric system CH3OBr/CH3BrO/BrCH2OH. The alcohol form BrCH2OH is much more stable by 33.9 and 106.2 kcal/mol than CH3OBr and its hypervalent bromine isomer CH3BrO isomer, respectively. The three isomers possess staggered structures with Cs symmetry.
The geometry differences are associated and focus on methyl torsions; the associated potential energy functions often exhibit significant skeletal flexing perturbations (couplings to stretching and bonding degrees of freedom) that modify an idealized rigid-rotor picture.
Figure 2 compares the geometric features of the rigid-rotor limit of idealized Pople–Gordon (PG) geometry (that is localized geometry in the valence bond natural bond orbital (NBO) basis) [
21] and DFT/B3LYP/aug-cc-pVTZ optimized structures in the delocalized Molecular orbital (MO) basis for the isomeric system CH
3OBr/CH
3BrO/BrCH
2OH. The optimized geometries deviate strikingly from idealized PG form particularly with respect to Br…H separation and the dihedral angle specifically in case of BrCH
2OH. For CH
3OBr the optimized C–O–Br angle open significantly 112.5° thereby partially relieve the apparent steric congestion. This is a result of the mutual repulsion of the lone pair electrons localized on both the Br and O atoms. The isomeric form, CH
3BrO is also enjoying a staggered conformation with the C–Br–O angle much smaller
ca. 103°. This smaller value of the C–Br–O angle suggests that there is a π-overlap between the lone-pair electrons of the bromine atom and that of the oxygen atom. This π-overlap imparts partial double bond character to the Br–O bond leading to decrease in its length to ~1.7 Å with the consequent decrease in electron repulsion in the carbon–bromine bond region. This point may be further clarified by NBO analysis of the total self-consistent field (SCF), deletion and delocalization energies of the tautomeric system. In these calculations, all the Rydberg and antibond orbitals, which are the non-Lewis NBO orbitals, that usually appear in the NBO analysis are deleted. The result of this deletion is the energy of the idealized NBO natural Lewis structure, with all Lewis NBOs doubly occupied. Unlike other deletions, in which there is a slight loss of variational self-consistency due to the redistributed occupancy of the deleted orbitals, the result of this deletion corresponds rigorously to the variational expectation value of the determinant of doubly occupied Lewis NBOs. This analysis is presented in
Table 2. For the tautomeric system IV, V and VI, IV is the most stable by 34.742 and 77.295 kcal/mol compared to V and VI, respectively. In an ideal Lewis Structure, V is more stable than IV and VI by 23.947 and 75.054 kcal/mol, respectively. The competitiveness of IV is due to the strong delocalization gaining 140.285 kcal/mol. The geometric features of IV reveal the origin of its competitiveness. Thus, the C–Br bond is now longer than 2.0 Å while the C–O bond approaches a double bond length with a value of 1.37 Å. The O–C–Br bond angle is 112°. The interaction in the C–O bond region is markedly increased at the expense of that in the C–Br. However, the repulsive interaction involving the lone pair electrons residing on the Br atom causes the observed enlargement of the Br–C–O angle.
Figure 2.
Comparison of the geometric features of the idealized PG and DFT optimized structure (Opt) for the isomeric system CH3OBr/CH3BrO/BrCH2OH.
Figure 2.
Comparison of the geometric features of the idealized PG and DFT optimized structure (Opt) for the isomeric system CH3OBr/CH3BrO/BrCH2OH.
Table 2.
NBO analyses of the total SCF energy (au), deletion (au) and delocalization (kcal/mol) energies of the studied bromine-containing species computed at the B3LYP/aug-cc-pVTZ level of theory.
Table 2.
NBO analyses of the total SCF energy (au), deletion (au) and delocalization (kcal/mol) energies of the studied bromine-containing species computed at the B3LYP/aug-cc-pVTZ level of theory.
| Substrate | Total SCF Energy | Deletion Energy | Delocalization Energy | Rotation Barrier, kcal/mol |
|---|
| I | −2688.09805 | −2682.98976 | 3205.503 | |
| II | −2688.08126 | −2683.11274 | 3117.798 | |
| III | −2688.19984 | −2687.94231 | 161.600 | |
| IV | −2689.41115 | −2689.18759 | 140.285 | 42.406 |
| V | −2689.35579 | −2689.22576 | 81.596 | 2.749 |
| VI | −2689.28797 | −2689.10615 | 114.097 | 0.763 |
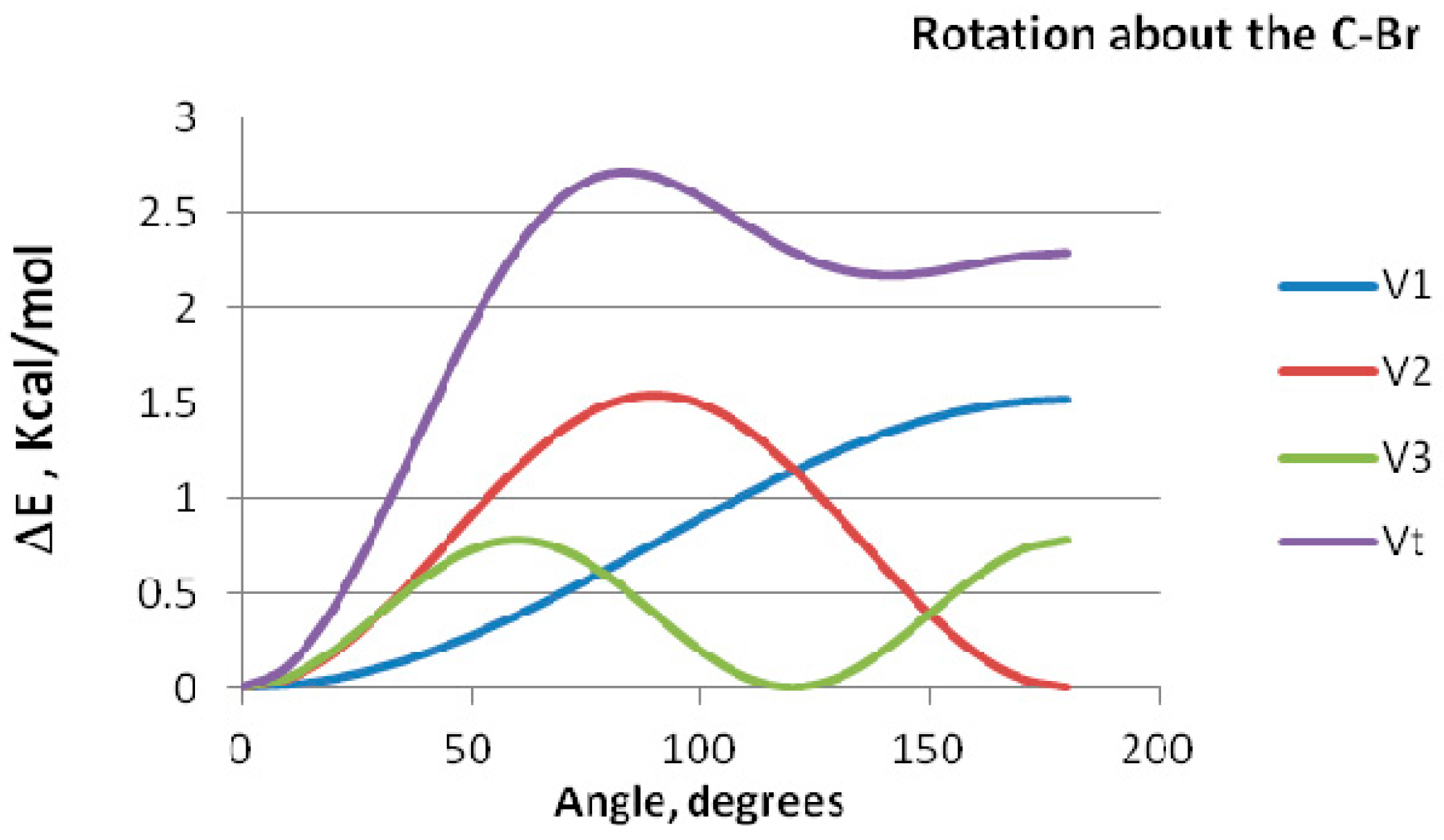
The basic issue is whether the stabilizing interactions in this tautomeric system should be primarily regarded as decrease of “steric repulsion” or increase of “bonding attraction”. Direct calculation of the potential energy function for rotation around the C–Br and the O–Br bonds and its Fourier transform analysis can be brought to bear on this issue.
The computed rotation barriers are depicted in
Table 2. The large difference in magnitude between the energy barriers for BrCH
2OH and that of the CH
3OBr and CH
3BrO indicate clearly that rotation about C–Br and O–Br bonds is of a completely different nature than that about the C–O bond. Delocalization interaction underlies the large rotation barrier for rotation about the C–O bond in BrCH
2OH. The origin of this rotational barrier can be identified by Fourier analysis of internal rotation function as detailed by Pople
et al. [
22,
23]. In this analysis, the potential energy is partitioned into components, namely bond dipoles, conjugative and electrostatic repulsion contributions The Fourier transform equation can be represented as V(φ) = V
1(φ) (1 − cos φ) + V
2(φ) (1 − cos2φ) + V
3(φ) (1 − cos3φ), where φ is the rotation angle, the V
1 represents the interaction of bond dipoles, V
2 represents the conjugative interaction, and V
3 is an electrostatic bond–bond repulsion term. Fourier decomposition of the potential function for CH
3BrO has been performed; results are displayed in
Figure 3.
Figure 3.
Fourier decomposition of the potential function for rotation about the C–Br bond in CH3BrO.
Figure 3.
Fourier decomposition of the potential function for rotation about the C–Br bond in CH3BrO.
All three components of the Fourier transform contribute significantly to the potential barrier. Thus, the one-fold term shows maximum repulsion between local dipoles at φ = 180°, and maximum attraction at φ = 0°. Conjugative stabilization, as reflected by the V2 term, is maximum at φ = 0°and 180°. The three fold electrostatic interaction component shows maximum stabilization at φ = 0° and 120° and is repulsive otherwise. The potential energy function of rotation for CH3OBr shows almost the same trend.
HCOBr is one of the decomposition products of the isomeric system CH
3OBr/CH
3BrO, which is believed to play a central role in O
3 depletion in the Arctic troposphere. This species can exit in a
cis(I) or a
trans(II) conformations. It should be mentioned that the optimization of this species always falls into the
cis-conformation, which seems to be the global minimum structure on the potential energy hypersurface. The
trans-conformation can only be obtained by freezing the dihedral angle during full optimization. The cis form (I) is more stable by 10.825 kcal/mol. The geometric features of this species shows the longest C–Br bond length of an average value of 2.5 Å, reflecting a minimal attractive interaction of the
d-orbitals of Br– with the
p-orbitals of carbon. The C–O bond length shows its minimum value in this species which reflects a tight binding and pronounced double bond character. This is further confirmed by a C–O–Br bond angle approaching 120°, a typical
sp2 hybridization scheme for the oxygen atom. The isoelectronic compound BrCHO (bromo formaldehyde) (III) shows the same bonding characteristics as the HCOBr species. Comparison of the NBO results for the total SCF, deletion and delocalization energies of the tautomeric system I, II and III is presented in
Table 2. It is apparent that the structure III is more stable than I and II by 63.877 and 74.410 kcal/mol, respectively. In a hypothetical Lewis structure, I is slightly more stable than III. The competitiveness of I and II are brought about by extensive hyperconjugation that gained them 3205.503 and 3117.798 kcal/mol, respectively.
Table 3 displays the vibration frequencies and intensities of the relevant Br–O and C–Br bonds and angles. For comparison, frequencies reported in the literature [
15,
20] are included in the same table. Data in
Table 3 reveal that there are two types of C–O bonds in the studied compounds. C=O bonds vibrate at higher frequencies (1820–1950 cm
−1 range) with much higher intensities than that of the C–O stretching vibrations which appear at lower frequencies (in the 1050–1100 cm
−1 range). The correspondence with the literature values are satisfactory bearing in mind that our frequency calculations are carried out within the harmonic approximation. In order to evaluate and assess the importance of anharmonic corrections of vibration modes, calculations have been performed taking in account the anharmonicity of the vibrations. The calculations were carried out in the gas phase using the VPT2 method as implemented by Barone [
24,
25] in the Gaussian program package [
26]. In all cases, the finest DFT integration grid was selected by using SCF = tight in the command line. Results are included in
Table 3. As a general trend, the anharmonic vibration frequencies are lower than the corresponding harmonic frequencies by few frequency units. There is also a clear but small effect on the computed anharmonic intensities. Furthermore, attachment of a Br atom to the carbon of a carbonyl group causes a blue shift and lowering of the intensity of the C–O stretching vibration. The presence of a C=O group shifts considerably the electron density away from the O–Br or the C–Br bond regions with a consequent blue shift of the O–Br and the C–Br stretching frequencies. Thus, in case of BrOCH the Br–O stretching mode appear at a very low frequency of 160–196 cm
−1 as compared to 620–730 cm
−1 range for this mode in case of CH
3OBr, its tautomer CH
3BrO and isomer BrCH
2OH. Calculation of the vibration spectrum of the OBr radical predicted the stretching vibration mode of the O–Br bond at 742 cm
−1. In case of the CH
3BrO where a methyl group is attached to the bromine, ν
Br–O shifts but slightly down field where as such a shift became more pronounced in case of the tautomeric compound CH
3OBr, where ν
Br–O appears at 625 cm
−1.
Table 3.
Vibrational Frequencies and Intensities (km/mol) for the studied bromine-containing compounds.
Table 3.
Vibrational Frequencies and Intensities (km/mol) for the studied bromine-containing compounds.
| Species | ν, cm−1 | Relative Intensity | νanharmonic, cm−1 | Relative Intensity (Anharmonic) | Assignment |
|---|
| cis-BrOCH (I) | 1924.6 (2068) a | 601.1 | 1905.218 | 613 | C–O str. |
| 196.8 (206) a | 65.3 | 197.912 | 59 | O–Br str. |
| 292.2 (348) a | 13.0 | 293.888 | 13 | Br–O–C angle bending |
| trans-BrOCH (II) | 1979.7 | 588.5 | 1957.464 | 590 | C–O str. |
| 161.6 | 71.44 | 157.284 | 62 | O–Br str. |
| 253.8 | 86.2 | 221.901 | 97 | Br–O–C angle bending |
| BrCHO (III) | 1851.0 (1799) a | 466 | 1836.751 | 484 | C–O str. |
| 633.1 (663) a | 168 | 626.148 | 172 | C–Br str. |
| 351.5 (370) a | 13 | 350.071 | 14 | O–C–Br angle bending |
| BrCH2OH (IV) | 1102.7 (1126) a | 286.1 | 1069.0 | 299 | C–O str. |
| 560.5 (625) a | 107 | 549.1 | 129 | C–Br str. |
| 284.0 (306) a | 37 | 290.0 | 22 | O–C–Br angle bendin |
| CH3OBr (V) | 1054.4 (1048) a | 63.2 | 1466.6 | 11 | C–O str. |
| 998,1 (581) a | 40 | 967.0 | 38 | O–Br str. |
| 307.1 (319) a | 3 | 303.5 | 4 | C–O–Br angle bending |
| CH3BrO (VI) | 657.3 (723) b | 40.1 | 648.6 | 31 | Br–O str. |
| 516.7 (530) b | 1.9 | 505.6 | 2 | C–Br str. |
| 207.7 (222) b | 5.5 | 208.0 | 6 | C–Br–O angle bending |
2.3. NBO-Based Quantification of Stereoelectronic Interactions
Table 4 lists the second order perturbation estimates of the hyperconjugative energies of I, II and III. The factors that contribute to the total stabilities of tautomers include the steric hindrance, electrostatic repulsion and hyperconjugation. As shown in
Figure 1, the steric hindrance and electrostatic repulsion are minimal in III compared to II and I. These two factors have led to the overall stability of III compared to I and II by 63.877 and 74.410 kcal/mol, respectively. It is clear that I experiences the maximum steric effect with comparable electrostatic repulsion compared to II. Despite this, I is more stable than II by 10.533 kcal/mol.
The competitiveness of I compared to II is due to the strong n2O4→σ*C1–Br3, σ*C1–Br3→σ*C1–O4, σ*C1–Br3→n2O4 and n1O4→n2O4 interactions that yielded 187.75, 11.16, 13.13 and 15.38 kcal/mol for the former and 195.27, 4.81, 6.93 and 1.23 kcal/mol for the later. It is clear that although the n2O4→σ*C1–Br3 interaction favours II by 7.52 kcal/mol; the later three interactions favour I by 26.70 kcal/mol.
As shown in
Table 4, II has been favoured by 77.172 kcal/mol due to the minimal steric hindrance, while I gained 87.704 kcal/mol compared to II as a result of the strong delocalization. That is, the overall preference of I of 10.532 kcal/mol is mainly due to hyperconjugation.
Tables S3 and S4 of the supplementary data show the details of preference of III compared to I (63.877 kcal/mol) and II (74.410 kcal/mol). This means that I, II and III have fairly comparable total energies. I and II have bridged the energy gap through extensive hyperconjugation.
Table 4.
Second order perturbation (E(2)) estimation of the hyperconjugative energies (kcal/mol) of cis-BrOCH (I), trans-BrOCH (II), and BrCHO (III) which were calculated using B3LYP/aug-cc-pVTZ level of theory.
Table 4.
Second order perturbation (E(2)) estimation of the hyperconjugative energies (kcal/mol) of cis-BrOCH (I), trans-BrOCH (II), and BrCHO (III) which were calculated using B3LYP/aug-cc-pVTZ level of theory.
| Interaction | I | II | III |
|---|
| σC1–H2→n2O4 | 6.58 | 9.74 | – |
| σC1–H2→σ*C1–Br3 | 7.71 | 8.27 | 3.52 |
| σC1–Br3→σ*C1–H2 | 3.82 | 4.18 | 1.90 |
| σ*C1–Br3→σ*C1–O4 | 11.16 | 4.81 | – |
| σ*C1–Br3→σC1–Br3 | 8.30 | 7.72 | 1.76 |
| σ*C1–Br3→n2O4 | 13.13 | 6.93 | – |
| n1O4→n2O4 | 15.38 | 1.23 | – |
| n1O4→σ*C1–H2 | 2.01 | – | 1.32 |
| n1O4→σ*C1–Br3 | 4.02 | – | – |
| n2O4→σ*C1–H2 | 5.69 | 7.92 | 20.31 |
| n2O4→σ*C1–Br3 | 3.17 | 12.06 | 52.67 |
| n2O4→σ*C1–Br3 | 187.75 | 195.27 | – |
| n2O4→σ*C1–O4 | 5.25 | 13.14 | – |
| n2Br3→σ*C1–Br3 | 2.52 | 3.03 | – |
| n2Br3→σ*C1–O4 | – | – | 5.03 |
| n3Br3→πC1–O4 | – | – | 16.55 |
| Total | 276.50 | 274.30 | 103.06 |
Table 5 depicts the second order perturbation estimates of the hyperconjugative energies of IV, V and VI. NBO analyses of the total SCF, deletion and delocalization energies (a.u.) of IV, V and VI are listed in
Tables S5–S8 of the supplementary data. It is clear that IV is more stable than V and VI by 34.741 and 77.295 kcal/mol, respectively. In an ideal Lewis Structure V becomes the most stable followed by IV despite the electrostatic repulsion between the negatively charged carbon and oxygen atoms. This is perhaps due to the proximity of the two Br and CH
3 bulky groups in both IV and VI. It is worth mentioning that V is more stable than VI by 42.553 kcal/mol. Not just that, also the ideal Lewis Structure of V is more stable than that of VI by 75.054 kcal/mol.
This stems from the strong steric hindrance between the two bulky CH3 and Br groups and the ineffective electrostatic repulsion between the negatively charged carbon and oxygen atoms and the attraction between the negatively charged carbon atom and positively charged bromine atom. However, the total energies of the two tautomers are comparable. The competitiveness of VI is due to hyperconjugation that yielded 32.501 kcal/mol compared to that of V.
The most influential hyperconjugative interactions (
cf.
Table 5) (n
2O4)n
2Br5→σ*
C1–Br6, (n
2O4)n
2Br5→σ*
C1–H3(σ*
C1–H5), (n
2Br6)n
3O6→σ*
C1–Br5(σ*
C1–O4) and n
3Br6→σ*
C1–O4, have contributed 21.09, 3.57, ˂0.5 and 5.25 kcal/mol respectively for IV; 5.61, 5.61, 1.48 and 1.61 kcal/mol respectively for V and 2.30, 2.19, ˂0.5 and 15.83 kcal/mol, respectively for VI. Therefore the origin of preference of IV is mainly due to the n
2O4→σ*
C1–Br6 and n
3Br6→σ*
C1–O4 donor–acceptor interactions; while the vicinal antiperiplanar σ
C1–H2→σ*
O4–Br6 and lone pairs-antibonding n
2O4→σ*
C1–H3 and n
2O4→σ*
C1–H5 interactions contribute mostly in V. However, the most influential hyperconjugation of VI was a lone pair donor and antibonding bond acceptor (n
3O6→σ*
C1–Br5).
Table 5.
Second order perturbation (E(2)) estimation of the hyperconjugative energies (kcal/mol) of BrCH2OH(IV), CH3OBr (V), and CH3BrO(VI) which were calculated using B3LYP/aug-cc-pVTZ level of theory.
Table 5.
Second order perturbation (E(2)) estimation of the hyperconjugative energies (kcal/mol) of BrCH2OH(IV), CH3OBr (V), and CH3BrO(VI) which were calculated using B3LYP/aug-cc-pVTZ level of theory.
| Interaction | IV | V | VI |
|---|
| σC1–H2→σ*O4–H5 (σ*O4–Br6)(σ*Br5–O6) | 3.09 | 6.63 | 1.42 |
| σO4–H5(σO4–Br6)→σ*C1–H2 | 2.16 | 2.25 | – |
| σC1–H2→σ*C1–Br5 | – | – | 1.63 |
| n1O4→σ*C1–H2 | 2.87 | 1.61 | – |
| σC1–H3→σ*C1–Br5 | – | – | 1.35 |
| σC1–H4→σ*C1–Br5 | – | – | 1.36 |
| (n2O4)n2Br5→σ*C1–Br6(σ*C1–H3)(σ*C1–H4) | 21.09 | 5.61 | 2.30 |
| (n2O4)n2Br5→σ*C1–H3(σ*C1–H5) | 3.57 | 5.61 | 2.19 |
| n1O4→σ*C1–H3 | 2.95 | – | – |
| (n2Br6)n3O6→σ*C1–Br5(σ*C1–O4) | – | 1.48 | 15.83 |
| n3Br6→σ*C1–O4 | 5.25 | 1.61 | – |
| σC1–Br5→σ*Br5–O6 | – | – | 1.11 |
| n2Br6→σ*C1–H2 | 1.55 | – | – |
| n2Br6→σ*C1–H3 | 1.71 | – | – |
| Total | 44.24 | 24.80 | 27.19 |
The C–F bond has been shown [
27] to impact the conformational preferences of organic compounds suggesting its potential utility as a molecular design tool. This impact of the conformational preferences is due to stereoelectronic effects, specifically the anomeric [
28] and gaughe effects [
29]. These effects may be defined as the hyperconjugative interaction of the fluorine lone pair with the σ framework. There is no corresponding studies on the C–Br bonds. It is thus interesting to examine, quantitatively, (1) whether such effects exists in the case of the C–Br and O–Br bonds; and (2) the extent to which it impacts the conformational preference of the studied compounds. To that end the interactions of the bromine lone pair (nBr) with the σ* C–H MO have been computed as a function of torsion angle of rotation about the C–Br and O–Br bonds. Results are displayed in
Figure 4a,b.
Figure 4 displays the calculated NBO energetic components for rotation about the C–Br and O–Br bonds as a function of torsion angle. Each of the two figures displays torsion variation of hyperconjugative interactions of the Br lone pairs n(2) and n(3) with the σ*C–H MO’s. These are the leading donor–acceptor contributions arising from vicinal hyperconjugative interactions with neighboring sigma-bond. In case of CH
3OBr an additional donor–acceptor interaction term results from the Br(n) interaction with the σ* acceptor orbitals of the C–O NBO. These interaction curves bear similarity to the potential function for rotation displayed in
Figure 2. It is clear from the similar vicinal environment in each of the two cases studied; the hyperconjugative details of methyl rotation are seen to be very similar. In case of CH
3BrO, the two Br(n)-σ*(C–H) interactions work in a reciprocal way but their effects are cooperative, in general. For the CH
3OBr case, the Br(n)-σ*CH and the Br(n)-σ*CO work cooperatively in a periodic manner, whereas, the second Br(n)-σ*CH interaction behave differently. It has a destabilizing effect in the 60°–120° region.
Figure 4.
Variation of the leading NBO donor–acceptor interaction energies with the torsion angle of rotation about (a) O–Br and (b) C–Br bonds.
Figure 4.
Variation of the leading NBO donor–acceptor interaction energies with the torsion angle of rotation about (a) O–Br and (b) C–Br bonds.
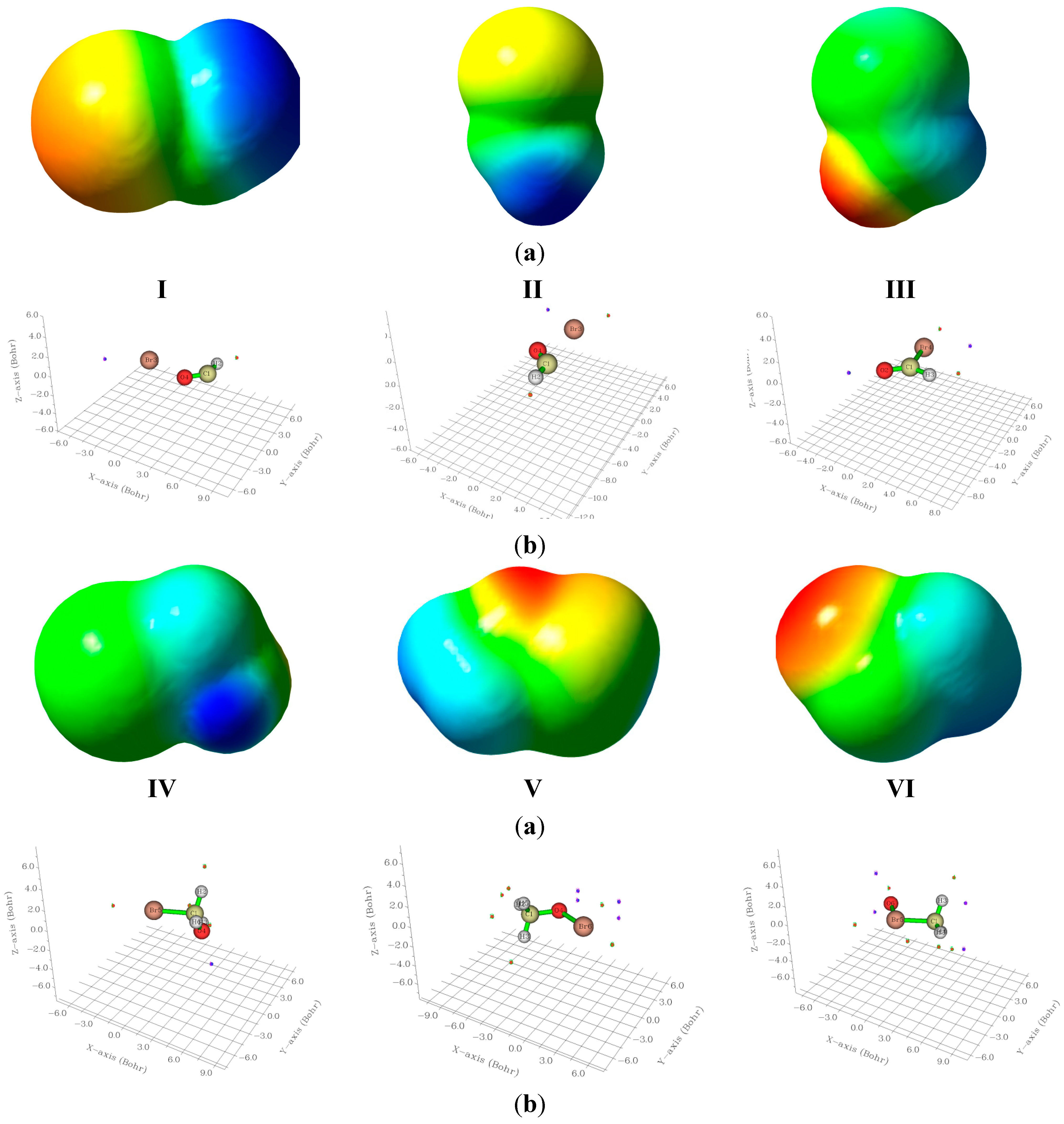
2.4. Electrostatic Potentials and Quantum Chemical Topology Analyses
The 3D electrostatic potential maps for the studied bromine species, are computed and visualized using GaussView 5.0 [
30] and are displayed in
Figure 5. The location of the surface maxima (red) and surface minima (blue) are computed and visualized using the Multiwfn software package [
31,
32]. In this software package the default isovalue is 0.001 au. The points so computed are illustrated in a 3D graphs in the same figure. The magnitudes of the most positive (V
S,max) and most negative (V
S,min), surface points are given in
Table 6. In all molecular species studied there exists a surface maximum (positive hole) along the C–Br and the O–Br bonds at the end region of the bromine atom. In all cases, these surface maxima are surrounded by an electroneutral region and surface local minimum point alongside the X–Br bond near the Br atom (
cf.
Figure 5). Such a bromine positive region is referred to as the σ-hole, and seems to be a characteristic of all C–halogen bonds [
33,
34,
35].
The bromine σ-hole in BrCH2OH (IV) shows its smallest value. This is most probably due to the large positive electrostatic potential of 78.902 kcal/mol surrounding the carbon atom. The Br σ-hole in BrCHO(III) is almost double that in the case of I although the bonding situations seem similar. In II, the positive potential is concentrated on H rather than the carbon atom. The isomeric system CH3OBr/CH3BrO shows also different trends for both the σ-holes and the surface minima around the oxygen atoms. Thus, the hypervalent bromine shows a smaller σ-hole and almost double negative potential at the oxygen atom. These represent global minimum on the surface, its large negative value is owing to the lone pair of oxygen. In case of BrOCH (cis)(I) the σ-hole is huge (107.88 kcal/mol) in contrast to its trans isomer (II) which shows a much smaller value.
Figure 5.
(a) Electrostatic potentials on the 0.001 au electron density surface; and (b) three dimensional graphs indicating the most positive maxima (red) and most negative minima (blue) surface points, for compounds I–VI.
Figure 5.
(a) Electrostatic potentials on the 0.001 au electron density surface; and (b) three dimensional graphs indicating the most positive maxima (red) and most negative minima (blue) surface points, for compounds I–VI.
Table 6 also shows some topological properties for the studied bromine species computed at the QTAIM level of theory. The theoretical basis of the theory has been detailed before and will not be repeated here [
36]. The Laplacian of the electron density at the bond critical points (BCP) of the isomeric system CH
3OBr/CH
3BrO/BrCH
2OH are all negative indicating accumulation of charge density in the X–Br bond region. However, their magnitudes vary considerably. Thus, while ∇
2ρ(r) at the O–Br BCP is remarkably large in case of CH
3OBr, its value is very small in case of the alcohol form. This behavior is consistent with the values of V
S,max reported in
Table 5 where IV shows the lowest value. The three species I, II and VII show positive ∇
2ρ(r) values, indicating depletion of charge density away from the bonding region. The ellipticity values are small indicating structure stability to varying extent; its relatively largest values are associated with the hypervalent bromine compound VI.
Table 6.
Computed electrostatic potential maxima (VS,max) and minima (VS,min) on the 0.001 a.u. electron-density contours (values are in kcal/mol), Laplacian bond order and the delocalization index DI(A,B), the electron density at the BCP’s and its Laplacian and ellipticity of the C–Br and O–Br bonds.
Table 6.
Computed electrostatic potential maxima (VS,max) and minima (VS,min) on the 0.001 a.u. electron-density contours (values are in kcal/mol), Laplacian bond order and the delocalization index DI(A,B), the electron density at the BCP’s and its Laplacian and ellipticity of the C–Br and O–Br bonds.
| Species | VS,max | VS,min | LBO | DI (A|B) | ρ (r) | ∇2ρ (r) | ε |
|---|
| I | 107.878 (Br) | −74.725 | 0.036 | 0.335 | 0.049 | 0.148 | 0.012 |
| II | 67.630 (H) | −32.094 | 0.012 | 0.037 | 0.480 | 0.094 | 0.053 |
| 21.672 (Br) | | | | | | |
| III | 20.693 (Br) | −56.076 | 0.298 | 1.160 | 0.122 | −0.015 | 0.066 |
| IV | 10.936 (Br) | −54.658 | 0.229 | 1.060 | 0.114 | −0.012 | 0.024 |
| V | 17.589 (Br) | −41.306 | 0.319 | 0.928 | 0.293 | −1.162 | 0.042 |
| | −41.305 | | | | | |
| VI | 26.623 (Br) | −21.026 | 0.123 | 0.996 | 0.105 | −0.122 | 0.105 |
| | −21.026 | | | | −0.122 * | 0.105 * |
| VII (HOBr) | 75.444 (H) | −68.255 | 0.181 | 1.249 | 0.142 | 0.187 | 0.046 |
| 52.797 (Br) | | | | | | |
The final topological parameter to be discussed in the present context is the Laplacian bond order (LBO) also included in
Table 5. LBO can be simply defined as ∇
2ρ(r) in fuzzy overlap space and may be given [
37] by LBO
A,B = −10 × ʃ W
A(r)W
B(r)∇
2ρ(r) dr, where the integration goes for negative values of ∇
2ρ(r) only (∇
2ρ(r) < 0) where w is a weighting function proposed by Becke representing the fuzzy space. A detailed discussion of LBO is given elsewhere [
36]. In case of fuzzy partitioning of the atomic space there are no clear boundaries between adjacent atoms. This is in contrast to the basis on which the QTAIM is based where it adopts a discrete partitioning of the atomic space between adjacent atoms. Therefore, no overlap spaces between atoms and consequently their atomic spaces are mutually exclusive. LBO values given in
Table 5 seems to describe bonding in the C–Br and O–Br regions better than ∇
2ρ(r). Linearly fitting the computed LBO values to νC–Br and νO–Br results in good correlations, with the former performing even better, with a correlation coefficient of 0.99. The computed LBO values reflects nicely the relative strength of the X–Br bonds studied in the present work. There exists a linear correlation of ∇
2ρ
BCP with the vibrational frequencies, although of much lower quality. Thus, for C–Br, the correlation coefficient is 0.74 and the fit invert sign of ∇
2ρ
BCP is at its high end and thus exhibits a wrong nature of the C–Br bond. In the case of O–Br, the fit is of a higher quality (r
2 = 0.81) and is the correct sign of the ∇
2ρ
BCP. The delocalization index DI(A,B) measures the average number of electrons delocalized (shared) between atoms A and B. If A and B are directly connected by a bond path then DI(A,B) is termed bond index. Values of DI(A,B) given in
Table 6 elaborate upon the above mentioned picture and show a wide variations ranging from 0.03 to 1.3. The O–Br bond seems illustrative in this respect. Thus, for the isomeric HOCBr system, the DI(A,B) index is 10 times as great for the
cis-over the trans form. On the other hand, DI(A,B) index for the BrCH
2OH/CH
3OBr/CH
3BrO system, show much fewer variations, and show almost constant accumulation of charge in the C–Br and O–Br bond regions. Ellipticity values given in
Table 6 elaborate on the covalent nature of both C–Br and O–Br but do not show a clear trend for their relative strength and give in general poor correlations with the vibrational frequency values.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



