
Nucleotide Salvage Deficiencies, DNA Damage and Neurodegeneration

Abstract
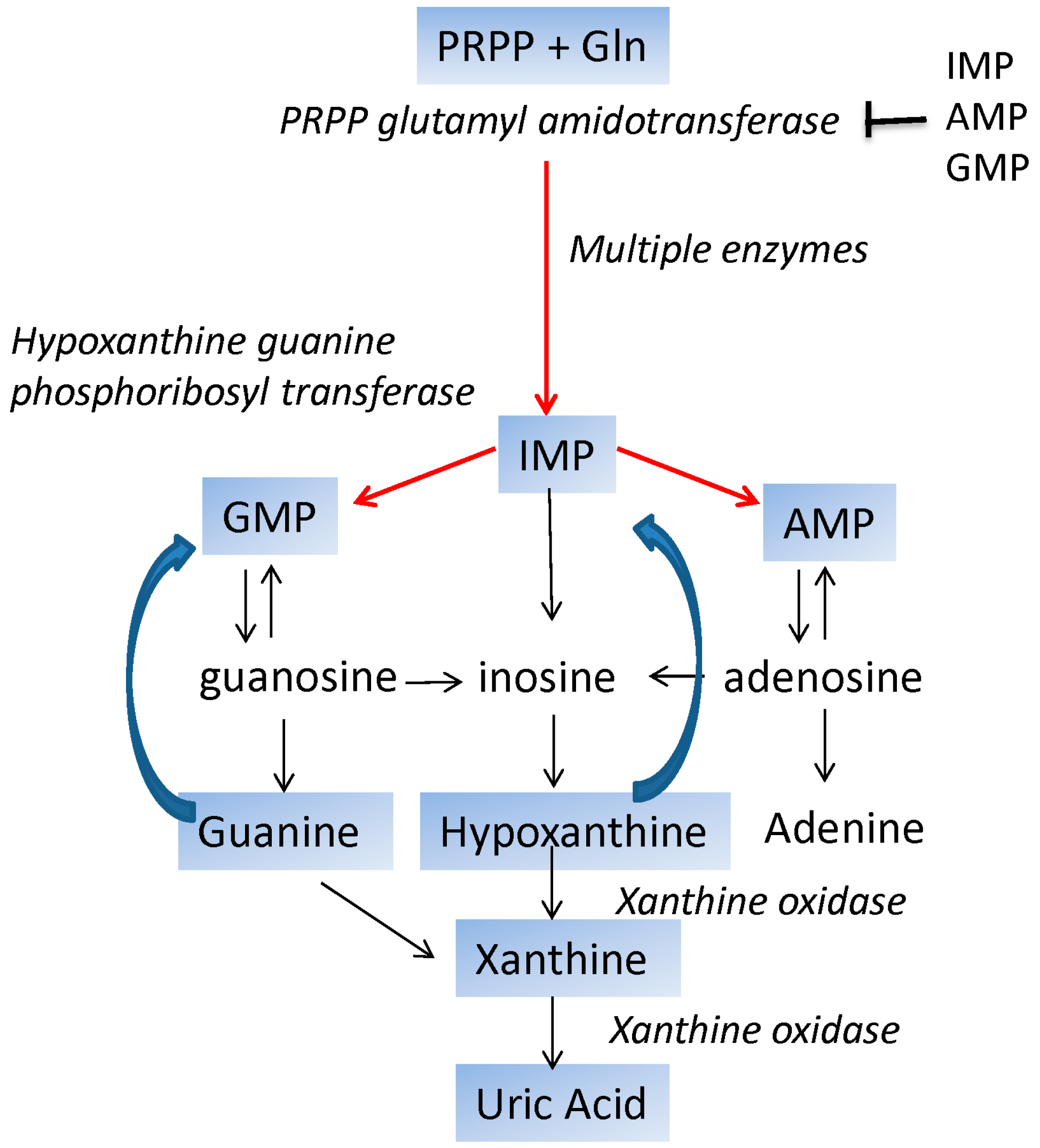
:1. Introduction

{kind=link}
{kind=link}
| Gene | Disease | Frequency | Mode of Inheritance | Enzymatic Defect | Neurophathy in Selected Patients | Effect on Mitochondria | Pathologies/Onset |
|---|---|---|---|---|---|---|---|
| HPRT1 | Lesch-Nyhan | Rare, >300 alleles 1/380,000 live births | X-linked | Hypoxanthine guanine phosphoribosyl transferase | Motor function | Indirect production of free radicals | Gout, diminished IQ, dystonia, Death due to hypotonia or renal failure |
| DGUOK | Mitochondrial depletion syndrome (MDS); hepatocerebral form | Extremely Rare, 22 different mutations described | Autosomal recessive | Deoxy-guanosine kinase | Hearing loss, nystagmus | Mitochondria depletion due to failure to produce substantial dGTP | Progressive liver failure, patients generally die of liver failure in early childhood |
| TK2 | MDS (myopathic form) | Rare | Autosomal recessive | Thymidine kinase II | Hypotonia, neurological features | Mitochondria depletion due to failure to salvage thymidine in mitochondria | Muscle weakness, extreme forms lead to respiratory failure and infantile death; mutations in less conserved aminoacids lead to progressive external ophthalmoplegia |
| Progressive external ophthalmoplegia (PEO) | |||||||
| TYMP (ECGF1) | Mitochondrial neurogastroIntestinal encephalomyopathy (MNGIE) | Rare, 50 different mutations have been described | Some domininant alleles | Thymidine phosphorylase | Peripheral neuropathy, ophthalmoparesis, leukoencephalopathy | Mitochondrial depletion | Gastrointestinal dysmotility, weight loss; onset within first 20 years, 37 years the median age of death |
| RRM2B | MDS (Encephalomyopathic form) | Rare | Recessive alleles lead to severe MDS or MNGIE; dominant truncations confer PEO | p53-induced ribonucleotide reductase B subunit (p53R2) | Microcephaly,sensorineural hearing loss, ophthalmoplegia | Mitochondria depletion; failure to repair UV damage | Severe skeletal depletion of mitochondria, hypotonia and muscle weakness, sensorineural hearing loss, failure to thrive, lactic acidosis; mutations that confer PEO or MGIE are adult onset |
| PEO | |||||||
| MNGIE | |||||||
| ATM | Ataxia Telangiectasia (A-T) | Rare | Autosomal recessive | ATM | Cerebellar ataxia | Mitochondria less-well formed | X-ray sensitivities, High frequencies of lymphoma/pulmonary infection |
2. Biochemistry of de Novo and Salvage Pathways for Nucleotide Biosynthesis in the Nervous System


3. Human Diseases Resulting from Defective Nucleotide Metabolism
3.1. Lesch-Nyhan Disease
3.2. Mitochondrial Depletion Syndromes (MDS)
3.3. Ataxia Telangietasia (A-T)
4. Animal Models
4.1. Mouse Models for Lesch-Nyhan Syndrome
Hprt−/− Mice
4.2. Mouse Models for MDS Linked to Thymidine Kinase Deficiency
Tk2−/− Mice
| Disease | Gene | Mouse Genotype | Phenotypes | ||
|---|---|---|---|---|---|
| Neurological | DNA Damage Sensitivity | Other | |||
| Lesch-Nyhan | HGPRT1 | Hprt−/− | None reported | None reported | Hyperuricemia |
| MDS | TK2 | Tk2−/− | None reported | None reported | Post-natal mortality (2–4 weeks); growth retardation; cellular mitochondrial defects; hypothermia due to the absence of subcutaneous adipose tissue; abnormal morphologies of brown adipocytes, myocardiocytes and hepatocytes |
| MDS | TK2 | TKH126N (knock-in) | Encelphalopathy; tremors; weakness; decreased activity; altered gait | None reported | Post-natal mortality with defects similar to that of Tk2−/− mice |
| MNGIE | TYMP (ECGF1) | TP−/−; UP−/− | Encephalopathy; abnormal myelin sheath morphology; mitochondrial DNA instability in the brain | None reported | Elevated plasma thymidine; defects in nucleotide homeostasis and enzyme/coenzyme metabolism |
| MDS | RRM2B | Rrm2b−/− | Abnormal sciatic nerve morphology | Higher rates of spontaneous mutation in the kidney | Renal organ failure at 14 weeks |
| Ataxia telangiectasia | ATM | Atm−/− | Abnormal neuronal cell morphologies; neuronal cell degeneration | Hypersensitivity to gamma radiation; abnormal cell cycle checkpoint response; spontaneous lymphomas | Growth retardation; premature death; decreased thymocyte numbers; infertility in both sexes |
4.3. Mouse Models for Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE)
TP−/− and UP−/− Mice
4.4. Mouse Models for p53R2 Deficiency and Over-Expression of Ribonucleotide Reductase (RNR) Subunits
Rrm2b−/− Mice
4.5. A-T Mouse Models and Neurodegeneration
5. Summary and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Micheli, V.; Camici, M.; Tozzi, M.G.; Ipata, P.L.; Sestini, S.; Bertelli, M.; Pompucci, G. Neurological disorders of purine and pyrimidine metabolism. Curr. Top. Med. Chem. 2011, 11, 923–947. [Google Scholar] [CrossRef] [PubMed]
- Chabes, A.; Bilyana, G.; Domkin, V.; Zhao, X.; Rothstein, R.; Thelander, L. Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell 2003, 112, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Fasullo, M.; Koudelik, J.; AhChing, P.; Giallanza, P.; Cera, C. Radiosensitive and mitotic recombination phenotypes of the Saccharomyces cerevisiae dun1 mutant defective in DNA damage-inducible gene expression. Genetics 1999, 152, 909–919. [Google Scholar]
- Fikus, M.U.; Mieczkowski, P.A.; Koprowski, P.; Rytka, J.; Sledziewska-Gójska, E.; Ciésla, Z. The product of the DNA damage-inducible gene of Saccharomyces cerevisiae, DIN7, specifically functions in mitochondria. Genetics 2000, 154, 73–81. [Google Scholar] [PubMed]
- Weinberg, G.; Ullman, B.; Martin, D.W. Mutator phenotypes in mammalian cell mutants with distinct biochemical defects and abnormal deoxyribonucleoside triphosphate pools. Proc. Natl. Acad. Sci. USA 1981, 78, 2447–2451. [Google Scholar] [CrossRef]
- Fu, R.; Ceballos-Picot, I.; Torres, R.J.; Larovere, L.E.; Yamada, Y.; Nguyen, K.V.; Hegde, M.; Visser, J.E.; Schretlen, D.J.; Nyhan, W.L.; et al.; Lesch-Nyhan Disease International Study Group Brain. Genotype–phenotype correlations in neurogenetics: Lesch-Nyhan disease as a model disorder. Brain 2014, 137, 1282–1303. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, C.; Almeida, L.S.; Nesti, C.; Pezzini, I.; Videira, A.; Vilarinho, L.; Santorelli, F.M. Syndromes associated with mitochondrial DNA depletion. Ital. J. Pediatr. 2014, 40, 34. [Google Scholar] [CrossRef] [PubMed]
- Copeland, W.C.; Longley, M.J. Mitochondrial genome maintenance in health and disease. DNA Repair 2014, 19, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Nishino, I.; Spinazzola, A.; Hirano, M. Thymidine phosphorylase gene mutations in MNGIE, a Human Mitochondrial Disorder. Science 1999, 283, 689–692. [Google Scholar] [CrossRef] [PubMed]
- Bunimovich, Y.L.; Nair-Gill, E.; Riedinger, M.; McCracken, M.N.; Cheng, D.; McLaughlin, J.; Radu, C.G.; Witte, O.N. Deoxycytidine kinase augments ATM-mediated DNA repair and contributes to radiation resistance. PLoS ONE 2014, 9, e104125. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.K.; Lebedeva, M.; Thomas, T.; Kovalenko, O.A.; Stumpf, J.D.; Shadel, G.S.; Santos, J.H. Intrinsic mitochondrial DNA repair defects in Ataxia Telangiectasia. DNA Repair 2014, 13, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, M.; Gatti, R.A. Pathogenesis of ataxia-telangiectasia: The next generation of ATM functions. Blood 2013, 121, 4036–4045. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.M.; Gatti, R.A. Clinical radiation sensitivity with DNA repair disorders: An overview. Int. J. Radiat. Oncol. Biol. Phys. 2009, 74, 1323–1331. [Google Scholar] [CrossRef] [PubMed]
- Federico, A.; Cardaioli, E.; da Pozzo, P.; Formichi, P.; Gallus, G.N.; Radi, E. Mitochondria, oxidative stress and neurodegeneration. J. Neurol. Sci. 2012, 322, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [PubMed]
- Furda, A.M.; Marrangoni, A.M.; Lokshin, A.; van Houten, B. Oxidants and not alkylating agents induce rapid mtDNA loss and mitochondrial dysfunction. DNA Repair 2012, 11, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Scheibye-Knudsen, M.; Fang, E.F.; Croteau, D.L.; Wilson, D.M., 3rd; Bohr, V.A. Protecting the mitochondrial powerhouse. Trends Cell Biol. 2014, 25, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Pursell, Z.F.; McDonald, J.T.; Mathews, C.K.; Kunkel, T.A. Trace amounts of 8-oxo-dGTP in mitochondrial dNTP pools reduce DNA polymerase gamma replication fidelity. Nucleic Acids Res. 2008, 36, 2174–2181. [Google Scholar] [CrossRef] [PubMed]
- Desler, C.; Lykke, A.; Rasmussen, L.J. The effect of mitochondrial dysfunction on cytosolic nucleotide metabolism. J. Nucleic Acids 2010, 2010, 701518. [Google Scholar] [CrossRef] [PubMed]
- Pontarin, G.; Ferraro, P.; Bee, L.; Reichard, P.; Bianchi, V. Mammalian ribonucleotide reductase subunit p53R2 is required for mitochondrial DNA replication and DNA repair in quiescent cells. Proc. Natl. Acad. Sci. USA 2012, 109, 13302–13307. [Google Scholar] [CrossRef] [PubMed]
- Nordlund, P.; Reichard, P. Ribonucleotide reductases. Annu. Rev. Biochem. 2006, 75, 681–706. [Google Scholar] [CrossRef] [PubMed]
- Abbracchio, M.P.; Burnstock, G.; Verkhratsky, A.; Zimmermann, H. Purinergic signalling in the nervous system: An overview. Trends Neurosci. 2009, 32, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Averaimo, S.; Nicol, X. Intermingled cAMP, cGMP and calcium spatiotemporal dynamics in developing neuronal circuits. Front. Cell. Neurosci. 2014, 8, 376. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, S.I.; Long, K.D.; Rosse, R.B.; Mastropaolo, J.; Eller, J. Hypothesized deficiency of guanine-based purines may contribute to abnormalities of neurodevelopment, neuromodulation, and neurotransmission in Lesch-Nyhan syndrome. Clin. Neuropharmacol. 2005, 28, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.L.; Lehninger, A.L.; Cox, M.M. Lehninger Principles of Biochemistry, 5th ed.; Ch WH Freeman and Company: New York, NY, USA, 2008; pp. 882–900. [Google Scholar]
- Ceballos-Picot, I.; Mockel, L.; Potier, M.C.; Dauphinot, L.; Shirley, T.L.; Torero-Ibad, R.; Fuchs, J.; Jinnah, H.A. Hypoxanthine-guanine phosphoribosyl transferase regulates early developmental programming of dopamine neurons: Implications for Lesch-Nyhan disease pathogenesis. Hum. Mol. Genet. 2009, 18, 2317–2327. [Google Scholar] [CrossRef] [PubMed]
- Rampazzo, C.; Miazzi, C.; Franzolin, E.; Pontarin, G.; Ferraro, P.; Frangini, M.; Reichard, P.; Bianchi, V. Regulation by degradation, a cellular defense against deoxyribonucleotide pool imbalances. Mutat. Res. 2010, 703, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Battelli, M.G.; Bolognesi, A.; Polito, L. Pathophysiology of circulating xanthine oxidoreductase: New emerging roles for a multi-tasking enzyme. Biochim. Biophys. Acta 2014, 1842, 1502–1517. [Google Scholar] [CrossRef] [PubMed]
- Steiger, S.; Harper, J.L. Mechanisms of spontaneous resolution of acute gouty inflammation. Curr. Rheumatol. Rep. 2014, 16, 392. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, L.J.; Mathews, C.K. Nucleoside triphosphate pool asymmetry in mammalian mitochondria. J. Biol. Chem. 2011, 286, 16992–16996. [Google Scholar] [CrossRef]
- Saada, A. Mitochondrial deoxyribonucleotide pools in deoxyguanosine kinase deficiency. Mol. Genet. Metab. 2008, 95, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, P.; Nicolosi, L.; Bernardi, P.; Reichard, P.; Bianchi, V. Mitochondrial deoxynucleotide pool sizes in mouse liver and evidence for a transport mechanism for thymidine monophosphate. Proc. Natl. Acad. Sci. USA 2006, 103, 18586–18991. [Google Scholar] [CrossRef] [PubMed]
- Martí, R.; Dorado, B.; Hirano, M. Measurement of mitochondrial dNTP pools. Methods Mol. Biol. 2012, 837, 135–148. [Google Scholar] [PubMed]
- Jinnah, H.A.; Ceballos-Picot, I.; Torres, R.J.; Visser, J.E.; Schretlen, D.J.; Verdu, A.; Laróvere, L.E.; Chen, C.J.; Cossu, A.; Wu, C.H.; et al.; Lesch-Nyhan Disease International Study Group Attenuated variants of Lesch-Nyhan disease. Brain 2010, 133 Pt 3, 671–689. [Google Scholar] [CrossRef] [PubMed]
- Mizunuma, M.; Fujimori, S.; Ogino, H.; Ueno, T.; Inoue, H.; Kamatani, N. A recurrent large Alu-mediated deletion in the hypoxanthine phosphoribosyltransferase (HPRT1) gene associated with Lesch-Nyhan syndrome. Hum. Mutat. 2001, 18, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Sampat, R.; Fu, R.; Larovere, L.E.; Torres, R.J.; Ceballos-Picot, I.; Fischbach, M.; de Kremer, R.; Schretlen, D.J.; Puig, J.G.; Jinnah, H.A. Mechanisms for phenotypic variation in Lesch-Nyhan disease and its variants. Hum. Genet. 2011, 129, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Camici, M.; Micheli, V.; Ipata, P.L.; Tozzi, M.G. Pediatric neurological syndromes and inborn errors of purine metabolism. Neurochem. Int. 2010, 56, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Ernst, M.; Zametkin, A.J.; Matochik, J.A.; Pascualvaca, D.; Jons, P.H.; Hardy, K.; Hankerson, J.G.; Doudet, D.J.; Cohen, R.M. Presynaptic dopaminergic deficits in Lesch-Nyhan disease. N. Engl. J. Med. 1996, 334, 1568–1572. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.F.; Harris, J.C.; Naidu, S.; Yokoi, F.; Marenco, S.; Ravert, H.T.; Yaster, M.; Evans, A.; Rousset, O.; Bryan, R.N.; et al. Dopamine transporter are markedly reduced in Lesch-Nyhan disease in vivo. Proc. Natl. Acad. Sci. USA 1996, 93, 5539–5543. [Google Scholar] [CrossRef] [PubMed]
- White, W.F.; Dichter, M.A.; Snodgrass, R. Benzodiazepine binding and interactions with the GABA receptor complex in living cultures of rat cerebral cortex. Brain Res. 1981, 215, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Göttle, M.; Prudente, C.N.; Fu, R.; Sutcliffe, D.; Pang, H.; Cooper, D.; Veledar, E.; Glass, J.D.; Gearing, M.; Visser, J.E.; et al. Loss of dopamine phenotype among midbrain neurons in Lesch-Nyhan disease. Ann. Neurol. 2014, 76, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Fairbanks, L.D.; Jacomelli, G.; Micheli, V.; Slade, T.; Simmonds, H.A. Severe pyridine nucleotide depletion in fibroblasts from Lesch-Nyhan patients. J. Neurochem. 1999, 72, 1139–1145. [Google Scholar] [PubMed]
- McCreanor, G.M.; Harkness, R.A. Lesch-Nyhan syndrome and its pathogenesis: Normal nicotinamide-adenine dinucleotide but reduced ATP concentrations that correlate with reduced poly(ADP-ribose) synthetase activity in HPRT-deficient lymphoblasts. J. Inherit. Metab. Dis. 1995, 18, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.H.; Park, Y.; Bader, J.S.; Friedmann, T. The housekeeping gene hypoxanthine guanine phosphoribosyltransferase (HPRT) regulates multiple developmental and metabolic pathways of murine embryonic stem cell neuronal differentiation. PLoS ONE 2013, 8, e74967. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, L.; Kim, J.E.; Miyanohara, A.; Kang, T.H.; Friedmann, T. Purinergic signaling in human pluripotent stem cells is regulated by the housekeeping gene encoding hypoxanthine guanine phosphoribosyltransferase. Proc. Natl. Acad. Sci. USA 2012, 109, 3377–3382. [Google Scholar] [CrossRef] [PubMed]
- Guibinga, G.H.; Murray, F.; Barron, N. HPRT-deficiency dysregulates cAMP-PKA signaling and phosphodiesterase 10A expression: Mechanistic insight and potential target for Lesch-Nyhan Disease? PLoS ONE 2013, 18, e63333. [Google Scholar] [CrossRef]
- Watts, R.W.; Harkness, R.A.; Spellacy, E.; Taylor, N.F. Lesch-Nyhan syndrome: Growth delay, testicular atrophy and a partial failure of the 11β-hydroxylation of steroids. J. Inherit. Metab. Dis. 1987, 10, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Copeland, W.C. Defects in mitochondrial DNA replication and human disease. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 64–74. [Google Scholar] [CrossRef]
- Tyynismaa, H.; Suomalainen, A. Mouse models of mitochondrial DNA defects and their relevance for human disease. EMBO Rep. 2009, 10, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Mandel, H.; Szargel, R.; Labay, V.; Elpeleg, O.; Saada, A.; Shalata, A.; Anbinder, Y.; Berkowitz, D.; Hartman, C.; Barak, M.; et al. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat. Genet. 2001, 29, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.; Karlsson, A. Cloning and expression of human deoxyguanosine kinase cDNA. Proc. Natl. Acad. Sci. USA 1996, 93, 7258–7262. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, S.; Wang, L. Molecular mechanisms of mitochondrial DNA depletion diseases caused by deficiencies in enzymes in purine and pyrimidine metabolism. Nucleosides Nucleotides Nucleic Acids 2008, 27, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Dimmock, D.P.; Zhang, Q.; Dionisi-Vici, C.; Carrozzo, R.; Shieh, J.; Tang, L.-Y.; Truong, C.; Schmitt, E.; Sifry-Platt, M.; Lucioli, S.; et al. Clinical and molecular features of mitochondrial DNA depletion due to mutations in deoxyguanosine kinase. Hum. Mutat. 2008, 29, 330–331. [Google Scholar] [CrossRef] [PubMed]
- Freisinger, P.; Fütterer, N.; Lankes, E.; Gempel, K.; Berger, T.M.; Spalinger, J.; Hoerbe, A.; Schwantes, C.; Lindner, M.; Santer, R.; et al. Hepatocerebral mitochondrial DNA depletion syndrome caused by deoxyguanosine kinase (DGUOK) mutations. Arch. Neurol. 2006, 63, 1129–1134. [Google Scholar] [CrossRef] [PubMed]
- Buchaklian, A.H.; Helbling, D.; Ware, S.M.; Dimmock, D.P. Recessive deoxyguanosine kinase deficiency causes juvenile onset mitochondrial myopathy. Mol. Genet. Metab. 2012, 107, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Tyynismaa, H.; Sun, R.; Ahola-Erkkilä, S.; Almusa, H.; Pöyhönen, R.; Korpela, M.; Honkaniemi, J.; Isohanni, P.; Paetau, A.; Wang, L.; et al. Thymidine kinase 2 mutations in autosomal recessive progressive external ophthalmoplegia with multiple mitochondrial DNA deletions. Hum. Mol. Genet. 2012, 21, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Kalko, S.G.; Paco, S.; Jou, C.; Rodríguez, M.A.; Meznaric, M.; Rogac, M.; Jekovec-Vrhovsek, M.; Sciacco, M.; Moggio, M.; Fagiolari, G.; et al. Transcriptomic profiling of TK2 deficient human skeletal muscle suggests a role for the p53 signalling pathway and identifies growth and differentiation factor-15 as a potential novel biomarker for mitochondrial myopathies. BMC Genomics 2014, 15, 91. [Google Scholar] [CrossRef] [PubMed]
- Giordano, C.; Sebastiani, M.; de Giorgio, R.; Travaglini, C.; Tancredi, A.; Valentino, M.L.; Bellan, M.; Cossarizza, A.; Hirano, M.; d’Amati, G.; et al. Gastrointestinal dysmotility in mitochondrial neurogastrointestinal encephalomyopathy is caused by mitochondrial DNA depletion. Am. J. Pathol. 2008, 173, 1120–1128. [Google Scholar] [CrossRef]
- Libernini, L.; Lupis, C.; Mastrangelo, M.; Carrozzo, R.; Santorelli, F.M.; Inghilleri, M.; Leuzzi, V. Mitochondrial neurogastrointestinal encephalomyopathy: Novel pathogenic mutations in thymidine phosphorylase gene in two Italian brothers. Neuropediatrics 2012, 43, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Valentino, M.L.; Martí, R.; Tadesse, S.; López, L.C.; Manes, J.L.; Lyzak, J.; Hahn, A.; Carelli, V.; Hirano, M. Thymidine and deoxyuridine accumulate in tissues of patients with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). FEBS. Lett. 2007, 581, 3410–3414. [Google Scholar] [CrossRef] [PubMed]
- González-Vioque, E.; Torres-Torronteras, J.; Andreu, A.L.; Martí, R. Limited dCTP availability accounts for mitochondrial DNA depletion in mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). PLoS Genet. 2011, 7, e1002035. [Google Scholar] [CrossRef] [PubMed]
- Filosto, M.; Scarpelli, M.; Tonin, P.; Lucchini, G.; Pavan, F.; Santus, F.; Parini, R.; Donati, M.A.; Cotelli, M.S.; Vielmi, V.; et al. Course and management of allogeneic stem cell transplantation in patients with mitochondrial neurogastrointestinal encephalomyopathy. J. Neurol. 2012, 259, 2699–2706. [Google Scholar] [CrossRef] [PubMed]
- Tyynismaa, H.; Carroll, C.J.; Raimundo, N.; Ahola-Erkkilä, S.; Wenz, T.; Ruhanen, H.; Guse, K.; Hemminki, A.; Peltola-Mjøsund, K.E.; Tulkki, V.; et al. Mitochondrial myopathy induces a starvation-like response. Hum. Mol. Genet. 2010, 19, 3948–3958. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, A.; Minai, L.; Serre, V.; Jais, J.P.; Sarzi, E.; Aubert, S.; Chrétien, D.; de Lonlay, P.; Paquis-Flucklinger, V.; Arakawa, H.; et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat. Genet. 2007, 39, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Pontarin, G.; Ferraro, P.; Rampazzo, C.; Kollberg, G.; Holme, E.; Reichard, P.; Bianchi, V. Deoxyribonucleotide metabolism in cycling and resting human fibroblasts with a missense mutation in p53R2, a subunit of ribonucleotide reductase. J. Biol. Chem. 2011, 286, 11132–11140. [Google Scholar] [CrossRef] [PubMed]
- Tyynismaa, H.; Ylikallio, E.; Patel, M.; Molnar, M.J.; Haller, R.G.; Suomalainen, A. A heterozygous truncating mutation in RRM2B causes autosomal-dominant progressive external ophthalmoplegia with multiple mtDNA deletions. Am. J. Hum. Genet. 2009, 85, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Eaton, J.S.; Lin, Z.P.; Sartorelli, A.C.; Bonawitz, N.D.; Shadel, G.S. Ataxia-telangiectasia mutated kinase regulates ribonucleotide reductase and mitochondrial homeostasis. J. Clin. Investig. 2007, 117, 2723–2734. [Google Scholar] [CrossRef] [PubMed]
- Hooper, M.; Hardy, K.; Handyside, A.; Hunter, S.; Monk, M. HPRT-deficient (Lesch-Nyhan) mouse embryos derived from germline colonization by cultured cells. Nature 1987, 326, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Jinnah, H.A.; Page, T.; Friedmann, T. Brain purines in a genetic mouse model of Lesch-Nyhan disease. J. Neurochem. 1993, 60, 2036–2045. [Google Scholar] [CrossRef] [PubMed]
- Visser, J.E.; Smith, D.W.; Moy, S.S.; Breese, G.R.; Friedmann, T.; Rothstein, J.D.; Jinnah, H.A. Oxidative stress and dopamine deficiency in a genetic mouse model of Lesch-Nyhan disease. Brain Res. Dev. Brain Res. 2002, 133, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Pelled, D.; Sperling, O.; Zoref-Shani, E. Abnormal purine and pyrimidine nucleotide content in primary astroglia cultures from hypoxanthine-guanine phosphoribosyltransferase-deficient transgenic mice. J. Neurochem. 1999, 72, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Jinnah, H.A.; de Gregorio, L.; Harris, J.C.; Nyhan, W.L.; O’Neill, J.P. The spectrum of inherited mutations causing HPRT deficiency: 75 new cases and a review of 196 previously reported cases. Mutat. Res. 2000, 463, 309–326. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Solaroli, N.; Bjerke, M.; Stewart, J.B.; Rozell, B.; Johansson, M.; Karlsson, A. Progressive loss of mitochondrial DNA in thymidine kinase 2-deficient mice. Hum. Mol. Genet. 2008, 17, 2329–2335. [Google Scholar] [CrossRef] [PubMed]
- Akman, H.O.; Dorado, B.; López, L.C.; García-Cazorla, A.; Vilà, M.R.; Tanabe, L.M.; Dauer, W.T.; Bonilla, E.; Tanji, K.; Hirano, M. Thymidine kinase 2 (H126N) knockin mice show the essential role of balanced deoxynucleotide pools for mitochondrial DNA maintenance. Hum. Mol. Genet. 2008, 17, 2433–2440. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.; Paredes, J.A.; Zhou, X.; Kuiper, R.V.; Curbo, S.; Karlsson, A. Long term expression of Drosophila melanogaster nucleoside kinase in thymidine kinase 2-deficient mice with no lethal effects caused by nucleotide pool imbalances. J. Biol. Chem. 2014, 288, 32835–32844. [Google Scholar] [CrossRef]
- Lopez, L.C.; Akman, H.O.; García-Cazorla, A.; Dorado, B.; Martí, R.; Nishino, I.; Tadesse, S.; Pizzorno, G.; Shungu, D.; Bonilla, E.; et al. Unbalanced deoxynucleotide pools cause mitochondrial DNA instability in thymidine phosphorylase-deficient mice. Hum. Mol. Genet. 2009, 18, 714–722. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Takeda, S.; Sagiya, Y.; Gotoh, M.; Nakamura, Y.; Arakawa, H. Impaired function of p53R2 in Rrm2b-null mice causes severe renal failure through attenuation of dNTP pools. Nat. Genet. 2003, 34, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Ylikallio, E.; Page, J.L.; Xu, X.; Lampinen, M.; Bepler, G.; Ide, T.; Tyynismaa, H.; Weiss, R.S.; Suomalainen, A. Ribonucleotide reductase is not limiting for mitochondrial DNA copy number in mice. Nucleic Acids Res. 2010, 38, 8208–8218. [Google Scholar] [CrossRef] [PubMed]
- Stern, N.; Hochman, A.; Zemach, N.; Weizman, N.; Hammel, I.; Shiloh, Y.; Rotman, G.; Barzilai, A. Accumulation of DNA damage and reduced levels of nicotine adenine dinucleotide in the brains of Atm-deficient mice. J. Biol. Chem. 2002, 277, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F. The appropriateness of the mouse model for ataxia-telangiectasia: Neurological defects but no neurodegeneration. DNA Repair 2013, 12, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Fasullo, M.; Tsaponina, O.; Sun, M.; Chabes, A. Elevated dNTP levels suppress hyper-recombination in Saccharomyces cerevisiae S-phase checkpoint mutants. Nucleic Acids Res. 2010, 38, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.D.; Zhang, H.; Eaton, J.S.; Rodeheffer, M.S.; Lebedeva, M.A.; O’Rourke, T.W.; Siede, W.; Shadel, G.S. The conserved Mec1/Rad53 nuclear checkpoint pathway regulates mitochondrial DNA copy number in Saccharomyces cerevisiae. Mol. Biol. Cell 2005, 16, 3010–3018. [Google Scholar] [CrossRef] [PubMed]
- Glick, N. Dramatic reduction in self-injury in Lesch-Nyhan disease following S-adenosylmethionine administration. J. Inherit. Metab. Dis. 2006, 29, 687. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.E.; Scaglia, F. Mitochondrial DNA depletion syndromes: Review and updates of genetic basis, manifestations, and therapeutic options. Neurotherapeutics 2013, 10, 186–198. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fasullo, M.; Endres, L. Nucleotide Salvage Deficiencies, DNA Damage and Neurodegeneration. Int. J. Mol. Sci. 2015, 16, 9431-9449. https://doi.org/10.3390/ijms16059431
Fasullo M, Endres L. Nucleotide Salvage Deficiencies, DNA Damage and Neurodegeneration. International Journal of Molecular Sciences. 2015; 16(5):9431-9449. https://doi.org/10.3390/ijms16059431
Chicago/Turabian StyleFasullo, Michael, and Lauren Endres. 2015. "Nucleotide Salvage Deficiencies, DNA Damage and Neurodegeneration" International Journal of Molecular Sciences 16, no. 5: 9431-9449. https://doi.org/10.3390/ijms16059431
APA StyleFasullo, M., & Endres, L. (2015). Nucleotide Salvage Deficiencies, DNA Damage and Neurodegeneration. International Journal of Molecular Sciences, 16(5), 9431-9449. https://doi.org/10.3390/ijms16059431