
Hyperactive RAS/PI3-K/MAPK Signaling Cascade in Migration and Adhesion of Nf1 Haploinsufficient Mesenchymal Stem/Progenitor Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
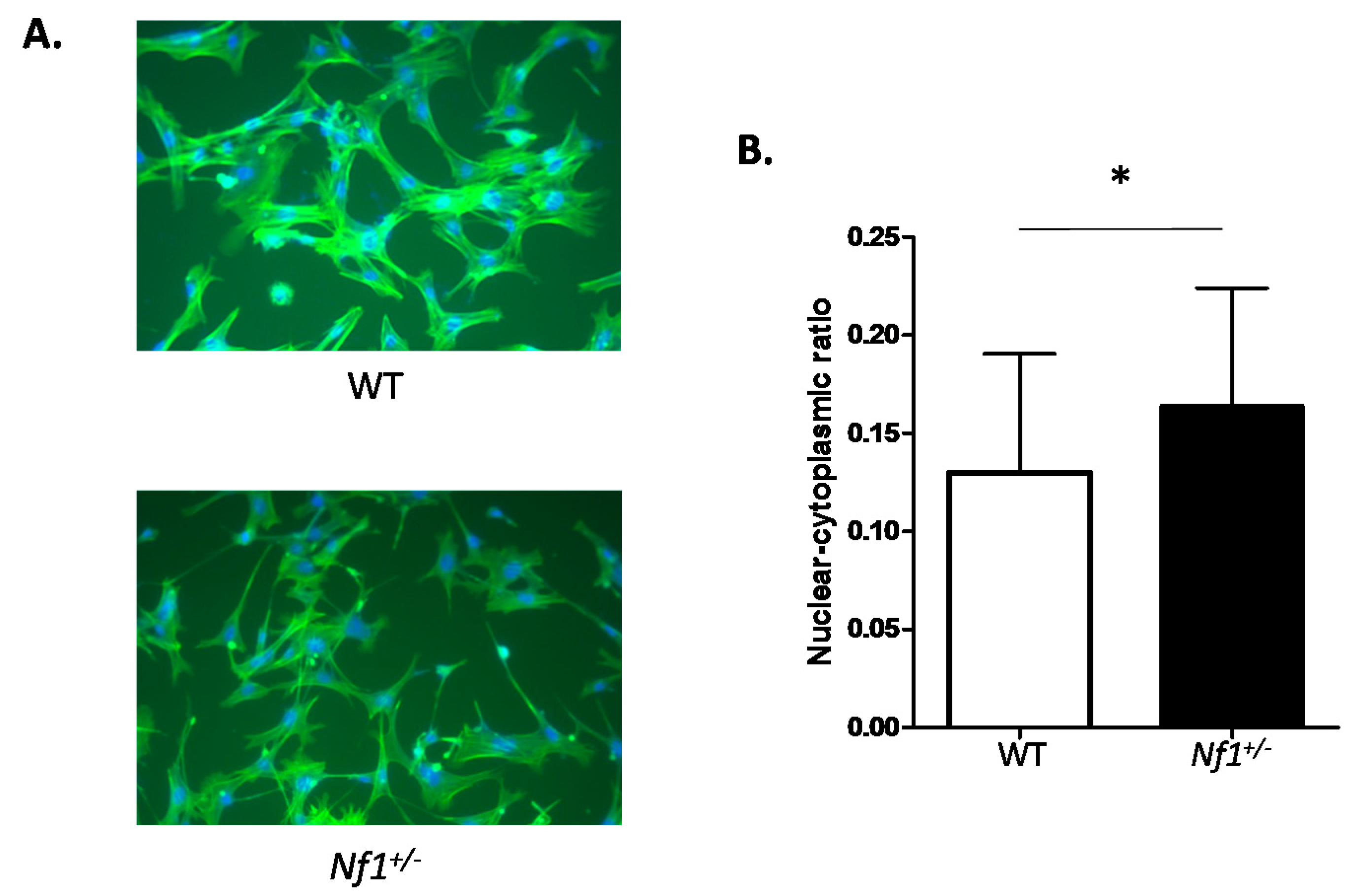
2.1. Nf1+/− MSPCs Have Increased Nuclear-to-Cytoplasmic Ratio

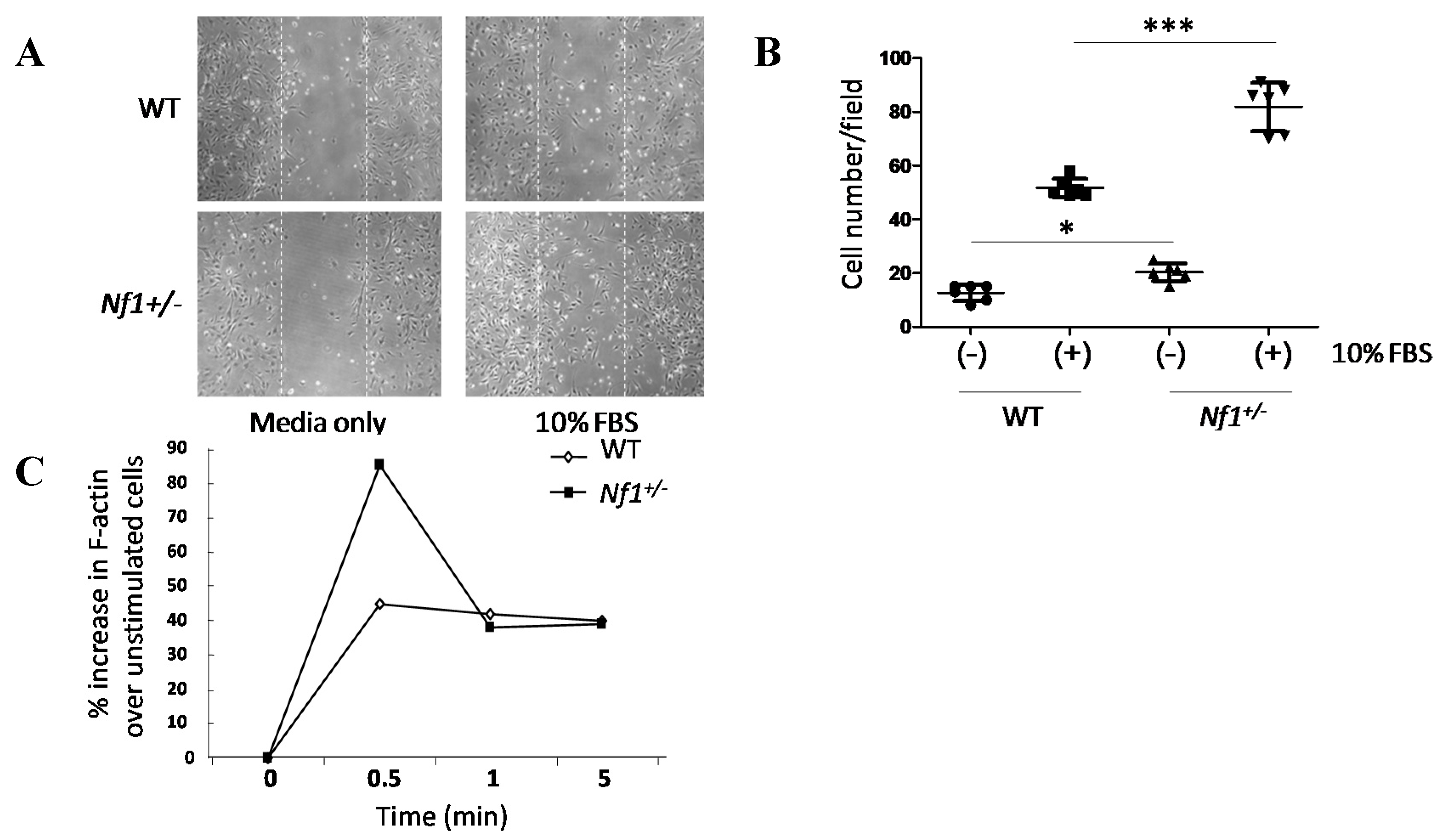
2.2. Nf1+/− MSPCs Have Increased Migratory Capacity

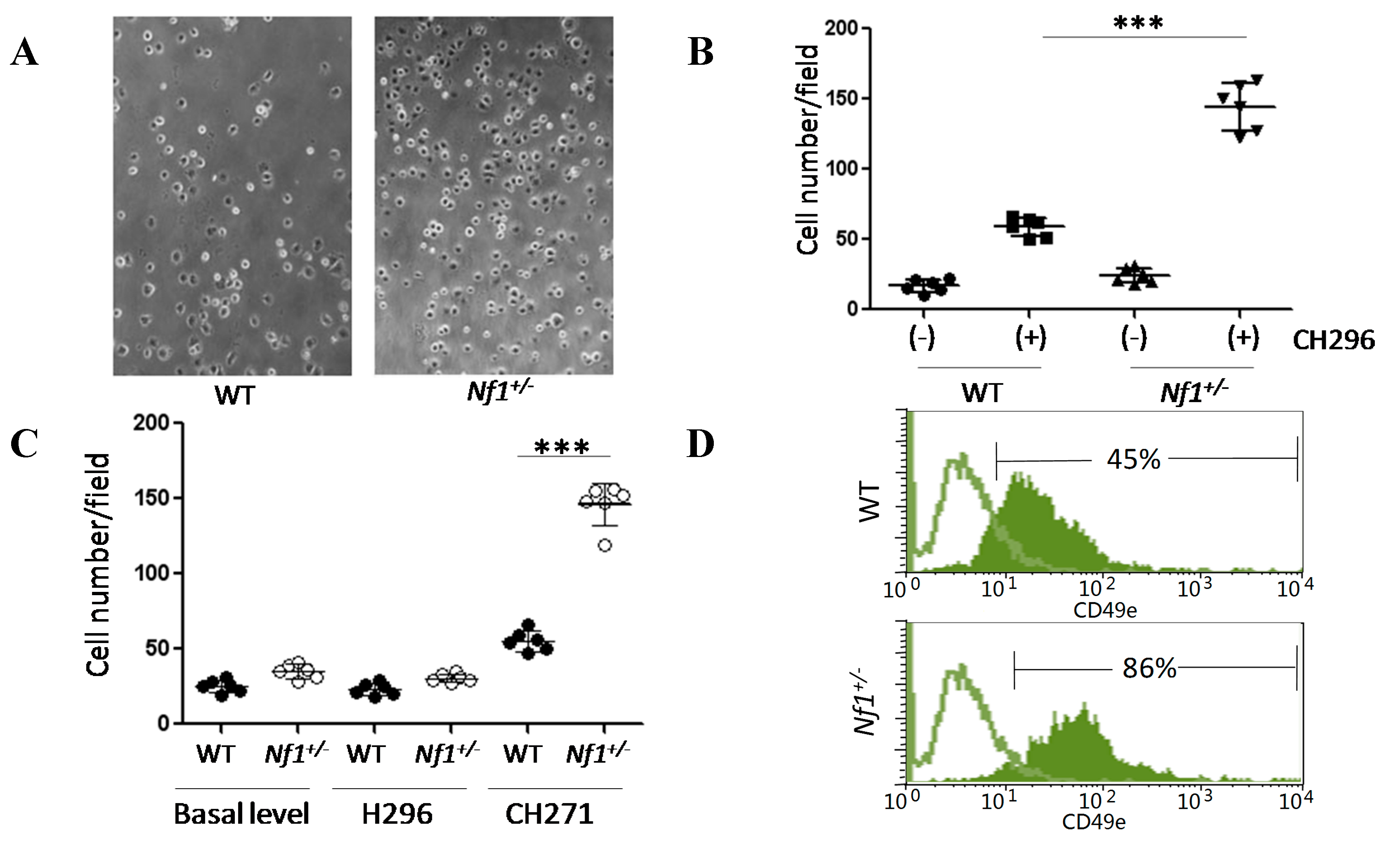
2.3. Nf1 Haploinsufficiency Enhances Cellular Affinity to CH271

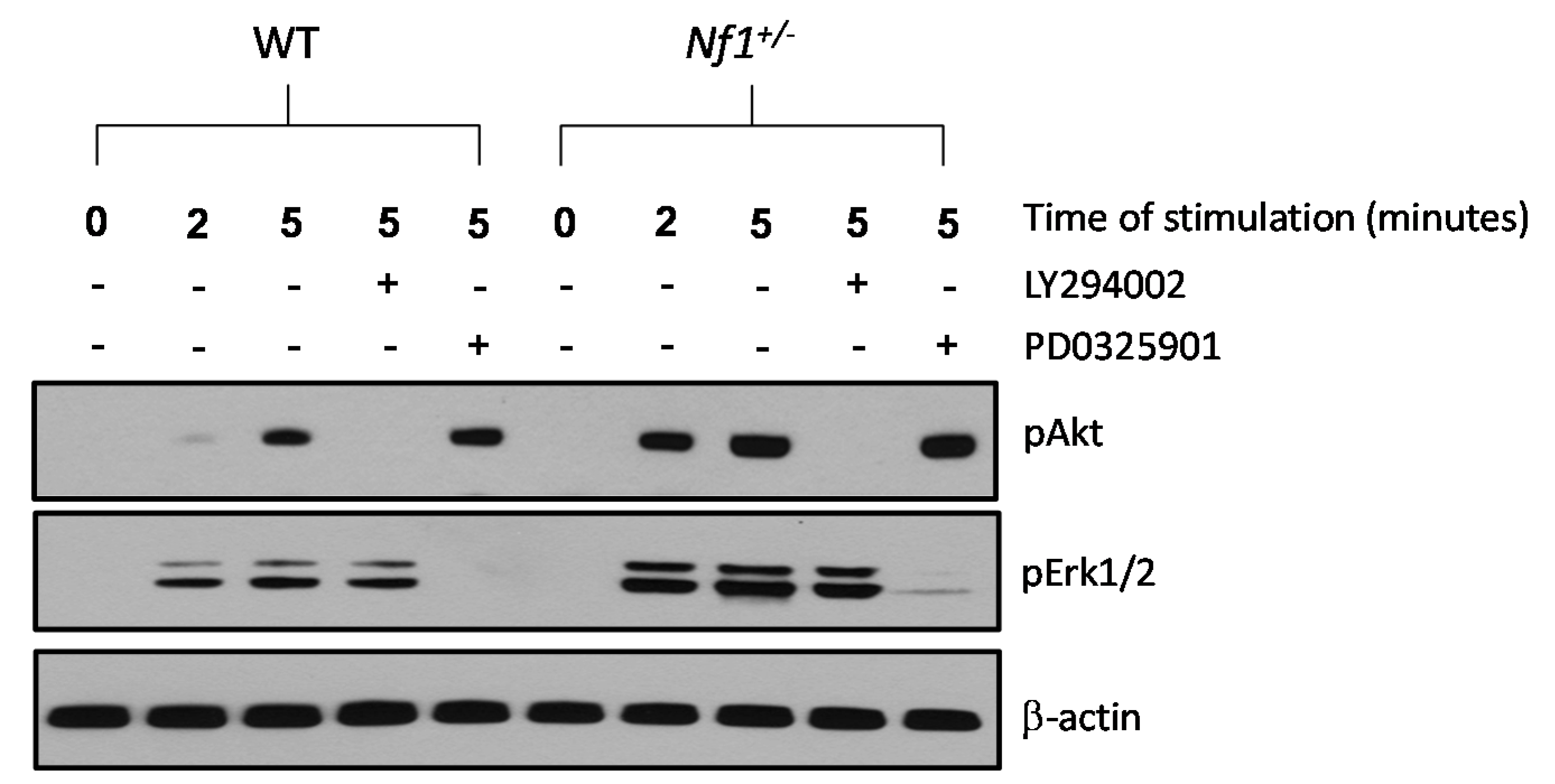
2.4. Hyper Activation of the PI3-K and MAPK Pathways in Nf1+/− MSPCs

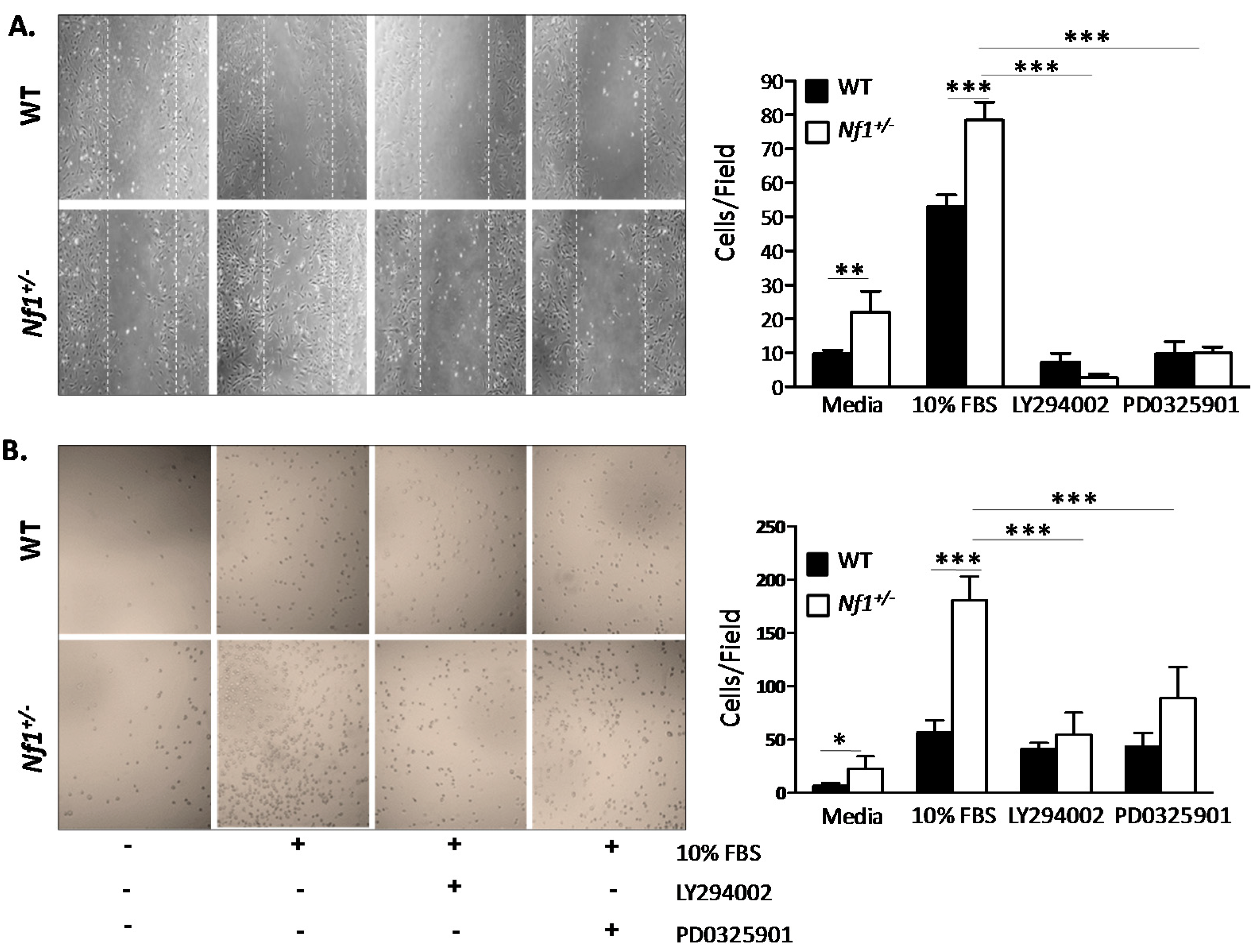
2.5. Enhanced Nf1+/− MSPCs Migration and Adhesion Is Rescued by LY294002 and PD0325901

3. Discussion
4. Experimental Section
4.1. Animals and Materials
4.2. Isolation and Expansion of MSPCs
4.3. Phenotypic Analysis of MSPCs
4.4. Cellular Morphology
4.5. Actin Polymerization
4.6. Adhesion Assay
4.7. Wound Healing Assay
4.8. Western Blot
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Friedman, J.M.; Birch, P.H. Type 1 neurofibromatosis: A descriptive analysis of the disorder in 1728 patients. Am. J. Med. Genet. 1997, 70, 138–143. [Google Scholar] [CrossRef]
- Szudek, J.; Birch, P.; Riccardi, V.M.; Evans, D.G.; Friedman, J.M. Associations of clinical features in neurofibromatosis 1 (NF1). Genet. Epidemiol. 2000, 19, 429–439. [Google Scholar] [CrossRef]
- Riccardi, V.M. Neurofibromatosis: Past, present, and future. N. Engl. J. Med. 1991, 324, 1283–1285. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, Y.; Dibbern, K.; Klein, J.; Riccardi, V.M.; Graham, J.M., Jr. Neurofibromatosis type 1—An update and review for the primary pediatrician. Clin. Pediatr. 1996, 35, 545–561. [Google Scholar] [CrossRef]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase superfamily: A conserved switch for diverse cell functions. Nature 1990, 348, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Hall, A. The cellular functions of small GTP-binding proteins. Science 1990, 249, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Friedenstein, A.J.; Chailakhjan, R.K.; Lalykina, K.S. The development of fibroblast colonies in monolayer cultures of guinea-pig bone marrow and spleen cells. Cell Tissue Kinet. 1970, 3, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Sacchetti, B.; Funari, A.; Michienzi, S.; di Cesare, S.; Piersanti, S.; Saggio, I.; Tagliafico, E.; Ferrari, S.; Robey, P.G.; Riminucci, M.; et al. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell 2007, 131, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; Macarthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’ayan, A.; Enikolopov, G.N.; Frenette, P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010, 466, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Bianco, P. Bone and the hematopoietic niche: A tale of two stem cells. Blood 2011, 117, 5281–5288. [Google Scholar] [CrossRef] [PubMed]
- Honczarenko, M.; Le, Y.; Swierkowski, M.; Ghiran, I.; Glodek, A.M.; Silberstein, L.E. Human bone marrow stromal cells express a distinct set of biologically functional chemokine receptors. Stem Cells 2006, 24, 1030–1041. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Xie, N.; Li, W.; Yuan, B.; Shi, Y.; Wang, Y. Immunobiology of mesenchymal stem cells. Cell Death Differ. 2014, 21, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Friedenstein, A.J. Marrow stromal fibroblasts. Calcif. Tissue Int. 1995, 56, S17. [Google Scholar] [CrossRef] [PubMed]
- Friedenstein, A.J.; Chailakhyan, R.K.; Gerasimov, U.V. Bone marrow osteogenic stem cells: In vitro cultivation and transplantation in diffusion chambers. Cell Tissue Kinet. 1987, 20, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Lennon, D.P.; Caplan, A.I.; DeChant, A.; Hecker, J.; Kranso, J.; Zaremba, A.; Miller, R.H. Hepatocyte growth factor mediates mesenchymal stem cell-induced recovery in multiple sclerosis models. Nat. Neurosci. 2012, 15, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Zhang, L.; Zhao, X.; Xu, G.; Zhang, Y.; Roberts, A.I.; Zhao, R.C.; Shi, Y. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell 2008, 2, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Kuorilehto, T.; Poyhonen, M.; Bloigu, R.; Heikkinen, J.; Vaananen, K.; Peltonen, J. Decreased bone mineral density and content in neurofibromatosis type 1: Lowest local values are located in the load-carrying parts of the body. Osteoporos. Int. 2005, 16, 928–936. [Google Scholar] [CrossRef] [PubMed]
- Illes, T.; Halmai, V.; de Jonge, T.; Dubousset, J. Decreased bone mineral density in neurofibromatosis-1 patients with spinal deformities. Osteoporos. Int. 2001, 12, 823–827. [Google Scholar] [PubMed]
- Lammert, M.; Kappler, M.; Mautner, V.F.; Lammert, K.; Storkel, S.; Friedman, J.M.; Atkins, D. Decreased bone mineral density in patients with neurofibromatosis 1. Osteoporos. Int. 2005, 16, 1161–1166. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Estwick, S.A.; Chen, S.; Yu, M.; Ming, W.; Nebesio, T.D.; Li, Y.; Yuan, J.; Kapur, R.; Ingram, D.; et al. Neurofibromin plays a critical role in modulating osteoblast differentiation of mesenchymal stem/progenitor cells. Hum. Mol. Genet. 2006, 15, 2837–2845. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.C.; Ingram, D.A.; Chen, S.; Hingtgen, C.M.; Ratner, N.; Monk, K.R.; Clegg, T.; White, H.; Mead, L.; Wenning, M.J.; et al. Neurofibromin-deficient Schwann cells secrete a potent migratory stimulus for Nf1+/− mast cells. J. Clin. Investig. 2003, 112, 1851–1861. [Google Scholar] [PubMed]
- Yang, F.C.; Chen, S.; Clegg, T.; Li, X.; Morgan, T.; Estwick, S.A.; Yuan, J.; Khalaf, W.; Burgin, S.; Travers, J.; et al. Nf1+/− mast cells induce neurofibroma like phenotypes through secreted TGF-β signaling. Hum. Mol. Genet. 2006, 15, 2421–2437. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, D.A.; Schwarz, E.L.; Viskochil, D.H.; Moyer-Mileur, L.J.; Murray, M.; Firth, S.D.; D’Astous, J.L.; Carey, J.C.; Pasquali, M. Evidence of increased bone resorption in neurofibromatosis type 1 using urinary pyridinium crosslink analysis. Pediatr. Res. 2008, 63, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Joseph, N.M.; Mukouyama, Y.S.; Mosher, J.T.; Jaegle, M.; Crone, S.A.; Dormand, E.L.; Lee, K.F.; Meijer, D.; Anderson, D.J.; Morrison, S.J. Neural crest stem cells undergo multilineage differentiation in developing peripheral nerves to generate endoneurial fibroblasts in addition to Schwann cells. Development 2004, 131, 5599–5612. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, T.; Boissy, Y.L.; Kombrinck, K.; Brannan, C.I.; Jenkins, N.A.; Copeland, N.G.; Ratner, N. Neurofibromin-deficient fibroblasts fail to form perineurium in vitro. Development 1995, 121, 3583–3592. [Google Scholar] [PubMed]
- Cichowski, K.; Jacks, T. NF1 tumor suppressor gene function: Narrowing the GAP. Cell 2001, 104, 593–604. [Google Scholar] [CrossRef]
- Son, B.R.; Marquez-Curtis, L.A.; Kucia, M.; Wysoczynski, M.; Turner, A.R.; Ratajczak, J.; Ratajczak, M.Z.; Janowska-Wieczorek, A. Migration of bone marrow and cord blood mesenchymal stem cells in vitro is regulated by stromal-derived factor-1-CXCR4 and hepatocyte growth factor-c-met axes and involves matrix metalloproteinases. Stem Cells 2006, 24, 1254–1264. [Google Scholar] [CrossRef] [PubMed]
- Forte, G.; Minieri, M.; Cossa, P.; Antenucci, D.; Sala, M.; Gnocchi, V.; Fiaccavento, R.; Carotenuto, F.; de Vito, P.; Baldini, P.M.; et al. Hepatocyte growth factor effects on mesenchymal stem cells: Proliferation, migration, and differentiation. Stem Cells 2006, 24, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Ball, S.G.; Shuttleworth, C.A.; Kielty, C.M. Vascular endothelial growth factor can signal through platelet-derived growth factor receptors. J. Cell Biol. 2007, 177, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, R.; Galvez, B.G.; Pusterla, T.; de Marchis, F.; Cossu, G.; Marcu, K.B.; Bianchi, M.E. Cells migrating to sites of tissue damage in response to the danger signal HMGB1 require NF-κB activation. J. Cell Biol. 2007, 179, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Cancer and the chemokine network. Nat. Rev. Cancer 2004, 4, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Takashima, Y.; Era, T.; Nakao, K.; Kondo, S.; Kasuga, M.; Smith, A.G.; Nishikawa, S. Neuroepithelial cells supply an initial transient wave of MSC differentiation. Cell 2007, 129, 1377–1388. [Google Scholar] [CrossRef] [PubMed]
- Mansilla, E.; Marin, G.H.; Drago, H.; Sturla, F.; Salas, E.; Gardiner, C.; Bossi, S.; Lamonega, R.; Guzman, A.; Nunez, A.; et al. Bloodstream cells phenotypically identical to human mesenchymal bone marrow stem cells circulate in large amounts under the influence of acute large skin damage: New evidence for their use in regenerative medicine. Transpl. Proc. 2006, 38, 967–969. [Google Scholar] [CrossRef] [PubMed]
- Ponte, A.L.; Marais, E.; Gallay, N.; Langonne, A.; Delorme, B.; Herault, O.; Charbord, P.; Domenech, J. The in vitro migration capacity of human bone marrow mesenchymal stem cells: Comparison of chemokine and growth factor chemotactic activities. Stem Cells 2007, 25, 1737–1745. [Google Scholar] [CrossRef] [PubMed]
- Kingsley, K.; Plopper, G.E. Platelet-derived growth factor modulates rat vascular smooth muscle cell responses on laminin-5 via mitogen-activated protein kinase-sensitive pathways. Cell Commun. Signal. 2005, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Muller, G.; Storz, P.; Bourteele, S.; Doppler, H.; Pfizenmaier, K.; Mischak, H.; Philipp, A.; Kaiser, C.; Kolch, W. Regulation of Raf-1 kinase by TNF via its second messenger ceramide and cross-talk with mitogenic signalling. EMBO J. 1998, 17, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.C.; Kapur, R.; King, A.J.; Tao, W.; Kim, C.; Borneo, J.; Breese, R.; Marshall, M.; Dinauer, M.C.; Williams, D.A. Rac2 stimulates Akt activation affecting BAD/Bcl-XL expression while mediating survival and actin function in primary mast cells. Immunity 2000, 12, 557–568. [Google Scholar] [CrossRef]
- Koivunen, J.; Karvonen, S.L.; Yla-Outinen, H.; Aaltonen, V.; Oikarinen, A.; Peltonen, J. NF1 tumor suppressor in epidermal wound healing with special focus on wound healing in patients with type 1 neurofibromatosis. Arch. Dermatol. Res. 2005, 296, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Stansfield, B.K.; Bessler, W.K.; Mali, R.; Mund, J.A.; Downing, B.D.; Kapur, R.; Ingram, D.A., Jr. Ras-Mek-Erk signaling regulates Nf1 heterozygous neointima formation. Am. J. Pathol. 2014, 184, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, D.A.; Yan, J.; He, Y.; Li, H.; Liu, Y.; Zhang, Q.; Jing, Y.; Guo, Z.; Zhang, W.; Yang, D.; et al. Multiple increased osteoclast functions in individuals with neurofibromatosis type 1. Am. J. Med. Genet. Part A 2011, 155A, 1050–1059. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Rangwala, F.; Fulkerson, P.C.; Ling, B.; Reed, E.; Cox, A.D.; Kamholz, J.; Ratner, N. Role of TC21/R-Ras2 in enhanced migration of neurofibromin-deficient Schwann cells. Oncogene 2004, 23, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Meirelles Lda, S.; Nardi, N.B. Murine marrow-derived mesenchymal stem cell: Isolation, in vitro expansion, and characterization. Br. J. Haematol. 2003, 123, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Loughran, G.; Healy, N.C.; Kiely, P.A.; Huigsloot, M.; Kedersha, N.L.; O’Connor, R. Mystique is a new insulin-like growth factor-I-regulated PDZ-LIM domain protein that promotes cell attachment and migration and suppresses Anchorage-independent growth. Mol. Biol. Cell 2005, 16, 1811–1822. [Google Scholar] [CrossRef] [PubMed]
- Javelaud, D.; Laboureau, J.; Gabison, E.; Verrecchia, F.; Mauviel, A. Disruption of basal JNK activity differentially affects key fibroblast functions important for wound healing. J. Biol. Chem. 2003, 278, 24624–24628. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; He, Y.; Sharma, R.; Xing, W.; Estwick, S.A.; Wu, X.; Rhodes, S.D.; Xu, M.; Yang, F.-C. Hyperactive RAS/PI3-K/MAPK Signaling Cascade in Migration and Adhesion of Nf1 Haploinsufficient Mesenchymal Stem/Progenitor Cells. Int. J. Mol. Sci. 2015, 16, 12345-12359. https://doi.org/10.3390/ijms160612345
Zhou Y, He Y, Sharma R, Xing W, Estwick SA, Wu X, Rhodes SD, Xu M, Yang F-C. Hyperactive RAS/PI3-K/MAPK Signaling Cascade in Migration and Adhesion of Nf1 Haploinsufficient Mesenchymal Stem/Progenitor Cells. International Journal of Molecular Sciences. 2015; 16(6):12345-12359. https://doi.org/10.3390/ijms160612345
Chicago/Turabian StyleZhou, Yuan, Yongzheng He, Richa Sharma, Wen Xing, Selina A. Estwick, Xiaohua Wu, Steven D. Rhodes, Mingjiang Xu, and Feng-Chun Yang. 2015. "Hyperactive RAS/PI3-K/MAPK Signaling Cascade in Migration and Adhesion of Nf1 Haploinsufficient Mesenchymal Stem/Progenitor Cells" International Journal of Molecular Sciences 16, no. 6: 12345-12359. https://doi.org/10.3390/ijms160612345
APA StyleZhou, Y., He, Y., Sharma, R., Xing, W., Estwick, S. A., Wu, X., Rhodes, S. D., Xu, M., & Yang, F.-C. (2015). Hyperactive RAS/PI3-K/MAPK Signaling Cascade in Migration and Adhesion of Nf1 Haploinsufficient Mesenchymal Stem/Progenitor Cells. International Journal of Molecular Sciences, 16(6), 12345-12359. https://doi.org/10.3390/ijms160612345