Abstract
Systemic lupus erythematosus (SLE) is a prototypic autoimmune disease that is characterized by the generation of immune responses to various nuclear components. Impaired clearance of apoptotic cells and loss of tolerance to self-antigens are involved both in the initiation and in the propagation of the disease. Dendritic cells (DCs) are key factors in the balance between autoimmunity and tolerance and play a role linking innate and adaptive immunity. DCs, particularly plasmacytoid DCs (pDCs), are the main source of type I interferon (IFN) cytokines, which contribute to the immunopathogenesis of SLE. There is accumulating evidence that pDCs and type I IFN cytokines take the leading part in the development of SLE. In this review, we discuss recent data regarding the role of pDCs and type I IFN cytokines in the pathogenesis of SLE and the potential for employing therapies targeting against aberrant regulation of the pDC-type I IFN axis for treating SLE.
1. Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease that affects multiple organs and has significant mortality. The etiology of SLE includes both genetic and environmental factors, but remains poorly understood. Various immunologic abnormalities contribute to the pathogenesis of SLE. These include disturbed clearance of apoptotic cells, loss of tolerance to self-antigens, aberrant activation of T and B cells, altered cytokine profiles, and pathogenic autoantibody production. The autoantibodies, which are serologic hallmarks of SLE, are mostly directed against nuclear antigens including double-stranded DNA (dsDNA), small nuclear ribonucleoproteins (snRNPs), and nucleosomes although autoantibodies are also directed against antigens located in the cytoplasm, those on the cell surface, and those secreted by the cell [1,2]. Numerous factors including abnormal apoptosis of immune cells, undisposed apoptotic cell debris, and aberrant interactions between hyperactive B and T cells are associated with autoantibody production, which can lead to the formation of immune complexes (ICs) in patients with SLE [3]. During the process of unraveling this complicated immunologic problem, clues that dendritic cells (DCs) and type I interferon (IFN) play a critical role in the pathogenesis of SLE have emerged over the past few decades. This review will focus on both the normal physiologic roles of DCs in the human immune system and our current understanding of the role of DCs and type I IFN in SLE. In addition, we will discuss recent data concerning new therapeutic approaches focused on DCs and type I IFN in SLE.
2. DCs and Immune Tolerance
2.1. DC Subsets & Characteristics
DCs were first identified by Steinman and Cohn based on their characteristic stellate morphology. Myeloid DCs (mDCs) and plasmacytoid DCs (pDCs) are two major subtypes of DCs [4]. Although DCs share the capacity to present antigens and regulate T and B cells, there are several distinctions between mDCs and pDCs. mDCs, which have a classical finger-like projection morphology, are professional antigen presenting cells (APCs) bearing various Toll-like receptors (TLRs) including TLR 1, 6, 8 and 10 [5]. Human mDCs are characterized by high expression levels of CD11c, CD1a and HLA-DR [6]. Unlike mDCs, pDCs, which have an appearance similar to that of plasma cells, release copious amounts of type I IFNs following activation and induce differentiation of B cells into antibody-producing plasma cells [7]. Although the pDC population is less than 1% of human peripheral blood mononuclear cells, they produce the majority of type I IFNs in the human body upon exposure to viral particles [8]. Human pDCs, which are negative for CD11c and CD1a, express IL-3 receptor (CD123), blood dendritic cell antigen (BDCA)-2 (CD303), neuropilin-1 (CD304 or BDCA-4), and relatively low level of HLA-DR [9]. pDCs can recognize pathogens through TLRs, such as TLR7 and TLR9 [10].
2.2. DCs as Guards against Autoimmunity
DCs, which link innate and adaptive immunity, serve as regulators of the immune system. They potentially promote immune tolerance and, on the contrary, might also initiate autoimmune reactions, depending on their circumstances. Steady-state DCs with an immature phenotype are essential for the induction and maintenance of peripheral immune tolerance. Although there are conflicting studies about the influence of the status of DCs on the maintenance of immune tolerance, activated DCs are universally considered to lose their ability to stabilize autoreactive T cells and induce regulatory T (Treg) cells [11,12].
At steady state, DCs contribute to immune tolerance. In the thymus, thymic DCs, along with medullary thymic epithelial cells (mTECs), present many tissue-specific self-antigens to developing thymocytes. Peripheral DCs also migrate into the thymus and present peripheral self-antigens to developing thymocytes to induce negative selection of autoreactive T cells [13]. DCs also promote the generation of Treg cells in the thymus [14]. In addition, DCs are considered to play an essential role in peripheral tolerance. In the normal milieu, antigen presentation by DCs induces an anergic state in effector T cells by overexpressing inhibitory molecules such as programmed cell death protein 1 (PD1) and cytotoxic T lymphocyte antigen 4 (CTLA4) on T cells [15]. Ligation of PD1 and PD1 ligand 1 (PDL1) or CTLA4 and CD80/86 is important for the induction of T cell tolerance [15]. DCs also seem to contribute to the induction and the maintenance of Treg cells in the periphery [16,17]. In the presence of transforming growth factor-β (TGF-β) and retinoic acid, DCs can induce Treg cells from naïve CD4+ T cells [12,18]. Expression of costimulatory molecules, CD80 and/or CD86 on DCs is also necessary for the maintenance of the number of Treg cells [19].
3. The Role of DCs in SLE
It has been widely demonstrated that dysregulations of both the innate and adaptive immune systems contribute to the pathogenesis of SLE. DCs, which link innate and adaptive immunity, are implicated in the development of SLE. Removal of DCs in MRL-Faslpr mice, a lupus-prone mouse model, ameliorated the disease and decreased T cell expansion as well as IgG/IgM autoantibody formation [20]. The specific ablation of CD95 (a death receptor also known as Fas) in DCs induces several disease manifestations such as production of antinuclear antibody and hyperimmunoglobulinemia in C57BL/6 mice [21]. DCs from SLE patients exhibit distinctive expression of co-stimulatory and inhibitory surface markers [22]. Among numerous previous studies that demonstrate the role of DCs in the development of SLE, pDCs and their product, type I IFN, are especially highlighted in the immunopathogenesis of SLE.
3.1. pDCs in SLE
Recent studies have provided circumstantial evidence that pDCs play an important role in the pathogenesis of SLE. Several reports discuss the altered population of pDCs in patients with SLE. Migita et al. demonstrated that circulating BDCA-2-positive pDCs are reduced in patients with SLE [23]. Fiore et al. and Tucci et al. reported that the number of pDCs in the blood of SLE patients is decreased, while pDC infiltrations are increased in the kidney tissues of patients with lupus nephritis, suggesting that activated pDCs might have migrated to the target organ [24,25]. Accumulation of pDCs in skin lesions has also been confirmed in SLE patients [26,27]. Phenotypic and functional changes of pDCs were investigated in SLE. Kwok et al. demonstrated that pDCs in SLE patients are exhausted and show a diminished response upon TLR9 stimulation [28]. Nie et al. revealed that bone marrow-derived pDCs of SLE patients have high expression levels of CD40 and CD86 and have a strong capacity to induce T cell proliferation [29]. Low expression of ChemR23, a chemokine receptor that is expressed on immature DCs, and high expression of CCL19, towards which mature DCs migrate, reflect the activation status of pDCs in patients with SLE [30,31,32]. Compared with pDCs from healthy subjects, circulating pDCs from SLE patients stimulated T cells more vigorously; however, they failed to induce Treg cells in the presence of apoptotic cells derived from polymorphonuclear cells [33]. Early depletion of pDCs in lupus-prone BXSB.DTR mice reduced lupus symptoms and signs including organomegaly, reactivity of T and B cells, production of autoantibodies, and severity of kidney pathology, supporting a pivotal role for pDCs in SLE [34].
In SLE, the disruption of innate tolerance to self-antigens is considered to be caused by the formation of ICs. Circulating ICs, containing self-nucleic acids and autoantibodies, trigger the activation of pDCs in SLE [35]. Self-DNA or RNA- containing ICs bind to the low-affinity Fc receptor for IgG (FcγRIIA, also known as CD32) and are then taken up into endosomes as a result of the engagement of TLR9 or TLR7 expressed in pDCs respectively [36]. Lupus-prone mice deficient in TLR7 exhibited an amelioration of the disease including a decrease in serum autoantibody production (e.g., anti-Sm/RNP autoantibody), negative regulation of lymphocyte activation, and reduced severity of lupus nephritis [37]. Overexpression of Tlr7 gene induced SLE-like phenomena in mice, suggesting that self-RNA-triggered TLR7 signaling contributes to the development of SLE [38]. On the other hand, TLR9 deficient lupus-prone mice exhibited an exacerbation of the disease with increased serum levels of IgG and IFNα [37]. The discordance in the contribution to the development of SLE between TLR7 and TLR9 remains poorly understood. In pDCs, TLR7/9 signaling is propagated via a MyD88-dependent pathway. MyD88 engagement ultimately leads to the expression of Type I/III IFN, proinflammatory cytokines such as IL-6 and TNF-α, and costimulatory molecules including CD40, CD80, and CD86 through the IRF5/7, NFκB, and MAPK pathway [39]. In particular, IRF7 is regarded as a master regulator of Type I IFN production [40]. The involvement of neutrophil has also been emphasized in TLR-7/9-mediated pDC activation in SLE. Compared with the neutrophils of healthy controls, those of SLE patients have a tendency to undergo more neutrophil death by neutrophil extracellular trap (NET) formation referred to as NETosis [41]. Increased NETosis in SLE is caused by ICs or IFNα, and it also results in an increased production of IFNα via pDC activation [41]. NETs extruded by neutrophils contain DNA as well as large amounts of LL37 and high-mobility group box 1 protein (HMGB1) [41,42]. Self-DNA uptaked by LL37, which is an antimicrobial peptide that is abundant in NETs released from neutrophils, can induce pDC activation via TLR9 [41,43]. HMGB1 released by damaged cells binds to nuclear DNA and enhances pDC activation through a TLR9-MyD88- dependent pathway [42].
3.2. Role of Type I IFN in SLE
The increased production of type I IFN by pDCs is involved in the pathogenesis of SLE [44]. IFNα is the most important type I IFN in SLE [45]. IFNα therapies sometime induce lupus-like phenomena in patients with malignancy or chronic hepatitis C [46,47]. The level of serum IFNα has been reported to have a positive correlation with lupus disease activity, in some patients [48]. Patients with SLE exhibit an IFN signature, a characteristic expression pattern of type I IFN-inducible genes, in peripheral leukocytes, that is associated with disease activity and severity [44,49,50]. T and B cell lymphopenia frequently found in SLE is sometimes explained by high level of IFNα [51]. Type I IFNs have also been reported to be associated with severe features of SLE such as positivity for anti-dsDNA and presence of nephritis by lowering serotonin synthesis [52].
Several mechanisms have been suggested for the pathogenic role of type I IFNs in the pathogenesis of SLE as shown in Figure 1. ICs containing self-nucleic acids, one of the hallmarks of SLE, activate DCs, thus producing abundant type I IFNs. Type I IFNs produced by pDCs stimulate DCs to initiate and maintain their maturation, which is essential for aberrant immune reactions in SLE [53]. Type I IFNs contribute to the generation of both CD4+ and CD8+ T cell responses via the activation of DCs [54,55]. Serum derived from the patients with SLE has increased T cell stimulatory function in mixed lymphocyte reaction study and the stimulatory function was dependent on serum level of IFNα, suggesting the ability of IFNα for stimulating autoreactive T cells [53]. Type I IFNs also promote the cytolytic activities of NK cells and cytotoxic T cells, which could increase tissue damage and antigen overloading, one of major contributors in the development of SLE [56,57]. Type I IFNs increase the production of B lymphocyte stimulator (BLyS) and a proliferation-inducing ligand (APRIL), which are involved in the survival of autoreactive B cells, by mDCs, and this contributes to B cell differentiation and Ig class switching, which are important for generating pathogenic autoantibodies in SLE [7,58,59]. Type I IFNs are also involved in the pathogenesis of SLE by supporting Th17 responses [60]. Type I IFNs promote Th17 responses by inducing the production of IL-6 and IL-23 by DCs [59]. IL-17, produced as a result of Th17 responses, alone or in conjunction with BLyS, can induce B cell hyperreactivity and differentiation into antibody-producing cells [61]. IL-17 can also cause tissue inflammation and organ damage by recruiting neutrophils, lymphocytes and macrophages [62]. In addition, Th17-associated cytokine, IL-21, contributes to breach of immune tolerance in SLE by promoting B cell responses and also by playing a role in the development of follicular helper T cell [63,64].
The analysis of the tissue from SLE patients provides direct evidence of a pathogenic role for type I IFNs in SLE. Synovial tissue from patients with SLE showed characteristic molecular signature, featured by the upregulation of IFN-inducible genes [65]. The IFN signature has also been observed in glomerular tissue from patients with SLE [66]. In a murine model of lupus, IFNα accelerated murine SLE and promoted the development of nephritis [67]. Type I IFNs caused glomerulosclerosis by inducing podocyte death while suppressing renal progenitor differentiation into mature podocytes [68]. Additional data about the effect on cardiovascular system support a pathogenic role for type I IFNs in SLE. Type I IFNs skewed the balance between vascular endothelial cell damage and repair, which led to the acceleration of atherosclerosis in both murine and human SLE [69,70]. A contribution of IFNα to the central nervous system manifestations of SLE is supported by the finding that cerebrospinal fluid from patients with neuropsychiatric lupus had a potency to induce large amounts of IFNα [71].
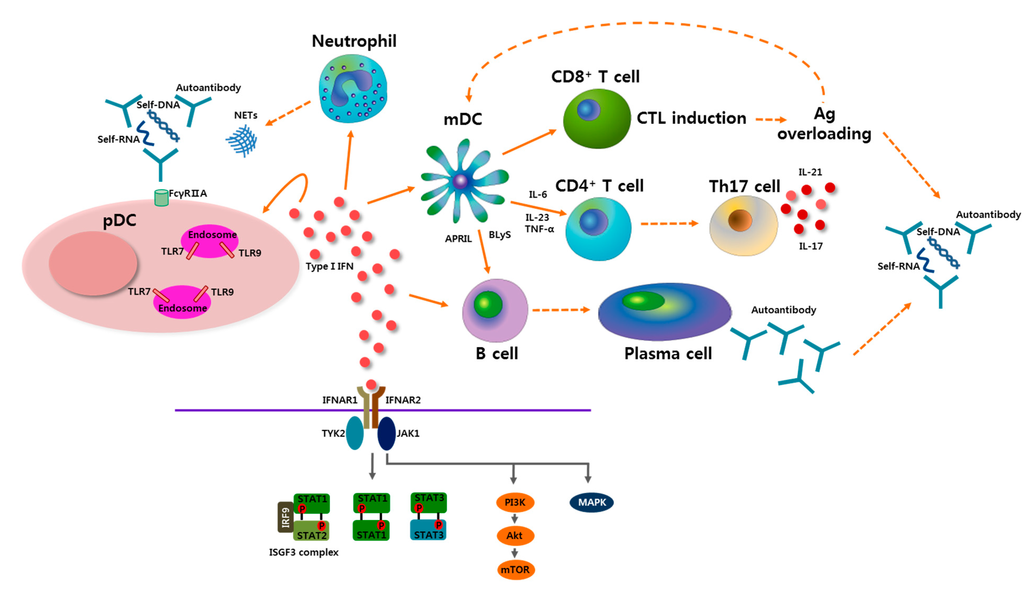
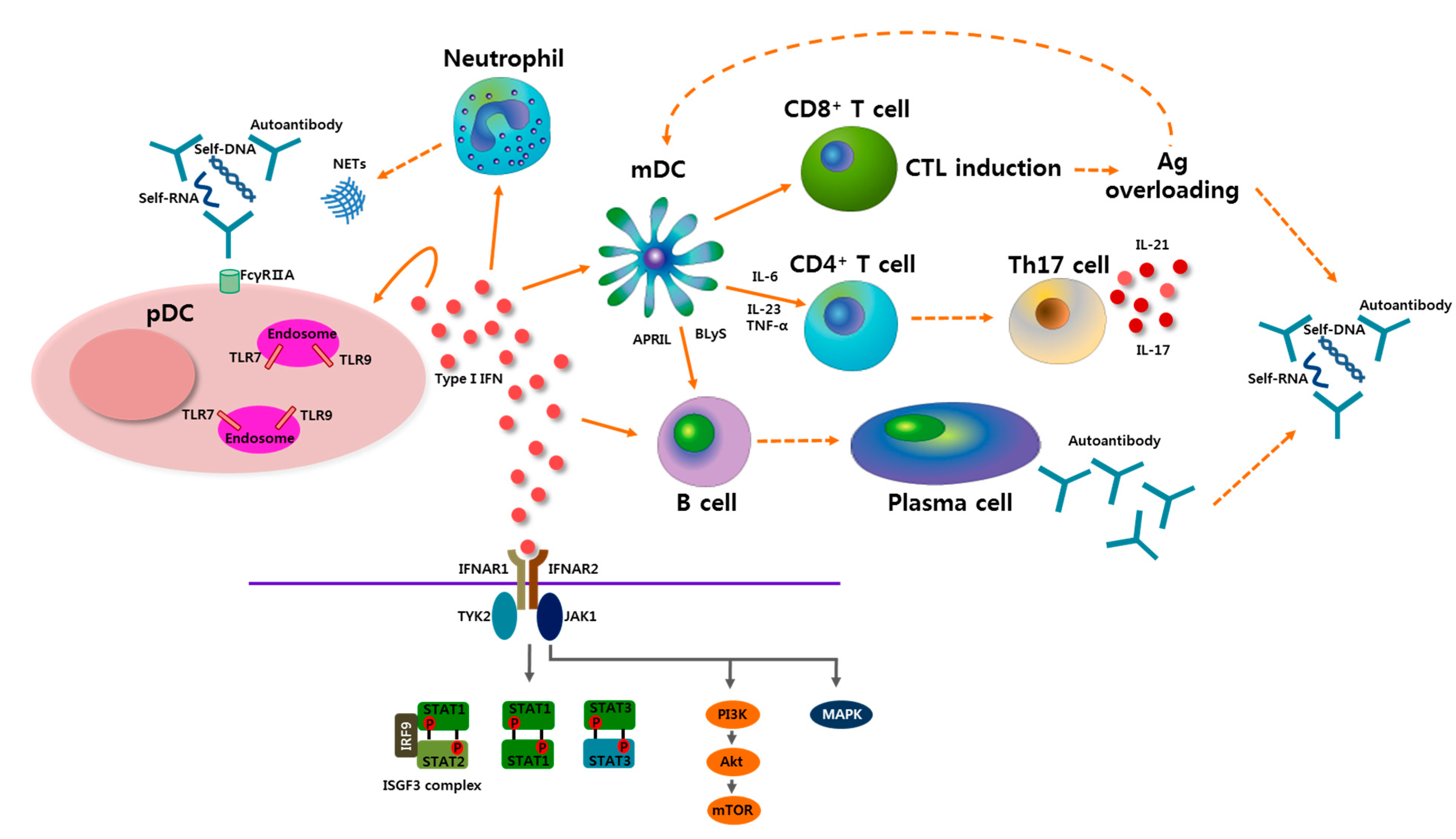
Figure 1.
Potential role of plasmacytoid dendritic cells (pDCs) and type I interferon (IFN) in the pathogenesis of systemic lupus erythematosus (SLE). Self-nucleic acids-containing immune complexes activate pDCs by transferring to endosomes after engagement of TLR7 or TLR9. Neutrophil extracellular traps (NETs), which are shed from neutrophils, induce activation of pDCs. Activated pDCs produce profuse type I IFN, which plays a central role in the pathogenesis of SLE. Type I IFN has an effect on many types of immunologic cells, resulting in diverse outcomes. Type I IFN lowers the activation threshold of T cells and B cells by aiding myeloid DCs (mDCs) to produce various stimulators including B lymphocyte stimulator (BLyS), a proliferation-inducing ligand (APRIL), interleukin-6 (IL-6), IL-23, and tumor necrosis factor-α (TNF-α). Th17 cell differentiation, which is supported by type I IFN, has an important role in SLE pathogenesis. IL-17 released by Th17 cells is involved in B cell hyperresponsiveness, autoantibody production, and target organ damage. IL-21, Th17-related cytokine, is also involved in B cell reactivity and follicular helper T cell development. Cytotoxic T lymphocyte (CTL) induction by type I IFN leads to overloading of antigen (Ag), which can be presented by mDCs to other immune cells, and also be a cause of immune complex formation as well as autoantibody production in SLE. After type I IFN binds to type I IFNα receptor (IFNAR), which is a heterodimeric receptor composed of IFNAR1 and IFNAR2, multiple downstream signaling pathways can be involved. Upon activating tyrosine kinase 2 (TYK2) and Janus activated kinase 1(JAK1), various signal transducer and activator of transcription (STAT) proteins are phosphorylated and form heterodimers or homodimers. A canonical IFN-stimulated gene factor 3 (ISGF3) signaling complex, which is a complex of STAT1-STAT2-IFN-regulator factor 9 (IRF9), leads to the induction of IFN-stimulated genes. Other STAT dimers also activate downstream molecules, and they are partially involved in inflammatory responses. Type I IFN signals can also propagate through the mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K) signaling pathways.
Figure 1.
Potential role of plasmacytoid dendritic cells (pDCs) and type I interferon (IFN) in the pathogenesis of systemic lupus erythematosus (SLE). Self-nucleic acids-containing immune complexes activate pDCs by transferring to endosomes after engagement of TLR7 or TLR9. Neutrophil extracellular traps (NETs), which are shed from neutrophils, induce activation of pDCs. Activated pDCs produce profuse type I IFN, which plays a central role in the pathogenesis of SLE. Type I IFN has an effect on many types of immunologic cells, resulting in diverse outcomes. Type I IFN lowers the activation threshold of T cells and B cells by aiding myeloid DCs (mDCs) to produce various stimulators including B lymphocyte stimulator (BLyS), a proliferation-inducing ligand (APRIL), interleukin-6 (IL-6), IL-23, and tumor necrosis factor-α (TNF-α). Th17 cell differentiation, which is supported by type I IFN, has an important role in SLE pathogenesis. IL-17 released by Th17 cells is involved in B cell hyperresponsiveness, autoantibody production, and target organ damage. IL-21, Th17-related cytokine, is also involved in B cell reactivity and follicular helper T cell development. Cytotoxic T lymphocyte (CTL) induction by type I IFN leads to overloading of antigen (Ag), which can be presented by mDCs to other immune cells, and also be a cause of immune complex formation as well as autoantibody production in SLE. After type I IFN binds to type I IFNα receptor (IFNAR), which is a heterodimeric receptor composed of IFNAR1 and IFNAR2, multiple downstream signaling pathways can be involved. Upon activating tyrosine kinase 2 (TYK2) and Janus activated kinase 1(JAK1), various signal transducer and activator of transcription (STAT) proteins are phosphorylated and form heterodimers or homodimers. A canonical IFN-stimulated gene factor 3 (ISGF3) signaling complex, which is a complex of STAT1-STAT2-IFN-regulator factor 9 (IRF9), leads to the induction of IFN-stimulated genes. Other STAT dimers also activate downstream molecules, and they are partially involved in inflammatory responses. Type I IFN signals can also propagate through the mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K) signaling pathways.
4. pDCs-Type I IFN Axis as a Therapeutic Target in SLE
Nucleic acid-sensing TLRs in pDCs and their product, type I IFN, have become major targets in the development of new therapeutic agents for SLE. Sifalimumab (MEDI-545), a fully humanized IgG1 monoclonal antibody against IFNα, exhibited good tolerability and inhibited the type I IFN signature in some patients with SLE [72,73]. Another humanized IgG1 monoclonal antibody against IFNα, rontalizumab (RG7415), demonstrated an acceptable safety profile, although further investigation is required to determine the clinical efficacy in patients with SLE [74]. Type I IFN priming in SLE was attempted using an IFNα kinoid (IFN-K), which is a synthetic compound composed of IFNα and a carrier protein, keyhole limpet hemocyanin [75]. Active immunization with IFN-K resulted in the production of anti-IFNα antibodies and improved disease activity markers in patients with SLE [75]. A few attempts at modulating TLR signaling in SLE suggest both promising safety and efficacy profiles [76].
5. Conclusions
Activation of the pDCs-type I IFN axis plays a central role in the pathogenesis of SLE, as evidenced by multiple experimental and observational studies. Although pDCs-type I IFN axis-based therapies for treating SLE are an opening stage, a better understanding of this axis in SLE would potentially enhance the development of new therapeutic agents for the management of SLE.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rahman, A.; Isenberg, D.A. Systemic lupus erythematosus. N. Engl. J. Med. 2008, 358, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Lisnevskaia, L.; Murphy, G.; Isenberg, D. Systemic lupus erythematosus. Lancet 2014, 384, 1878–1888. [Google Scholar] [CrossRef]
- Munoz, L.E.; Lauber, K.; Schiller, M.; Manfredi, A.A.; Herrmann, M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat. Rev. Rheumatol. 2010, 6, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Satpathy, A.T.; Wu, X.; Albring, J.C.; Murphy, K.M. Re(de)fining the dendritic cell lineage. Nat. Immunol. 2012, 13, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, N.; Ho, S.; Antonenko, S.; Malefyt, R.W.; Kastelein, R.A.; Bazan, F.; Liu, Y.J. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J. Exp. Med. 2001, 194, 863–869. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, K.P.; Munster, D.J.; Clark, G.J.; Dzionek, A.; Schmitz, J.; Hart, D.N. Characterization of human blood dendritic cell subsets. Blood 2002, 100, 4512–4520. [Google Scholar] [CrossRef] [PubMed]
- Jego, G.; Palucka, A.K.; Blanck, J.P.; Chalouni, C.; Pascual, V.; Banchereau, J. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity 2003, 19, 225–234. [Google Scholar] [CrossRef]
- Siegal, F.P.; Kadowaki, N.; Shodell, M.; Fitzgerald-Bocarsly, P.A.; Shah, K.; Ho, S.; Antonenko, S.; Liu, Y.J. The nature of the principal type I interferon-producing cells in human blood. Science 1999, 284, 1835–1837. [Google Scholar] [CrossRef] [PubMed]
- Shortman, K.; Liu, Y.J. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2002, 2, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Kaisho, T. Pathogen sensors and chemokine receptors in dendritic cell subsets. Vaccine 2012, 30, 7652–7657. [Google Scholar] [CrossRef] [PubMed]
- Hawiger, D.; Inaba, K.; Dorsett, Y.; Guo, M.; Mahnke, K.; Rivera, M.; Ravetch, J.V.; Steinman, R.M.; Nussenzweig, M.C. Dendritic cells induce peripheral t cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 2001, 194, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Kretschmer, K.; Apostolou, I.; Hawiger, D.; Khazaie, K.; Nussenzweig, M.C.; von Boehmer, H. Inducing and expanding regulatory t cell populations by foreign antigen. Nat. Immunol. 2005, 6, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Bonasio, R.; Scimone, M.L.; Schaerli, P.; Grabie, N.; Lichtman, A.H.; von Andrian, U.H. Clonal deletion of thymocytes by circulating dendritic cells homing to the thymus. Nat. Immunol. 2006, 7, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Proietto, A.I.; van Dommelen, S.; Zhou, P.; Rizzitelli, A.; D’Amico, A.; Steptoe, R.J.; Naik, S.H.; Lahoud, M.H.; Liu, Y.; Zheng, P.; et al. Dendritic cells in the thymus contribute to T-regulatory cell induction. Proc. Natl. Acad. Sci. USA 2008, 105, 19869–19874. [Google Scholar] [CrossRef] [PubMed]
- Probst, H.C.; McCoy, K.; Okazaki, T.; Honjo, T.; van den Broek, M. Resting dendritic cells induce peripheral CD8+ T cell tolerance through PD-1 and CTLA-4. Nat. Immunol. 2005, 6, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Sela, U.; Olds, P.; Park, A.; Schlesinger, S.J.; Steinman, R.M. Dendritic cells induce antigen-specific regulatory T cells that prevent graft vs. host disease and persist in mice. J. Exp. Med. 2011, 208, 2489–2496. [Google Scholar] [CrossRef] [PubMed]
- Suffner, J.; Hochweller, K.; Kuhnle, M.C.; Li, X.; Kroczek, R.A.; Garbi, N.; Hammerling, G.J. Dendritic cells support homeostatic expansion of Foxp3+ regulatory t cells in Foxp3.Lucidtr mice. J. Immunol. 2010, 184, 1810–1820. [Google Scholar] [CrossRef] [PubMed]
- Coombes, J.L.; Siddiqui, K.R.; Arancibia-Carcamo, C.V.; Hall, J.; Sun, C.M.; Belkaid, Y.; Powrie, F. A functionally specialized population of mucosal cd103+ dcs induces Foxp3+ regulatory T cells via a TGF-β and retinoic acid-dependent mechanism. J. Exp. Med. 2007, 204, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Bar-On, L.; Birnberg, T.; Kim, K.W.; Jung, S. Dendritic cell-restricted CD80/86 deficiency results in peripheral regulatory T-cell reduction but is not associated with lymphocyte hyperactivation. Eur. J. Immunol. 2011, 41, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Teichmann, L.L.; Ols, M.L.; Kashgarian, M.; Reizis, B.; Kaplan, D.H.; Shlomchik, M.J. Dendritic cells in lupus are not required for activation of T and B cells but promote their expansion, resulting in tissue damage. Immunity 2010, 33, 967–978. [Google Scholar] [CrossRef] [PubMed]
- Stranges, P.B.; Watson, J.; Cooper, C.J.; Choisy-Rossi, C.M.; Stonebraker, A.C.; Beighton, R.A.; Hartig, H.; Sundberg, J.P.; Servick, S.; Kaufmann, G.; et al. Elimination of antigen-presenting cells and autoreactive T cells by fas contributes to prevention of autoimmunity. Immunity 2007, 26, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Carreno, L.J.; Pacheco, R.; Gutierrez, M.A.; Jacobelli, S.; Kalergis, A.M. Disease activity in systemic lupus erythematosus is associated with an altered expression of low-affinity Fc γ receptors and costimulatory molecules on dendritic cells. Immunology 2009, 128, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Migita, K.; Miyashita, T.; Maeda, Y.; Kimura, H.; Nakamura, M.; Yatsuhashi, H.; Ishibashi, H.; Eguchi, K. Reduced blood BDCA-2+ (lymphoid) and CD11c+ (myeloid) dendritic cells in systemic lupus erythematosus. Clin. Exp. Immunol. 2005, 142, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Fiore, N.; Castellano, G.; Blasi, A.; Capobianco, C.; Loverre, A.; Montinaro, V.; Netti, S.; Torres, D.; Manno, C.; Grandaliano, G.; et al. Immature myeloid and plasmacytoid dendritic cells infiltrate renal tubulointerstitium in patients with lupus nephritis. Mol. Immunol. 2008, 45, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Tucci, M.; Quatraro, C.; Lombardi, L.; Pellegrino, C.; Dammacco, F.; Silvestris, F. Glomerular accumulation of plasmacytoid dendritic cells in active lupus nephritis: Role of interleukin-18. Arthritis Rheum. 2008, 58, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Farkas, L.; Beiske, K.; Lund-Johansen, F.; Brandtzaeg, P.; Jahnsen, F.L. Plasmacytoid dendritic cells (natural interferon-α/β-producing cells) accumulate in cutaneous lupus erythematosus lesions. Am. J. Pathol. 2001, 159, 237–243. [Google Scholar] [CrossRef]
- Blomberg, S.; Eloranta, M.L.; Cederblad, B.; Nordlin, K.; Alm, G.V.; Ronnblom, L. Presence of cutaneous interferon-α producing cells in patients with systemic lupus erythematosus. Lupus 2001, 10, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Kwok, S.K.; Lee, J.Y.; Park, S.H.; Cho, M.L.; Min, S.Y.; Park, S.H.; Kim, H.Y.; Cho, Y.G. Dysfunctional interferon-α production by peripheral plasmacytoid dendritic cells upon toll-like receptor-9 stimulation in patients with systemic lupus erythematosus. Arthritis Res. Ther. 2008, 10. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.J.; Mok, M.Y.; Chan, G.C.; Chan, A.W.; Jin, O.U.; Kavikondala, S.; Lie, A.K.; Lau, C.S. Phenotypic and functional abnormalities of bone marrow-derived dendritic cells in systemic lupus erythematosus. Arthritis Res. Ther. 2010, 12. [Google Scholar] [CrossRef] [PubMed]
- Gerl, V.; Lischka, A.; Panne, D.; Grossmann, P.; Berthold, R.; Hoyer, B.F.; Biesen, R.; Bruns, A.; Alexander, T.; Jacobi, A.; et al. Blood dendritic cells in systemic lupus erythematosus exhibit altered activation state and chemokine receptor function. Ann. Rheum. Dis. 2010, 69, 1370–1377. [Google Scholar] [CrossRef] [PubMed]
- Wittamer, V.; Franssen, J.D.; Vulcano, M.; Mirjolet, J.F.; le Poul, E.; Migeotte, I.; Brezillon, S.; Tyldesley, R.; Blanpain, C.; Detheux, M.; et al. Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. J. Exp. Med. 2003, 198, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Vermi, W.; Riboldi, E.; Wittamer, V.; Gentili, F.; Luini, W.; Marrelli, S.; Vecchi, A.; Franssen, J.D.; Communi, D.; Massardi, L.; et al. Role of chemr23 in directing the migration of myeloid and plasmacytoid dendritic cells to lymphoid organs and inflamed skin. J. Exp. Med. 2005, 201, 509–515. [Google Scholar] [CrossRef]
- Jin, O.; Kavikondala, S.; Mok, M.Y.; Sun, L.; Gu, J.; Fu, R.; Chan, A.; Yeung, J.; Nie, Y.; Lau, C.S. Abnormalities in circulating plasmacytoid dendritic cells in patients with systemic lupus erythematosus. Arthritis Res. Ther. 2010, 12. [Google Scholar] [CrossRef] [PubMed]
- Rowland, S.L.; Riggs, J.M.; Gilfillan, S.; Bugatti, M.; Vermi, W.; Kolbeck, R.; Unanue, E.R.; Sanjuan, M.A.; Colonna, M. Early, transient depletion of plasmacytoid dendritic cells ameliorates autoimmunity in a lupus model. J. Exp. Med. 2014, 211, 1977–1991. [Google Scholar] [CrossRef] [PubMed]
- Barrat, F.J.; Meeker, T.; Gregorio, J.; Chan, J.H.; Uematsu, S.; Akira, S.; Chang, B.; Duramad, O.; Coffman, R.L. Nucleic acids of mammalian origin can act as endogenous ligands for toll-like receptors and may promote systemic lupus erythematosus. J. Exp. Med. 2005, 202, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Lovgren, T.; Eloranta, M.L.; Bave, U.; Alm, G.V.; Ronnblom, L. Induction of interferon-α production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis Rheum. 2004, 50, 1861–1872. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.R.; Shupe, J.; Nickerson, K.; Kashgarian, M.; Flavell, R.A.; Shlomchik, M.J. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 2006, 25, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Deane, J.A.; Pisitkun, P.; Barrett, R.S.; Feigenbaum, L.; Town, T.; Ward, J.M.; Flavell, R.A.; Bolland, S. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity 2007, 27, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Karrich, J.J.; Jachimowski, L.C.; Uittenbogaart, C.H.; Blom, B. The plasmacytoid dendritic cell as the swiss army knife of the immune system: Molecular regulation of its multifaceted functions. J. Immunol. 2014, 193, 5772–5778. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Yanai, H.; Negishi, H.; Asagiri, M.; Sato, M.; Mizutani, T.; Shimada, N.; Ohba, Y.; Takaoka, A.; Yoshida, N.; et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005, 434, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Romo, G.S.; Caielli, S.; Vega, B.; Connolly, J.; Allantaz, F.; Xu, Z.; Punaro, M.; Baisch, J.; Guiducci, C.; Coffman, R.L.; et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Avalos, A.M.; Mao, S.Y.; Chen, B.; Senthil, K.; Wu, H.; Parroche, P.; Drabic, S.; Golenbock, D.; Sirois, C.; et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and rage. Nat. Immunol. 2007, 8, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Lande, R.; Ganguly, D.; Facchinetti, V.; Frasca, L.; Conrad, C.; Gregorio, J.; Meller, S.; Chamilos, G.; Sebasigari, R.; Riccieri, V.; et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Baechler, E.C.; Batliwalla, F.M.; Karypis, G.; Gaffney, P.M.; Ortmann, W.A.; Espe, K.J.; Shark, K.B.; Grande, W.J.; Hughes, K.M.; Kapur, V.; et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA 2003, 100, 2610–2615. [Google Scholar] [CrossRef] [PubMed]
- Reizis, B.; Colonna, M.; Trinchieri, G.; Barrat, F.; Gilliet, M. Plasmacytoid dendritic cells: One-trick ponies or workhorses of the immune system? Nat. Rev. Immunol. 2011, 11, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Schilling, P.J.; Kurzrock, R.; Kantarjian, H.; Gutterman, J.U.; Talpaz, M. Development of systemic lupus erythematosus after interferon therapy for chronic myelogenous leukemia. Cancer 1991, 68, 1536–1537. [Google Scholar] [CrossRef]
- Ho, V.; McLean, A.; Terry, S. Severe systemic lupus erythematosus induced by antiviral treatment for hepatitis C. J. Clin. Rheumatol. Pract. Rep. Rheum. Musculoskelet. Dis. 2008, 14, 166–168. [Google Scholar] [CrossRef] [PubMed]
- Dall’era, M.C.; Cardarelli, P.M.; Preston, B.T.; Witte, A.; Davis, J.C., Jr. Type I interferon correlates with serological and clinical manifestations of SLE. Ann. Rheum. Dis. 2005, 64, 1692–1697. [Google Scholar] [CrossRef] [PubMed]
- Postal, M.; Sinicato, N.A.; Pelicari, K.O.; Marini, R.; Lavras Costallat, L.T.; Appenzeller, S. Clinical and serological manifestations associated with interferon-α levels in childhood-onset systemic lupus erythematosus. Clinics 2012, 67, 157–162. [Google Scholar] [CrossRef]
- Bennett, L.; Palucka, A.K.; Arce, E.; Cantrell, V.; Borvak, J.; Banchereau, J.; Pascual, V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 2003, 197, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Dong, C.; Cooper, M.D. Impairment of t and B cell development by treatment with a type I interferon. J. Exp. Med. 1998, 187, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Lood, C.; Tyden, H.; Gullstrand, B.; Klint, C.; Wenglen, C.; Nielsen, C.T.; Heegaard, N.H.; Jonsen, A.; Kahn, R.; Bengtsson, A.A. Type I interferon-mediated skewing of the serotonin synthesis is associated with severe disease in systemic lupus erythematosus. PLoS ONE 2015, 10, e0125109. [Google Scholar] [CrossRef] [PubMed]
- Blanco, P.; Palucka, A.K.; Gill, M.; Pascual, V.; Banchereau, J. Induction of dendritic cell differentiation by IFN-α in systemic lupus erythematosus. Science 2001, 294, 1540–1543. [Google Scholar] [CrossRef] [PubMed]
- Longhi, M.P.; Trumpfheller, C.; Idoyaga, J.; Caskey, M.; Matos, I.; Kluger, C.; Salazar, A.M.; Colonna, M.; Steinman, R.M. Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J. Exp. Med. 2009, 206, 1589–1602. [Google Scholar] [CrossRef] [PubMed]
- Le Bon, A.; Etchart, N.; Rossmann, C.; Ashton, M.; Hou, S.; Gewert, D.; Borrow, P.; Tough, D.F. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat. Immunol. 2003, 4, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Curtsinger, J.M.; Valenzuela, J.O.; Agarwal, P.; Lins, D.; Mescher, M.F. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J. Immunol. 2005, 174, 4465–4469. [Google Scholar] [CrossRef] [PubMed]
- Biron, C.A.; Nguyen, K.B.; Pien, G.C.; Cousens, L.P.; Salazar-Mather, T.P. Natural killer cells in antiviral defense: Function and regulation by innate cytokines. Ann. Rev. Immunol. 1999, 17, 189–220. [Google Scholar] [CrossRef] [PubMed]
- Le Bon, A.; Schiavoni, G.; D’Agostino, G.; Gresser, I.; Belardelli, F.; Tough, D.F. Type I interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity 2001, 14, 461–470. [Google Scholar] [CrossRef]
- Yasuda, K.; Richez, C.; Maciaszek, J.W.; Agrawal, N.; Akira, S.; Marshak-Rothstein, A.; Rifkin, I.R. Murine dendritic cell type I IFN production induced by human IgG-RNA immune complexes is IFN regulatory factor (IRF) 5 and IRF7 dependent and is required for ILl–6 production. J. Immunol. 2007, 178, 6876–6885. [Google Scholar] [CrossRef] [PubMed]
- Ambrosi, A.; Espinosa, A.; Wahren-Herlenius, M. IL-17: A new actor in IFN-driven systemic autoimmune diseases. Eur. J. Immunol. 2012, 42, 2274–2284. [Google Scholar] [CrossRef] [PubMed]
- Doreau, A.; Belot, A.; Bastid, J.; Riche, B.; Trescol-Biemont, M.C.; Ranchin, B.; Fabien, N.; Cochat, P.; Pouteil-Noble, C.; Trolliet, P.; et al. Interleukin 17 acts in synergy with B cell-activating factor to influence B cell biology and the pathophysiology of systemic lupus erythematosus. Nat. Immunol. 2009, 10, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L. An overview of IL-17 function and signaling. Cytokine 2008, 43, 402–407. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, K.; Spolski, R.; Ettinger, R.; Kim, H.P.; Wang, G.; Qi, C.F.; Hwu, P.; Shaffer, D.J.; Akilesh, S.; Roopenian, D.C.; et al. Regulation of B cell differentiation and plasma cell generation by IL-21, a novel inducer of Blimp-1 and Bcl-6. J. Immunol. 2004, 173, 5361–5371. [Google Scholar] [CrossRef] [PubMed]
- Vogelzang, A.; McGuire, H.M.; Yu, D.; Sprent, J.; Mackay, C.R.; King, C. A fundamental role for interleukin-21 in the generation of t follicular helper cells. Immunity 2008, 29, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Nzeusseu Toukap, A.; Galant, C.; Theate, I.; Maudoux, A.L.; Lories, R.J.; Houssiau, F.A.; Lauwerys, B.R. Identification of distinct gene expression profiles in the synovium of patients with systemic lupus erythematosus. Arthritis Rheum. 2007, 56, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- Peterson, K.S.; Huang, J.F.; Zhu, J.; D’Agati, V.; Liu, X.; Miller, N.; Erlander, M.G.; Jackson, M.R.; Winchester, R.J. Characterization of heterogeneity in the molecular pathogenesis of lupus nephritis from transcriptional profiles of laser-captured glomeruli. J. Clin. Investig. 2004, 113, 1722–1733. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Bethunaickan, R.; Huang, W.; Lodhi, U.; Solano, I.; Madaio, M.P.; Davidson, A. Interferon-α accelerates murine systemic lupus erythematosus in a T cell-dependent manner. Arthritis Rheum. 2011, 63, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Migliorini, A.; Angelotti, M.L.; Mulay, S.R.; Kulkarni, O.O.; Demleitner, J.; Dietrich, A.; Sagrinati, C.; Ballerini, L.; Peired, A.; Shankland, S.J.; et al. The antiviral cytokines IFN-α and IFN-β modulate parietal epithelial cells and promote podocyte loss: Implications for IFN toxicity, viral glomerulonephritis, and glomerular regeneration. Am. J. Pathol. 2013, 183, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Denny, M.F.; Thacker, S.; Mehta, H.; Somers, E.C.; Dodick, T.; Barrat, F.J.; McCune, W.J.; Kaplan, M.J. Interferon-α promotes abnormal vasculogenesis in lupus: A potential pathway for premature atherosclerosis. Blood 2007, 110, 2907–2915. [Google Scholar] [CrossRef] [PubMed]
- Thacker, S.G.; Zhao, W.; Smith, C.K.; Luo, W.; Wang, H.; Vivekanandan-Giri, A.; Rabquer, B.J.; Koch, A.E.; Pennathur, S.; Davidson, A.; et al. Type I interferons modulate vascular function, repair, thrombosis, and plaque progression in murine models of lupus and atherosclerosis. Arthritis Rheum. 2012, 64, 2975–2985. [Google Scholar] [CrossRef] [PubMed]
- Santer, D.M.; Yoshio, T.; Minota, S.; Moller, T.; Elkon, K.B. Potent induction of IFN-α and chemokines by autoantibodies in the cerebrospinal fluid of patients with neuropsychiatric lupus. J. Immunol. 2009, 182, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Merrill, J.T.; Wallace, D.J.; Petri, M.; Kirou, K.A.; Yao, Y.; White, W.I.; Robbie, G.; Levin, R.; Berney, S.M.; Chindalore, V.; et al. Safety profile and clinical activity of sifalimumab, a fully human anti-interferon alpha monoclonal antibody, in systemic lupus erythematosus: A phase I, multicentre, double-blind randomised study. Ann. Rheum. Dis. 2011, 70, 1905–1913. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.; Wallace, D.J.; Spindler, A.; Chindalore, V.; Kalunian, K.; Mysler, E.; Neuwelt, C.M.; Robbie, G.; White, W.I.; Higgs, B.W.; et al. Sifalimumab, a human anti-interferon-α monoclonal antibody, in systemic lupus erythematosus: A phase I randomized, controlled, dose-escalation study. Arthritis Rheum. 2013, 65, 1011–1021. [Google Scholar] [CrossRef] [PubMed]
- McBride, J.M.; Jiang, J.; Abbas, A.R.; Morimoto, A.; Li, J.; Maciuca, R.; Townsend, M.; Wallace, D.J.; Kennedy, W.P.; Drappa, J. Safety and pharmacodynamics of rontalizumab in patients with systemic lupus erythematosus: Results of a phase I, placebo-controlled, double-blind, dose-escalation study. Arthritis Rheum. 2012, 64, 3666–3676. [Google Scholar] [CrossRef] [PubMed]
- Lauwerys, B.R.; Hachulla, E.; Spertini, F.; Lazaro, E.; Jorgensen, C.; Mariette, X.; Haelterman, E.; Grouard-Vogel, G.; Fanget, B.; Dhellin, O.; et al. Down-regulation of interferon signature in systemic lupus erythematosus patients by active immunization with interferon α-kinoid. Arthritis Rheum. 2013, 65, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, X.; Zhang, F.; Yin, H. Toll-like receptors as therapeutic targets for autoimmune connective tissue diseases. Pharmacol. Ther. 2013, 138, 441–451. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).