1. Introduction
Age-related macular degeneration (AMD) is the main cause of blindness in the elderly in industrialized countries. The dry form of AMD is characterized by the disease-associated formation of drusen, which represent debris accumulating between the Bruch’s membrane and the basal lamina of the retinal pigment epithelium (RPE). The resulting geographic atrophy of the RPE layer is followed by vision loss due to photoreceptor degeneration in the overlying region of the sensory retina, the so called macula. Currently, no treatment is available to regain lost vision in patients suffering from dry AMD. The disease mechanisms are poorly understood, thus aggravating the search for potential therapeutic approaches.
The sodium iodate (NaIO
3) model of pharmacologically induced retinal degeneration, known to display AMD-associated features, was widely used to further understand the cell-death mechanisms in RPE and photoreceptors (PRC) [
1,
2,
3]. In this model, the progression and onset of the degeneration can be modulated, facilitating the design of a potential therapeutic strategy to repopulate the damaged RPE layer [
4,
5]. NaIO
3 is an oxidizing compound that has been shown to be specifically toxic for RPE cells [
6]. Under physiological conditions, RPE cells exhibit various crucial retinal functions and the undisturbed unit of RPE and PRCs provides the basis for a normal visual perception (see review [
7]). Therefore, without functional underlying RPE cells, the photoreceptors are unable to survive resulting in vision loss [
8].
The NaIO
3 mouse model has been well characterized with respect to the loss of RPE cells and the death of PRCs, resulting in a thinning of the outer nuclear layer (ONL) and a reduction in visual functions [
9]. To date, it is controversially discussed whether NaIO
3 cytotoxicity acts exclusively on RPE cells with PRCs dying secondarily as a consequence of the loss of functional RPE or whether it directly affects both retinal cell types. Previous reports have supported a direct effect on RPE cells, in which the basal plasma membrane is destroyed following NaIO
3 administration, resulting in damage to intracellular organelles [
10]. Additionally, it has been suggested that NaIO
3 can cross-react with melanin, increasing the turnover of glyoxylate, which is cytotoxic to RPE cells [
11]. NaIO
3 is also known to have an inhibitory action on the activity of crucial enzymes (triose phosphate dehydrogenases, lactate dehydrogenase) [
12]. Other reports described an altered adhesion between the neurosensory retina and the RPE layer in response to NaIO
3 intoxication [
13,
14]. Recent studies have indicated a direct effect of NaIO
3 on the sensory retina [
15,
16]. NaIO
3 was thereby demonstrated to alter gene expression
in vivo by down-regulation of RPE cell-specific markers as early as 24 h post-injection, concomitantly with an increase in the expression of the pro-apoptotic
Bax gene within the neurosensory retina. Furthermore, it was demonstrated that NaIO
3 treatment
in vitro increased levels of reactive oxygen species (ROS) in the 661W cone photoreceptor cell line [
17,
18]. However, no report to date has defined whether caspase-dependent or caspase-independent cell-death pathways are involved in NaIO
3-induced RPE and PRC death
in vivo; this knowledge is crucial for the design of a experimental strategy to intervene with NaIO
3-induced photoreceptor cell loss.
Apoptosis is the conventional form of cell death, in which caspases are activated, resulting in nuclear fragmentation and chromosome condensation. To date, growing evidence exists supporting the contribution of nonconventional (caspase-independent) cell-death mechanisms to the progression of neurodegenerative diseases [
19,
20]. Activation of calpains (calcium-dependent proteases) and cathepsins (lysosomal proteases) in fact has been reported in degenerative processes [
21,
22]. Calpains are ubiquitously expressed and highly activated under stress conditions in response to an increased influx of ions through cGMP-gated cation channels. They have been reported to be involved in PRC cell death in the
rd1 mouse model, or retinitis pigmentosa [
23], as well as in P23H and S334ter rhodopsin mutant rats [
24]. The underlying mechanism can be either caspase-dependent or caspase-independent. Necrotic cell death (necrosis), on the other hand, is a less defined and uncontrolled death mechanism that does not involve the activation of conventional cell death key players.
In the presented study, with the aim to characterize the NaIO3 model that displays AMD-associated features, we assessed retinal changes following the administration of NaIO3 in vivo and in vitro. Thereby, it could be shown that, in PRCs of NaIO3-injected animals, caspases as well as calpain proteases were upregulated on the protein level. Furthermore, we provide evidence that, in RPE cells, NaIO3 treatment resulted in necrosis in the absence of caspase-dependent conventional cell death, whereas it triggered caspase-dependent apoptosis in the cone photoreceptor cell line 661W. Consistently, incubation of NaIO3-treated RPE cells with necrostatin 1 (Nec-1), an inhibitor of nonconventional necrosis, was cytoprotective, but the compound did not prevent NaIO3-induced PRC cell death. In summary, NaIO3 triggered both RPE and PRC degeneration via different pathways involved in cell death induction and execution. Therefore, a combinatory treatment might be the most straightforward approach to modulate this complex model of retinal degeneration, opening new possibilities to use the NaIO3 mouse model to design a therapeutic strategy to prevent vision loss.
3. Discussion
The NaIO
3 animal model of retinal degeneration is widely used in vision research to investigate retinal cell death. Treatment with NaIO
3 results in reduced vision and is followed by RPE damage with concomitant photoreceptor degeneration [
17]. However, the death mechanisms that are involved are still not fully elucidated. To identify additional details, we assessed the PRC death pathways
in vivo and conducted an
in vitro assessment of the effect of NaIO
3 on both RPE cells and PRCs. Here, we report that NaIO
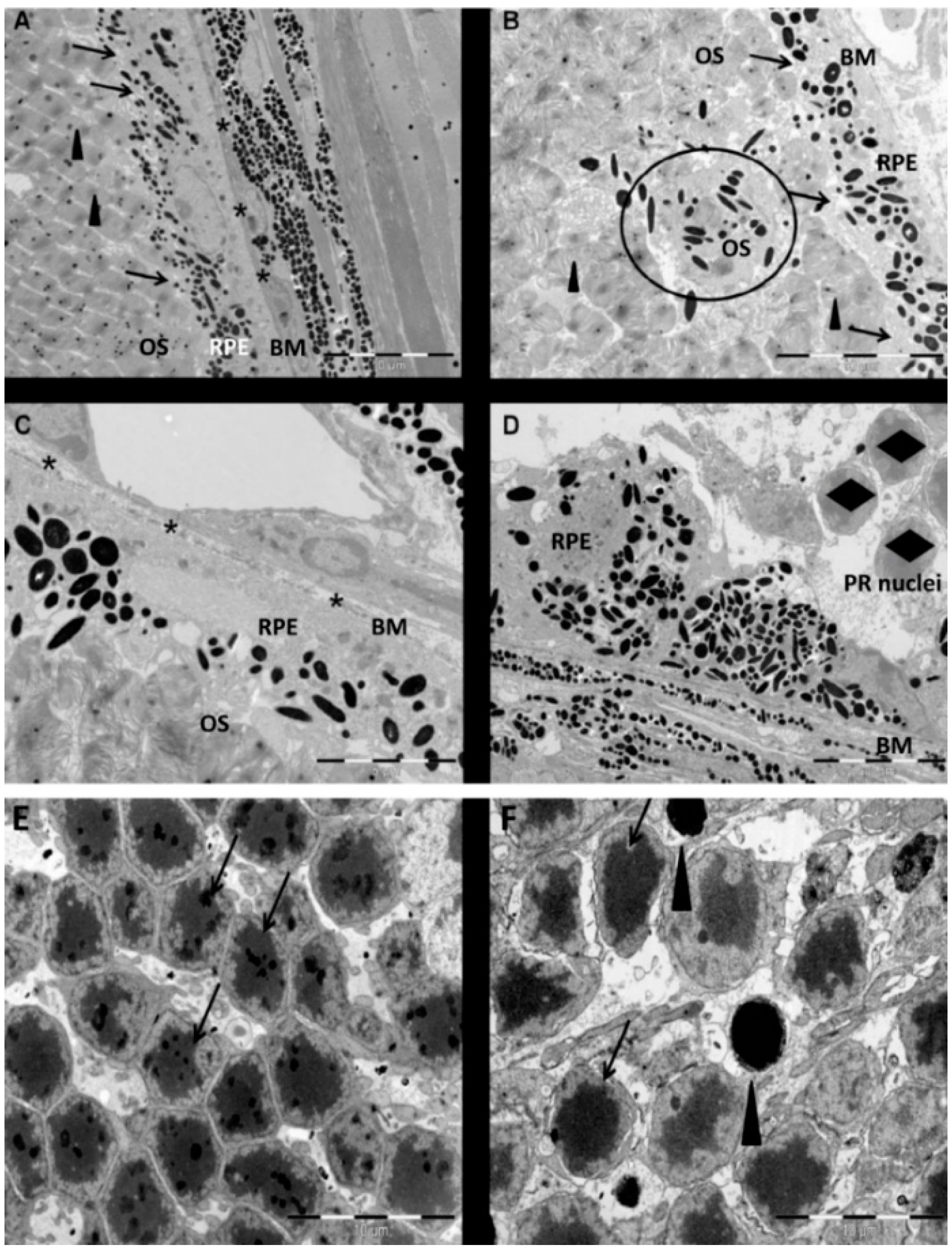
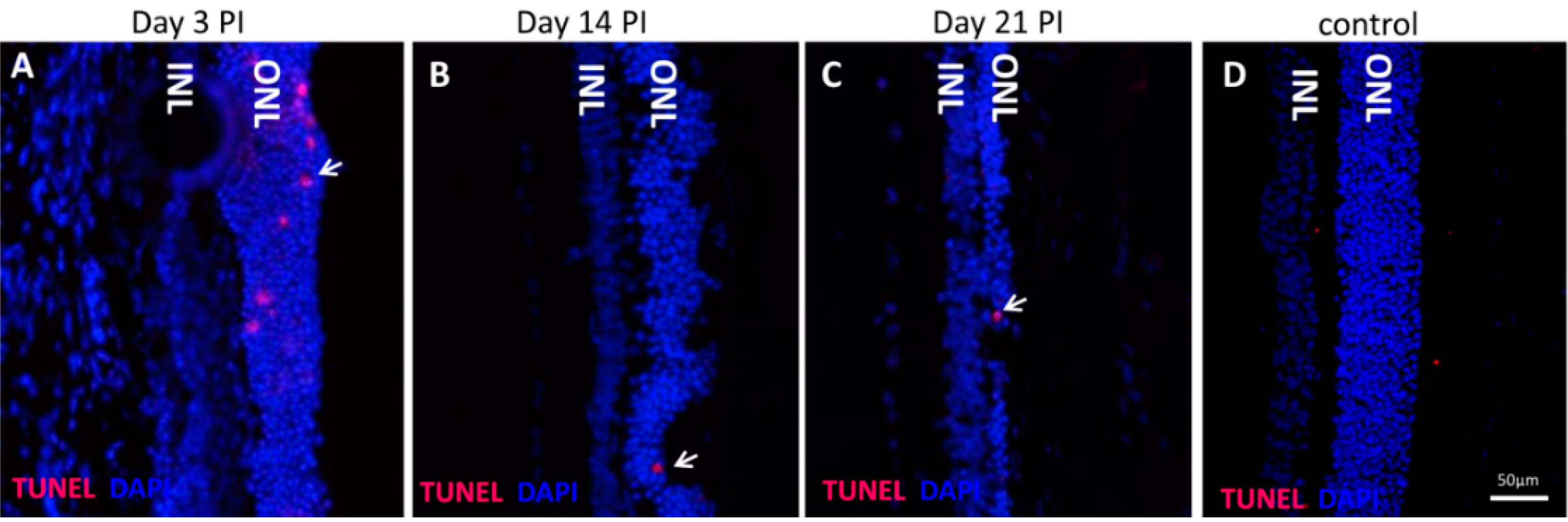
3 in vivo induced rapid and pronounced degeneration of the RPE as well as a moderate and slow degeneration of the PRCs with a low number of TUNEL-positive cells in the ONL, starting at three days PI. We did not found TUNEL positivity in other retinal layers, for example the ganglion cell or the inner nuclear layer; nevertheless, we cannot exclude an effect of NaIO
3 on those layers. The damaged PRCs
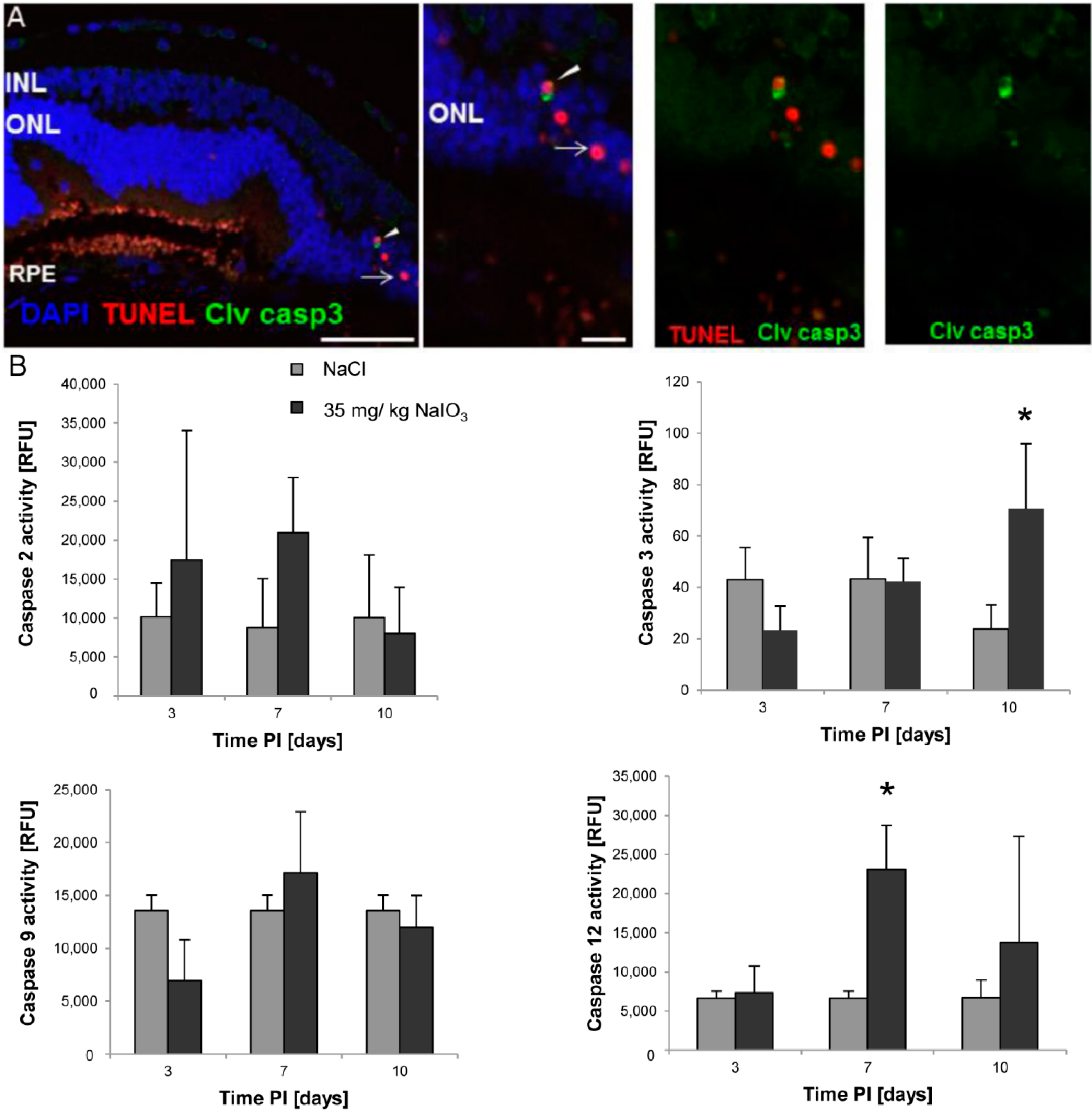
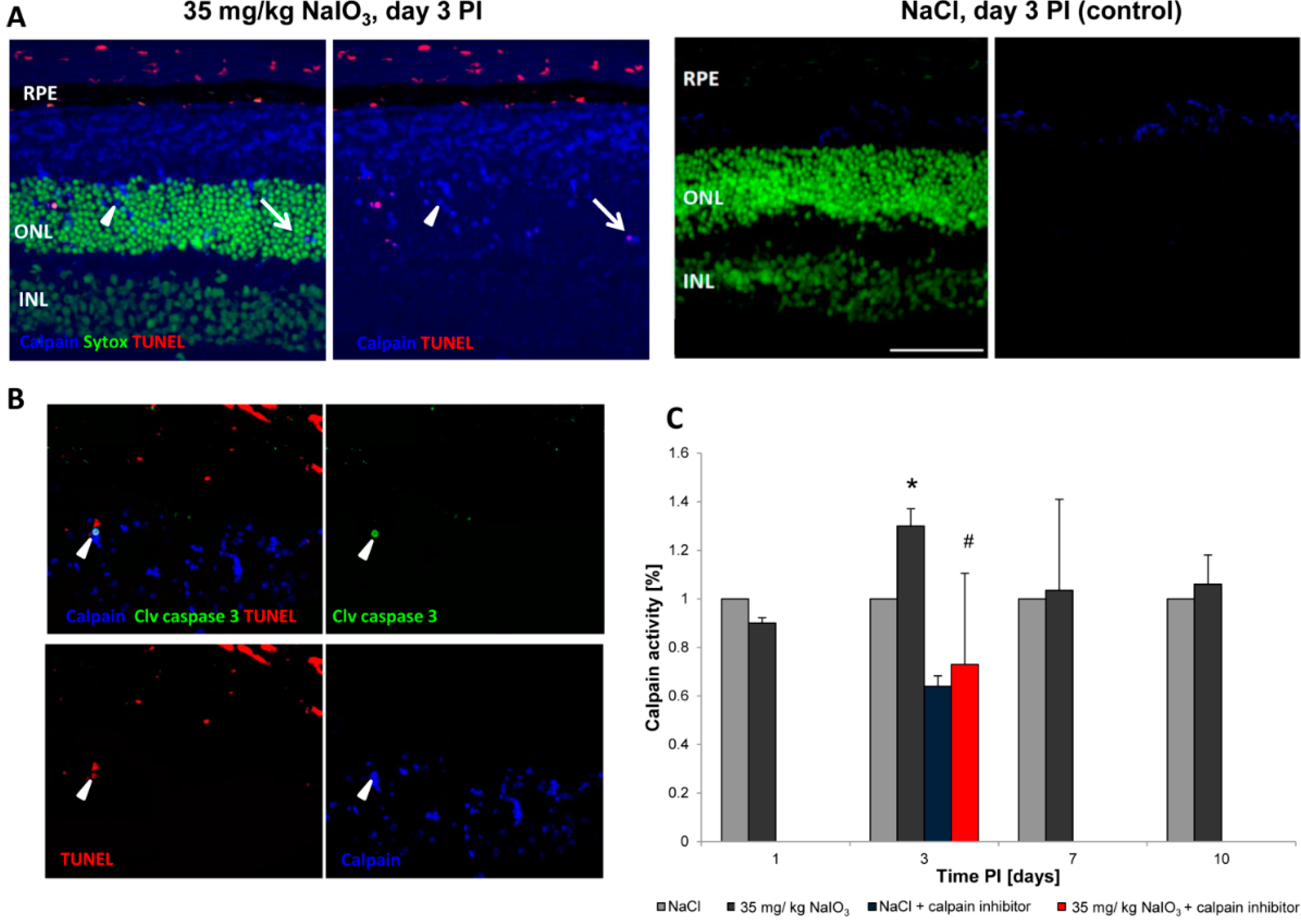
in vivo were positive for cleaved caspase-3 as well as for activated calpain. Calpain is a calcium-dependent, non-lysosomal cysteine protease and was previously reported to be activated in 661W cells upon calcium overload in response to a nitric oxide donor [
29]. In some photoreceptors of NaIO
3-injected mice, calpain-positive cells co-expressed caspase-3, indicating that both cell-death modes occur concomitantly. That might represent the default pathway of PRC death, as similar results have also been observed in other retinal degeneration models
in vitro [
29] and
in vivo [
24]. A different explanation could be that calpain mediates caspase-dependent cell death in the PRCs by inducing cleavage and, thereby, activation of caspases.
In vitro, cell-death pathways were assessed in detail in RPE and 661W cells after NaIO
3 treatment in order to differentiate between primary necrosis and conventional caspase-dependent apoptosis. An unambiguous identification of necrotic cell death is challenging, because of the absence of biochemical markers both
in vitro and
in vivo. However, the rupture of the plasma membrane with absence of caspase-3 positivity is a main indicator for ongoing necrotic cell death. This approach was pursued using the ApoToxGlo assay, which, besides viability and caspase-dependent cell death, measures the so-called dead-cell protease activity upon necrosis-induced loss of membrane integrity. Herein, we could provide evidence that NaIO
3 treatment triggers apoptotic caspase-dependent cell death in the 661W cone-derived photoreceptor cell line, whereas necrotic cell death was induced in NaIO
3-treated RPE cells. The observed direct effect on PRCs is supported by a recently published study using a NaIO
3 concentration in the range of 250–750 µg/mL [
16]. In our experiments, the lowest concentration used (6 mM or 1182 µg/mL) also resulted in 661W cell death. In contrast, we did not see a significant difference in vulnerability to NaIO
3 when comparing primary RPE and 661W cells, only with respect to the mode of cell death. The fact that a high level of apoptosis is triggered in 661W cells, whereas only a few PRCs were apoptotic
in vivo, might be related to the route of delivery. Although the
in vitro administration resulted in direct exposure of the cells to the toxin,
in vivo-administered NaIO
3 reaches the PRCs through the blood–retina barrier (BRB) and the RPE, as the most adjacent cell type. The
Zonulae occludentes, which are the anatomical basis of the BRB function of the RPE, were reported to be severely impaired by NaIO
3 treatment [
30].
Interestingly, primary PRCs seem to be less susceptible to the toxin than 661W cells, as their detected loss of viability upon NaIO
3 treatment reached only approximately 50%, compared to 12.5% in the latter. This also supports a direct effect of NaIO
3 on (rod) PRCs, but we were not able to distinguish the executed cell-death modes. However, as PRCs die
in vitro if they lose contact to other retinal cells, our data using the primary PRC culture must be interpreted carefully, as ongoing cell death was already seen in untreated control cells. Differences in terms of the loss of cell viability between rod and cone PRCs have also been described in a recent study, suggesting that NaIO
3 triggered the rapid and irreversible destruction of cones, whereas a decrease in the rod-evoked ERG response was subsequently rescued [
4].
In conclusion, we herewith identified necrosis as the main pathway involved in NaIO
3-induced RPE cell death. These results support earlier studies, observing morphological changes in the retina by OCT and electron microscopy [
31,
32]. The used intermediate concentration of 12 mM NaIO
3 (2376 μg/mL) is comparable to the recently published data by Wang
et al. [
16], who demonstrated a cytotoxic effect of NaIO
3 on ARPE19 cells, an immortalized human RPE cell line that displays RPE features
in vitro [
33]. As it is possible that apoptotic cells undergo necrosis in a secondary stage
in vitro, owing to the lack of macrophages, we performed an analysis on NaIO
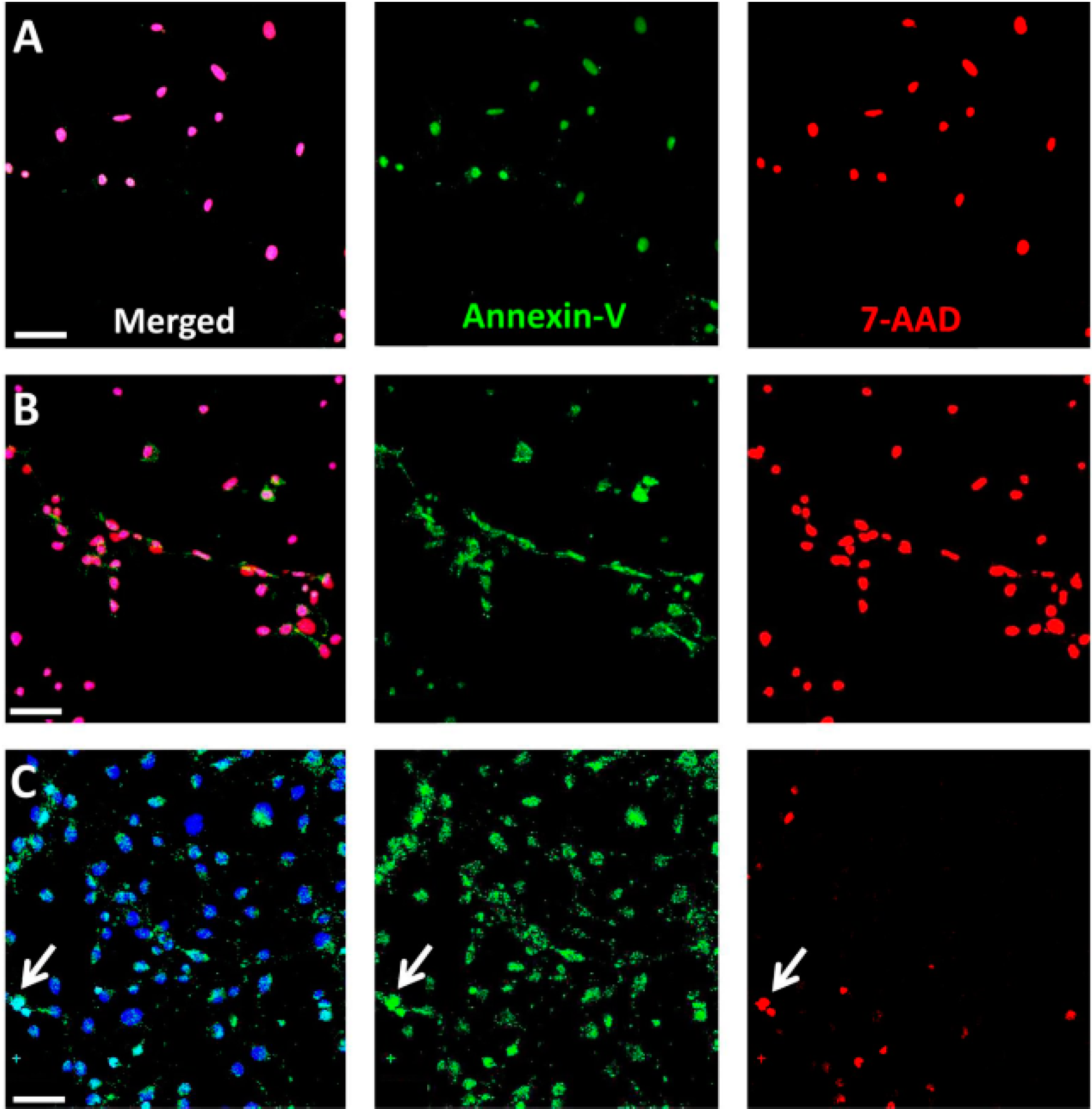
3-treated RPE cells at early time points post-exposure, confirming necrotic cell death by the concomitant presence of Annexin-V and 7-AAD. Consistently, NaIO
3-induced RPE degeneration
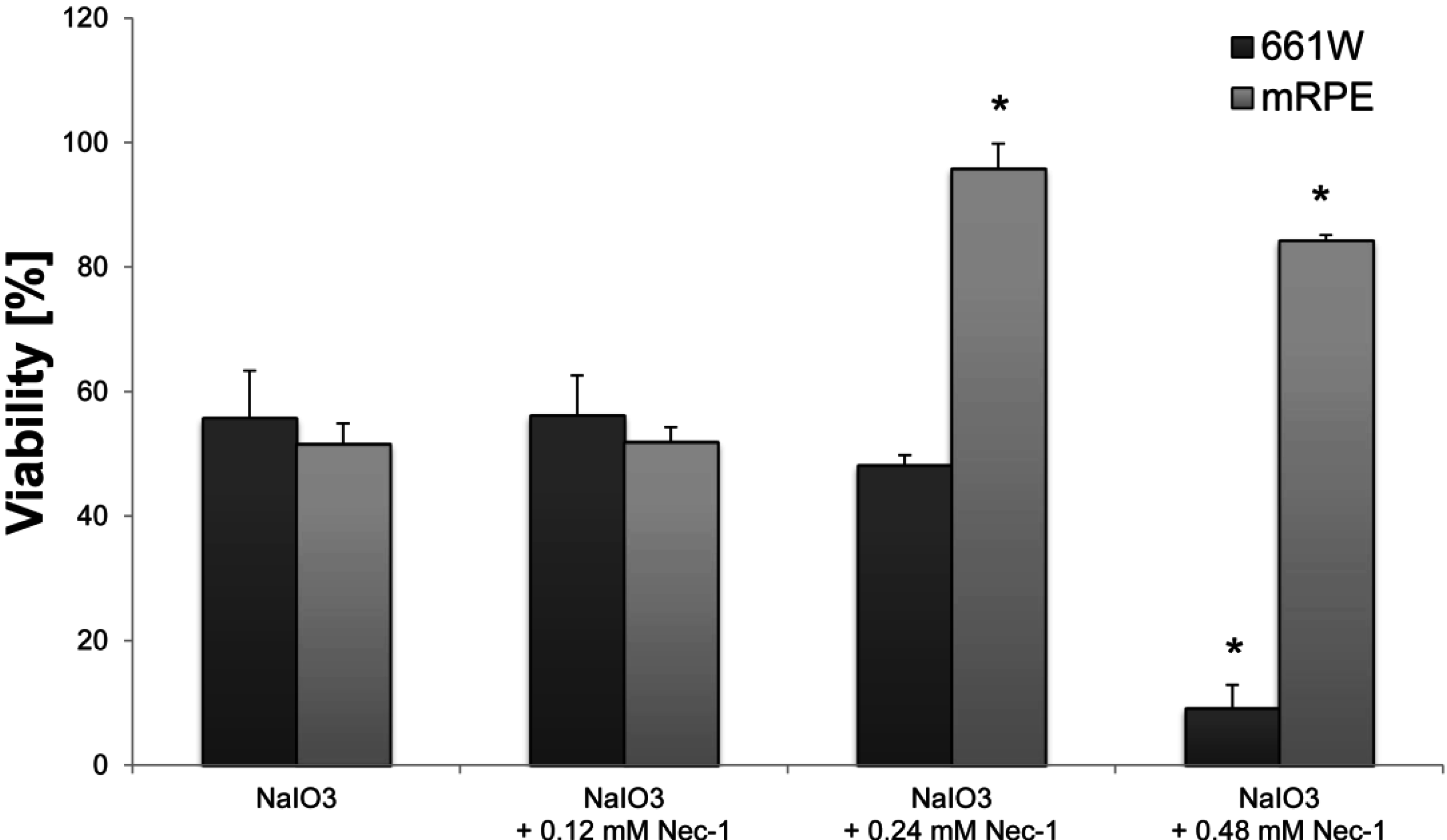
in vitro was significantly reduced by Nec-1, an inhibitor of the nonconventional, necrotic-like cell death under caspase-compromised conditions [
34]. Nec-1 has been shown to decrease the production of reactive oxygen species (ROS) [
35], levels of which have been reported to be elevated in NaIO
3-treated RPE cells [
18]. Protection against ROS formation could, therefore, contribute to the significant rescue effect of Nec-1 observed in NaIO
3-treated RPE cells. However, further investigations will be necessary in order to shed light on the exact mechanisms by which Nec-1 is able to rescue NaIO
3-treated RPE cells. Alternative effects of NaIO
3 on the RPE, in addition to the induction of oxidative stress [
18], could involve mitochondrial dysfunction and upregulation of p62 [
36].
4. Experimental Section
4.1. Animal Treatment
C57BL/6J mice (4–6 weeks old, Charles River, Germany) were treated according to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research, following approval from the Commission for Animal Experimentation of the Canton of Bern, Switzerland. A single intravenous injection of sterile 1% NaIO3 (35 mg/kg bodyweight) in 0.9% NaCl was performed. Control animals received a single intravenous injection of 0.9% NaCl. Animals were sacrificed at different time points PI, and the eyes were then enucleated for electron microscopy (EM), immunohistochemistry (IHC), and ELISA-based protein activity assays.
4.2. Cell Culture
To assess the effect of NaIO
3 on retinal cells
in vitro, the murine photoreceptor cell line 661W (kindly provided by M. Al-Ubaidi, University of Oklahoma, Oklahoma City, OK, USA) was used [
37]. The cells were propagated under normal culture conditions in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with GlutaMAX and 1% antibiotic/antimycotic + 10% fetal bovine serum (FBS; Life Technologies, Carlsbad, CA, USA).
Primary mouse RPE cells were enzymatically isolated from 10-day-old wild-type (WT) mice. Briefly, enucleated eyes were incubated with 2% dispase (Life Technologies) in phosphate-buffered salt solution (PBS; Life Technologies) with gentle shaking at 37 °C for 25 min. The anterior half of the eye and the vitreous part were discarded. The retina was gently floated off of the RPE with PBS and removed by cutting the optic nerve. The RPE cells were mechanically harvested, centrifuged, and re-suspended in DMEM + 10% FBS. The epithelial origin of the prepared cells was determined by staining for the tight-junction protein ZO-1 and F-actin (see
Figure S2). RPE cells were used at low passage up to passage 6.
The primary culture of photoreceptors was prepared from retinal tissue of postnatal day 4–8 WT animals using the Papain Dissociation System (Worthington Biochemical, Lakewood, NJ, USA), according to the manufacturer’s instruction. After the last centrifugation step, the cells were re-suspended in neurobasal medium supplemented with 2% B27, 1% N2, 1% antibiotic/antimycotic as well as 0.8 mM Glutamine (Life Technologies).
4.3. Electron Microscopy
Eyes were enucleated and fixed with Karnowsky fixation medium (1% paraformaldehyde (PFA); Sigma-Aldrich, Buchs, Switzerland), 3% sodium cacodylate–HCl (Science Services, Munich, Germany), and 3% glutaraldehyde (Sigma-Aldrich) for 24 h. Following the removal of the lens, post-fixation, and washing in EM buffer (2.5% glutaraldehyde (Sigma-Aldrich) and 0.1 M sodium cacodylate–HCl (Science Services)) for 15 min, the eyes were post-fixed in 4% osmium tetroxide (Science Services) for 15 min. The tissue was dehydrated, washed with a resin/1,2-propylene oxide mixture (Merck, Darmstadt, Germany) three times for 20 min each, and infiltrated with resin overnight. The resin-embedded tissue was then trimmed and cut into 80-nm thick sections (Ultracut, Reichert Microscope Services, Depew, NY, USA) using an Ultra-45 °C diamond knife (Diatome, Biel, Switzerland). Sections were applied to copper grids (G100H-C3; Science Services) and contrasted with 0.1% lead citrate (Science Services). EM analyses were performed on a CM 12 electron microscope (Philips Applied Technologies, Eindhoven, The Netherlands).
4.4. Immunohistochemistry
Caspase detection: Fixed and dehydrated eyes of treated and untreated mice were embedded in paraffin. Tissue sections with a thickness of five µm were cut. Antigen retrieval was performed using sodium citrate buffer (10 mM sodium citrate, 0.05% Tween-20, pH 6.0; Sigma-Aldrich) in a pressure cooker for five min. Sections were then permeabilized using 0.2% Triton X-100 in Tris-buffered saline (TBS; Sigma-Aldrich) for 20 min, and blocked 1% bovine serum albumin (BSA; Sigma-Aldrich) and 10% normal goat serum (NGS; DAKO, Baar, Switzerland) for 1 h before an overnight incubation with the primary antibody (rabbit anti-cleaved-caspase-3; 1:100; Cell signaling, Leiden, The Netherlands) at 4 °C. After washing, incubation in a secondary antibody (goat anti-rabbit Alexa Fluor® 594 or 488 (Life Technologies), 1:500) was performed for 45 min. DAPI (Vector Laboratories, Burlingame, CA, USA) or Sytox Green (Life Technologies) was used to counterstain the cell nuclei.
Calpain activity: Unfixed eyes were snap-frozen in Tissue-Tek O.C.T Compound (Sakura, Alphen aan den Rijn, The Netherlands) and 7-μm cryosections were processed to visualize calpain activity, according to Paquet-Durand,
et al., 2006 [
23]. All cell outlines are labelled, with calpain activity-positive cells displaying a bright labelling in the nucleus and cytosol. A fluorescent calpain substrate 7-amino-4-chloromethylcoumarin,
t-BOC-
l-leucyl-
l-methionine amide (CMAC,
t-BOC-Leu-Met; Life Technologies), was added at a final concentration of 2 µm, and then incubated in the dark for 1 h at 35 °C. Sytox Green (Life Technologies) was used as a nuclear counterstain. Calpain-positive cells were counted manually in the visual field by the cell-counter plug-in for ImageJ, which was calculated as a percentage of positive cells with respect to all photoreceptor cells in the ONL. To assess TUNEL and calpain or caspase-3 and calpain activity in co-staining, sections stained for calpain activity were subsequently fixed in freshly prepared 4% PFA at RT for 15 min before applying the TUNEL kit or performing antibody staining, respectively.
RPE-specific markers: Mouse primary RPE cells (50,000 cells/well) grown on fibronectin-coated (10 µg/mL) eight-well chamber slides (Lab-Tek™ II, Sigma-Aldrich) were fixed with 4% PFA for 15 min, washed in PBS, and blocked with 1% BSA in PBS supplemented with 10% NGS for 1 h. Immunohistochemistry using rabbit anti-ZO-1 and Texas Red conjugated phalloidin was performed as described above.
TUNEL assay: Staining was performed using the in situ cell-death detection kit, TMRed (Roche Applied Sciences, Basel, Switzerland) on either cryo- or paraffin sections. Staining was carried out according to the manufacturer’s instructions. Confocal microscopy was performed, either by the Zeiss scanning laser microscope (Zeiss LSM710; Carl Zeiss Microscopy, Jena, Germany) or a Leica SP2 (Leica Microsystems, Heerbrugg, Switzerland).
4.5. Calpain and Caspase Activity Assays
The calpain and caspase activity was assessed on protein lysates of treated and untreated retinas using enzyme activity assay kits (BioVision, Milpitas, CA, USA) according to the manufacturer’s instructions. Animals were sacrificed at days 3, 7, and 10 PI of 35 mg/kg NaIO3, and the retinas were harvested and pooled (n ≥ 4). Following homogenization of the retina and subsequent cell lysis in RIPA buffer (150 mM NaCl, 1.0% IGEPAL®, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0; Sigma-Aldrich) supplemented with inhibitors to prevent protein degradation through non-caspase family proteases (Complete Mini; Roche Applied Science) on ice for 30 min, the lysates were centrifuged at 13,000 rpm at 4 °C for 20 min. In the supernatant, the protein content was assessed using the DC protein assay (BioRad, Cressier, Switzerland), and 50 µg of retinal lysate proteins (triplicates) were incubated in reaction buffer in a well of a 96-well plate. The samples were incubated for 1 h with the specific substrate, which emits a yellow–green fluorescence upon cleavage in the presence of the corresponding caspase/calpain. The cleaved substrate was then fluorometrically measured at 505 nm using a multimode plate reader (Infinite 200PRO; Tecan, Männedorf, Switzerland). Activity data from NaIO3-treated animals were plotted against control values obtained from NaCl-injected control mice for each specific time point. Results are expressed as fold increase in enzyme activity. In order to confirm a specific activity after NaIO3 treatment, three independent retinal lysate samples of NaIO3-treated mice were supplemented with 100 µM of the inhibitor Z-LLY-FMK (BioVision, Milpitas, CA, USA) for 1 h before incubation with the substrate.
4.6. ApoTox Glo Triplex Assay
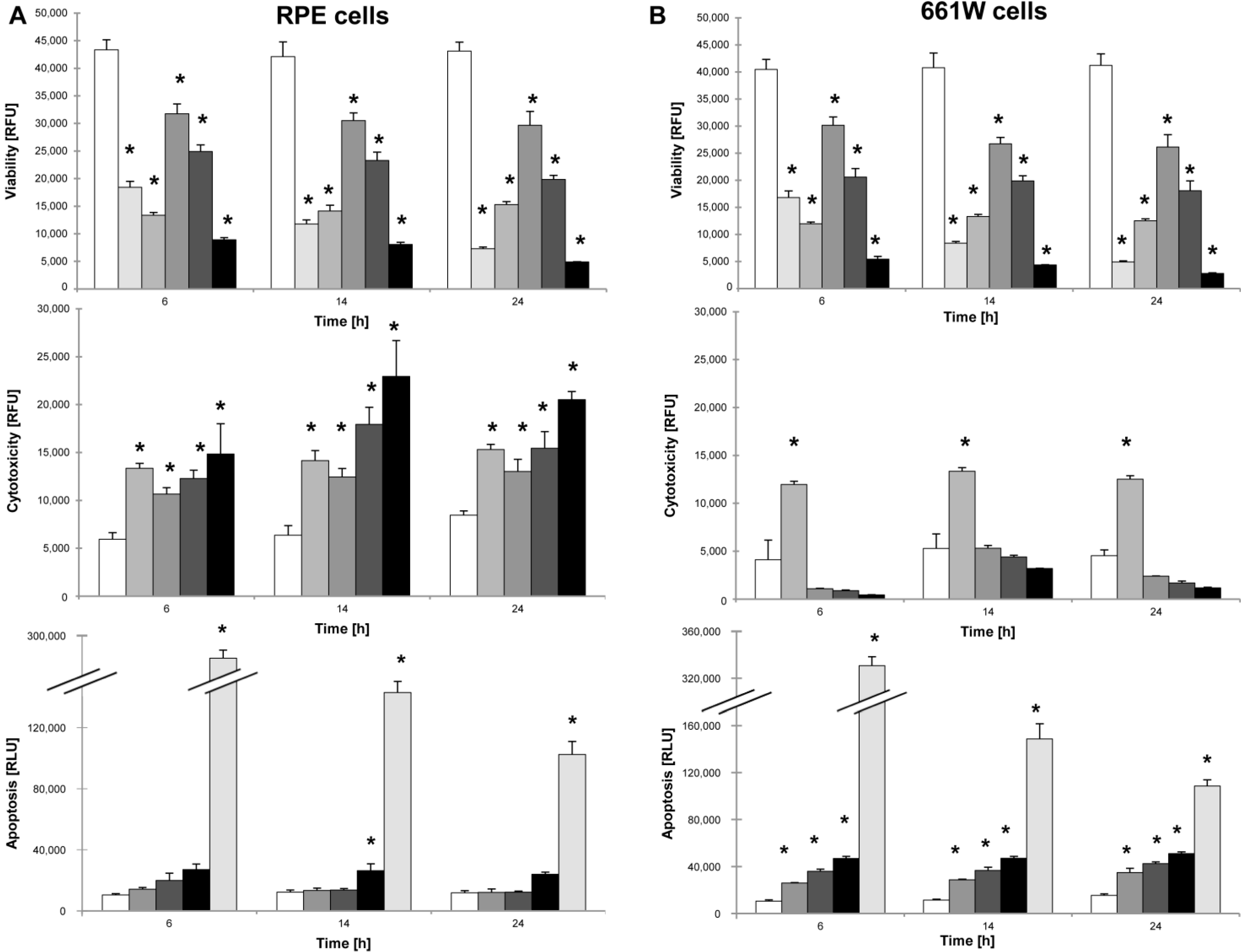
The ApoTox Glo Triplex assay (Promega, Madison, WI, USA) was used to simultaneously assess viability, cytotoxicity, and caspase-dependent apoptosis in response to various concentrations of NaIO3 in vitro. A 1 µM staurosporine solution (Sigma-Aldrich) was used as a positive control for apoptosis, and cells were sonicated prior to plating in order to rupture the cell membrane as positive control for necrosis. For the experimental setup, 661W photoreceptors and primary RPE cells were seeded at a density of 10,000 cells/well (30,000 cells/cm2). The next day, the cells were sonicated or treated with either staurosporine (1 μM), different concentrations of NaIO3 (6, 12, or 48 mM), or left untreated to act as controls. At different time points post-treatment (6, 14, and 24 h), the ApoTox Glo assay was performed according to the manufacturer’s instructions. Cell viability (wavelength sets: 400Ex/505Em) and cytotoxicity (wavelength sets: 485Ex/520Em) were measured using a microplate reader (Tecan Infinite 200Pro). At each time point, a second measurement followed, using the Caspase-Glo® Assay Technology (Promega), for caspase-dependent cell death by adding a luminogenic caspase-3/-7 substrate to the cell culture medium. Following incubation at room temperature for 1 h, the luminescence was measured with an integration time setting of between 0.5 and 1 s, using the Tecan Infinite 200Pro reader.
4.7. Annexin-V and 7-AAD Staining
To confirm primary necrosis in RPE cells in vitro, we used a staining kit for 7-AAD and Annexin-V. Necrotic cells are positive for both markers at any stage, whereas early apoptotic cells include a stage at which cells are exclusively Annexin-V positive. RPE and 661W cells were seeded on fibronectin-coated (10 µg/mL) chamber slides (Lab-Tek™ II, Sigma-Aldrich; 20,000 cells/well) for 24 h before treatment with NaIO3 (48 mM). Staurosporine (1 µM) and ionomycin (0.1 mM; Sigma-Aldrich) treatment was employed as positive controls. Cells were washed and incubated in the 7-AAD/Annexin-V reaction mix and incubated for 15 min before fixation with 2% PFA. Counterstaining was performed using the Hoechst stain NucBlue® Live Ready Probes Reagent® (Life Technologies).
4.8. Necrostatin-1 Treatment
Nec-1 (Enzo Life Sciences (Farmingdale, NY, USA)) was dissolved in DMSO according to the manufacturer and further diluted in DMEM to achieve the desired concentrations (0.12, 0.24, 0.48 mM). DMSO was also added to control cells (at the dissolvent concentration used) in order to exclude a cytotoxic effect of the solvent. Nec-1 was administered concomitantly with the NaIO3 and the XTT viability assay (Roche Life Science) was performed 24 h later according to the manufacturer’s instructions. Blank values with Nec-1 but without cells were subtracted in order to correct for its color-related absorbance.
4.9. Statistical Analysis
Three individual experiments were performed (mean ± SD) and a statistical evaluation using analysis of variance (ANOVA) was executed in SPSS software 20 (IBM, Hampshire, UK), followed by a multiple-comparisons post hoc test to determine the significant differences between the mean values. Differences were considered significant at p ≤ 0.05.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




 cells,
cells,  + 1 μM staurosporine,
+ 1 μM staurosporine,  + 100% sonication,
+ 100% sonication,  + 6 mM NaIO3,
+ 6 mM NaIO3,  + 12 mM NaIO3,
+ 12 mM NaIO3,  + 48 mM NaIO3); * p < 0.05.
+ 48 mM NaIO3); * p < 0.05.


