Abstract
The inhibitory effects on the mycelial growth of plant pathogen Botritys cinerea have been evaluated for a series of geranylphenols substituted with one, two and three methoxy groups in the aromatic ring. The results show that the antifungal activity depends on the structure of the geranylphenols, increasing from 40% to 90% by increasing the number of methoxy groups. On the other hand, the acetylation of the –OH group induces a change of activity that depends on the number of methoxy groups. The biological activity of digeranyl derivatives is lower than that exhibited by the respective monogeranyl compound. All tested geranylphenols have been synthesized by direct coupling of geraniol and the respective phenol. The effect of solvent on yields and product distribution is discussed. For monomethoxyphenols the reaction gives better yields when acetonitrile is used as a solvent and AgNO3 is used as a secondary catalyst. However, for di- and trimethoxyphenols the reaction proceeds only in dioxane.
1. Introduction
Prenylated compounds with one or more prenyl moieties are common natural products that have been isolated predominantly from marine organisms and plants [1,2,3,4]. These compounds have attracted much attention because they often possess antimicrobial, antioxidant, anti-inflammatory, antiviral and anticancer activities [5,6,7,8]. Various attempts to establish structure–activity relationships (SARs) relating to these biological activities with structural features of metabolites and their derivatives have been made [1,6]. The results indicate that these activities depend on the length of the side chain and the nature and relative position of substituent groups on the aromatic ring. The most active compounds are those having two isoprene units in the prenyl chain, and the chemical structures of some active geranylphenols are shown in Figure 1.
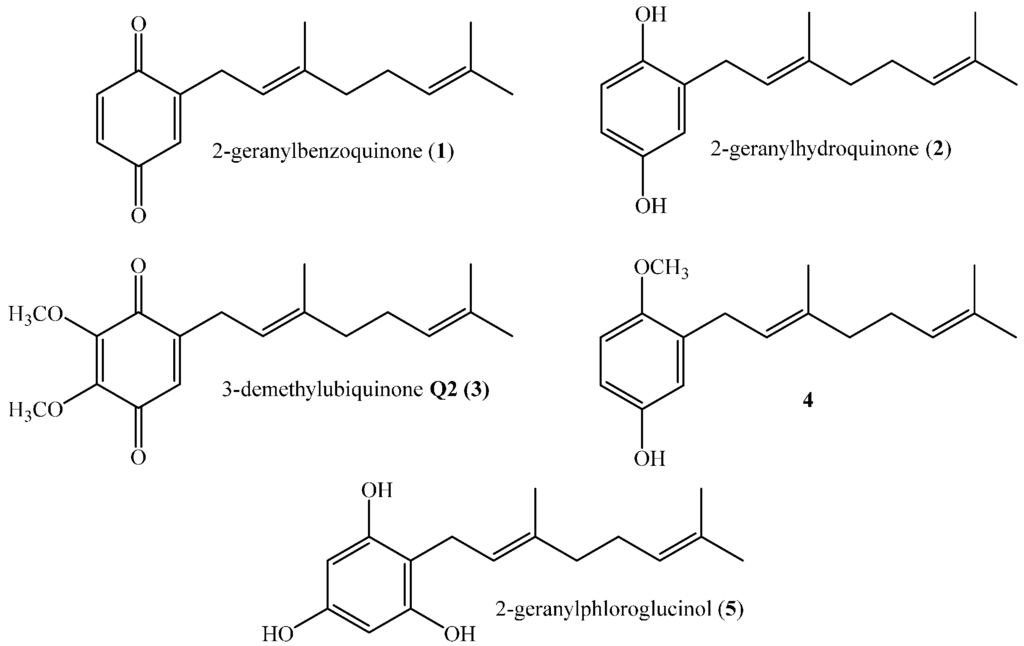
Figure 1.
Structures of some active linear geranylphenols.
Figure 1.
Structures of some active linear geranylphenols.
Based on these antecedents, we have undertaken a study of the effect of a series of geranylphenols on the mycelial growth inhibition of plant pathogen Botrytis cinerea as a function of molecular structure [9]. This knowledge might be useful to design and synthesize new substances to control such kinds of pathogens. Thus, the purpose of this work is to increase the comprehension of the structural features determining the antifungal activity of these compounds. With this aim in mind, the synthesis, structure determination, and inhibitory activity of a series of 15 geranylphenols with different numbers of methoxy groups in the aromatic ring are reported in this work.
The effect of these compounds on the mycelial growth of plant pathogen B. cinerea has been evaluated and the results are discussed in terms of their molecular structure. The effect of the structure of the starting phenol over the yield and nature of products obtained with both methods is also assessed.
2. Results and Discussion
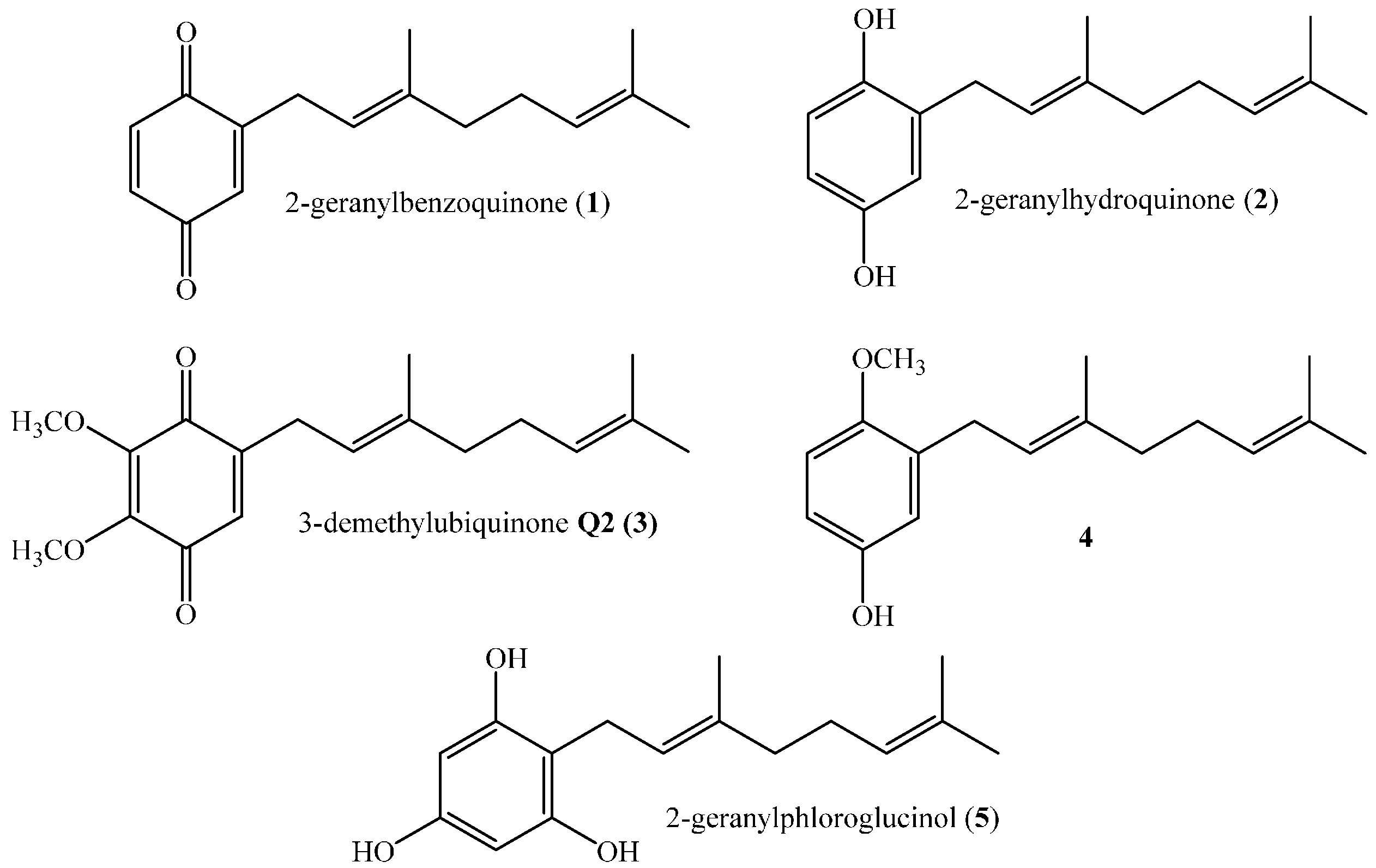
Compounds 6–24 (see Figure 2) were synthesized through the electrophilic aromatic substitution reaction (EAS), which proceeds by the slow addition of geraniol to a solution of phenol derivatives in dioxane and in the presence of BF3·Et2O which acts as catalyst [1,6,10,11]. This reaction was chosen, between many different synthetic pathways for obtaining prenylated compounds [1,11,12,13,14], because it has been previously used to synthesize a series of synthetic analogs of 3-demethylubiquinone (3) having one, two, or three methoxy groups in different positions of the quinone moiety [6]. Alternatively, the reaction was also carried out in acetonitrile instead of dioxane, using AgNO3 as a secondary catalyst [9]. The results indicate that both yields and products depend on the solvent.
Figure 2.
Structures of linear geranylphenols 6–24 tested in this work.
Figure 2.
Structures of linear geranylphenols 6–24 tested in this work.
2.1. Synthesis
The direct coupling between geraniol and commercially available 4-methoxyphenol using acetonitrile/AgNO3 gives compound 6 as a unique product with 18% yield, which is almost twice the yield reported for the same reaction in dioxane (10%) [15]. However, this yield is much lower than that reported for the reaction of 4-methoxyphenol with geranyl bromide in the presence of NaH, which leads to 6 with a 56% yield [16]. On the other hand, the reaction between geraniol and guaiacol (2-methoxyphenol) generates compounds 8, 10 and 11 according to Scheme 1. The yields of 8, 10 and 11 in acetonitrile/AgNO3 were 4.5%, 6.3% and 0.6%, respectively, which are relatively higher than those obtained using dioxane, i.e., 2.5%, 4.1% and 0.3%, respectively [11].
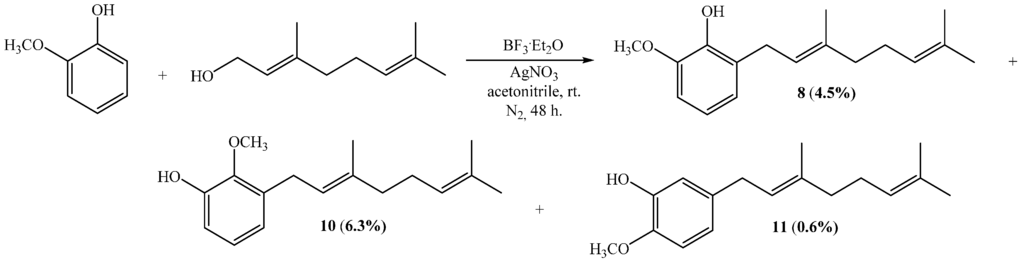
Scheme 1.
Synthesis of compounds 8, 10 and 11.
Scheme 1.
Synthesis of compounds 8, 10 and 11.
Compounds 12 and 13 were obtained by the coupling of geraniol and 3-methoxyphenol (3-hydroxyanisol) in acetonitrile, with yields of 7.4% and 4.2%, respectively (Scheme 2). Both substances are new compounds and their structure characterization is described in the next section.
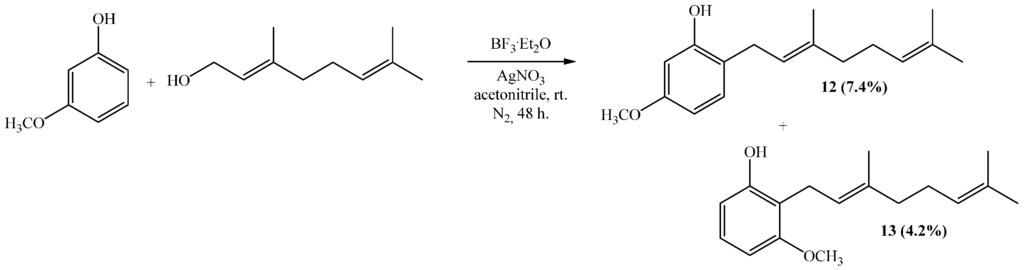
Scheme 2.
Synthesis of compounds 12 and 13.
Scheme 2.
Synthesis of compounds 12 and 13.
Thus, the geranylation of monomethoxy phenols is more efficient when it is carried out in the presence of acetonitrile/AgNO3, with yields depending on the position of the methoxy substituent group. The reaction yield follows the order: para > metha ≈ ortho. This result can be explained in terms of ring activation by hydroxyl and methoxy groups, which is higher for the 4-methoxyphenol.
The acetylated derivatives 7 and 9 were obtained by a standard acetylation reaction of 6 and 8 with 95% and 93% yields, respectively. The NMR data of compounds 6–8, 10 and 11 were consistent with those previously reported [11], whereas the NMR of compound 9 confirm the presence of the acetylated derivative (see below).
The coupling reaction between 2,4-dimethoxyphenol and geraniol leads to compounds 14, 15 and 16, with 1.0%, 1.2% and 3.0% yields, respectively (Scheme 3), when dioxane is used as a solvent.
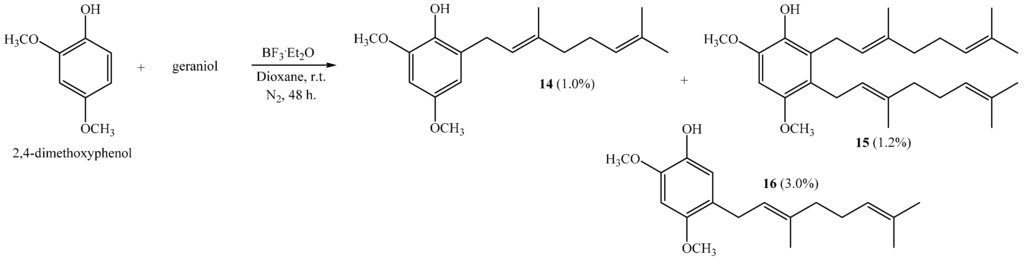
Scheme 3.
Preparation of dimethoxygeranylphenols 14–16.
Scheme 3.
Preparation of dimethoxygeranylphenols 14–16.
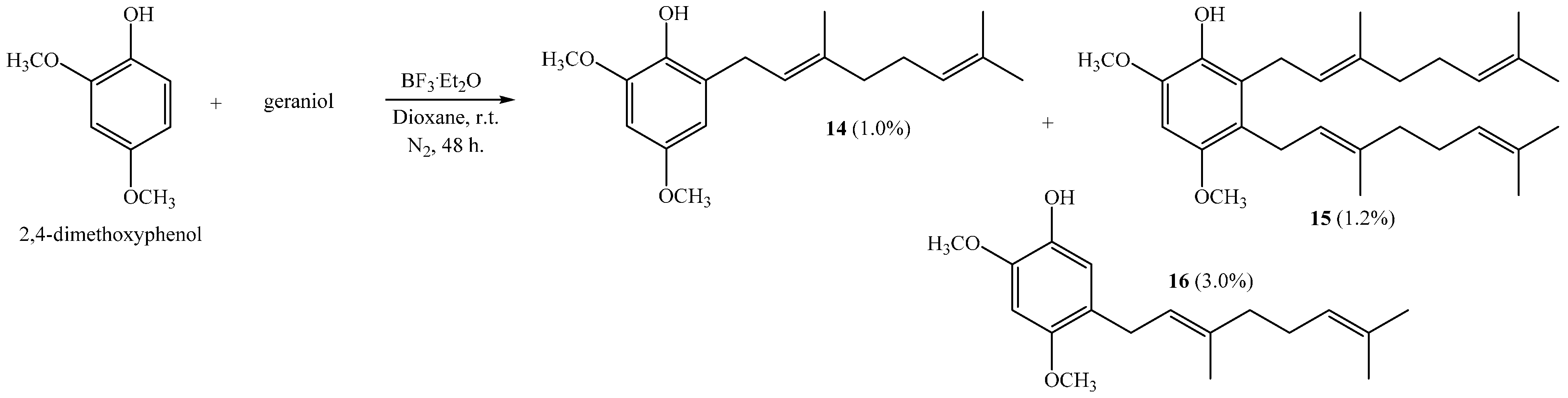
Interestingly, a digeranylphenol 15 is obtained with very low yield. The coupling of two prenyl chains has been described for the reactions of 2,3,4-trimethoxyphenol with geraniol; 2,4-dimethoxyphenol with farnesol; and 1,3,5-trihydroxybenzene with geraniol [6,11]. In acetonitrile there is no coupling reaction, whereas compound 14 is the only product when ether is the solvent [6]. This compound is obtained with 46% by a reaction of 2,4-dimethoxyphenol and geranyl bromide [17]. The NMR data of compounds 14 and 16 were consistent with those previously reported [6,10].
The coupling reaction between trimethoxyphenols and geraniol follows a more complex pattern than that exhibited by dimethoxyphenols [6,10]. For all of them, the coupling reaction using dioxane as a solvent leads to different products depending on the phenol structure, whereas in the presence of acetonitrile/AgNO3 there is no reaction (see Table 1). Three isomers of trimethoxyphenol, namely 2,3,4-trimethoxyphenol, 2,4,5-trimethoxyphenol and 2,4,6-trimethoxyphenol, were synthesized by Baeyer-Villiger oxidation of the respective trimethoxybenzaldehyde, which was commercially available, and the subsequent saponification of the resulting trimethoxyphenyl formate [10].
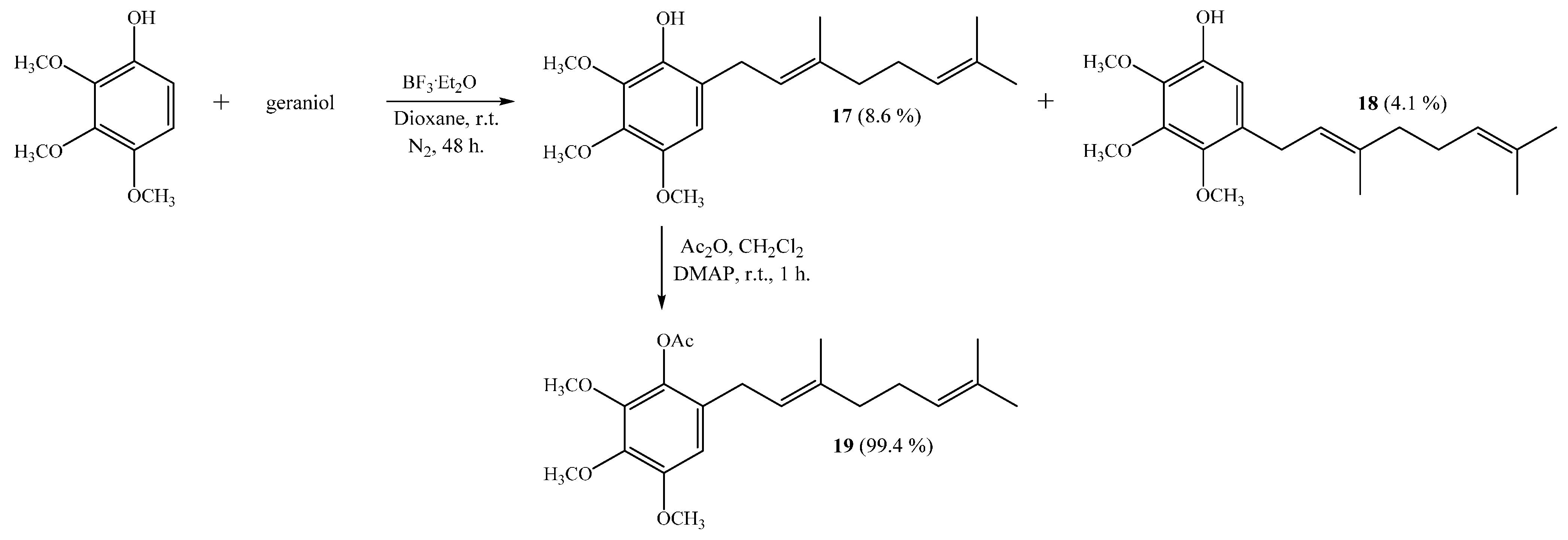
The coupling of 2,3,4-trimethoxyphenol with geraniol leads to monogeranyl compounds 17 and 18 with 8.6% and 4.1% yields, respectively (Scheme 4). Subsequently, compound 19 was obtained from 17 with a 99.4% yield by standard acetylation. The NMR data of compound 17 were consistent with those previously reported [6].
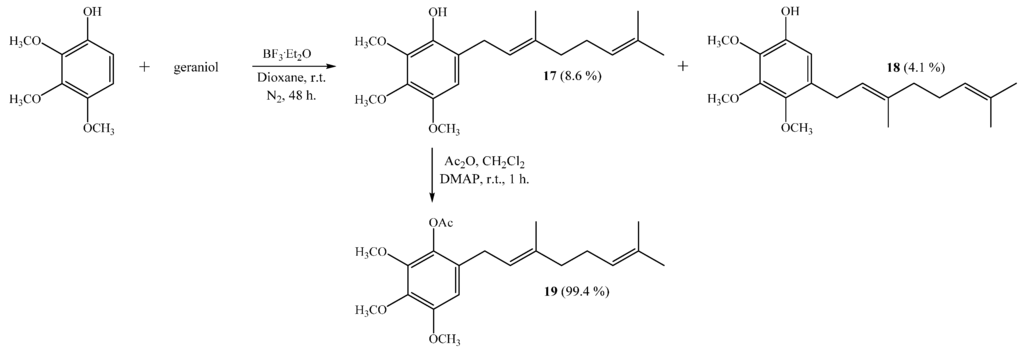
Scheme 4.
Synthesis of compounds 17–18 by the coupling reaction of 2,3,4-trimethoxyphenol with geraniol in dioxane. Compound 19 was obtained by acetylation of 17.
Scheme 4.
Synthesis of compounds 17–18 by the coupling reaction of 2,3,4-trimethoxyphenol with geraniol in dioxane. Compound 19 was obtained by acetylation of 17.
The reaction of 2,4,6-trimethoxyphenol with geraniol gives compound 20 and with digeranyl gives compound 21 with 16.4% and 14.0% yields, respectively (see Scheme 5). Both compounds are new and their chemical structures were determined by NMR (see structure determination section).
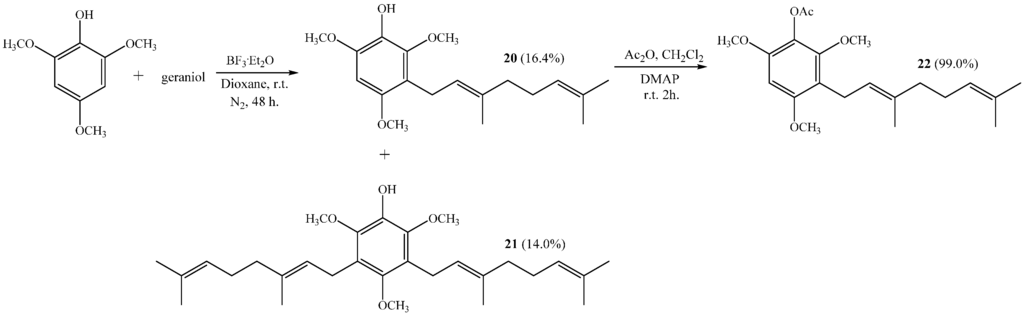
Scheme 5.
Synthesis of geranylmethoxyphenols 20, 21 and acetylated derivative 22.
Scheme 5.
Synthesis of geranylmethoxyphenols 20, 21 and acetylated derivative 22.
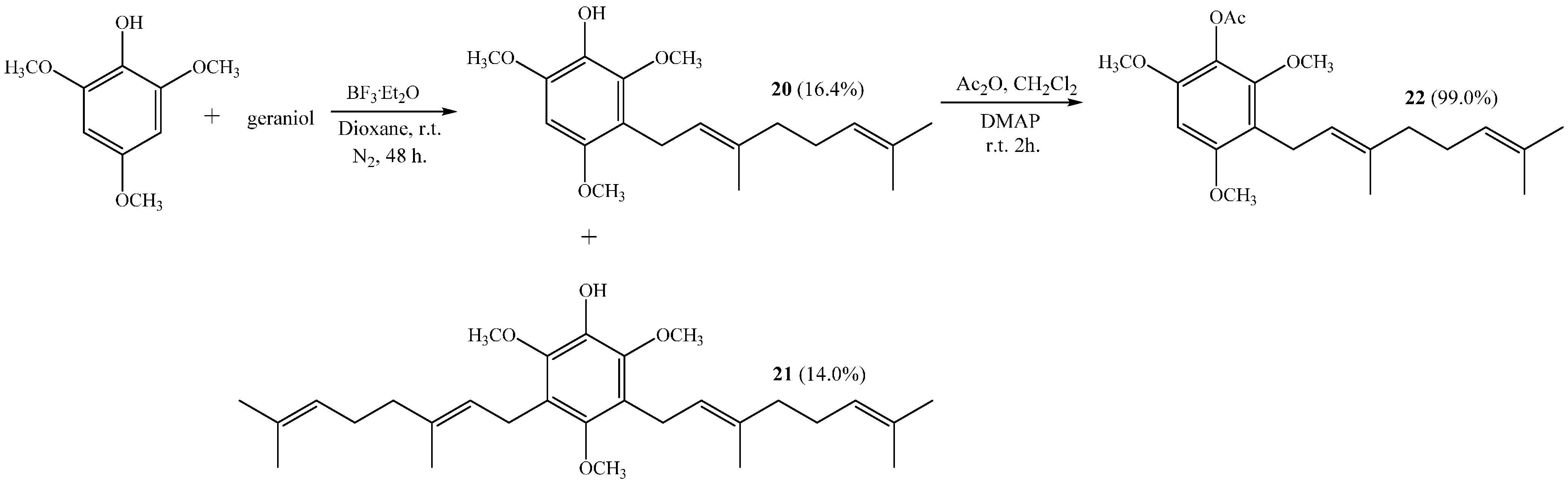
Compound 22 was obtained from 20 with a 99.0% yield by standard acetylation.
Finally, the direct geranylation of 2,4,5-trimethoxyphenol leads unexpectedly to compounds 16 and 23 with 17.3% and 18.7% yields, respectively, whereas the mono-geranylated compound 25 was not obtained (see Scheme 6).
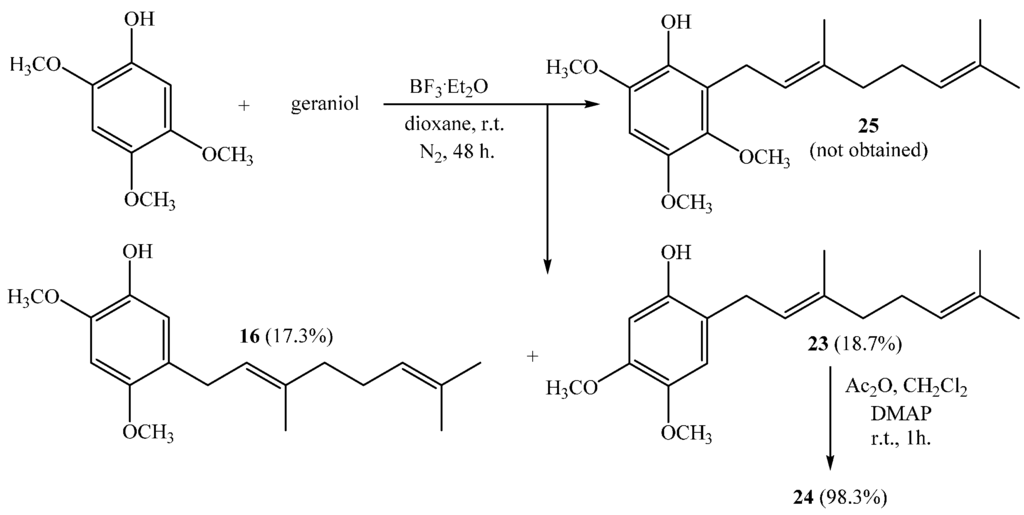
Scheme 6.
Obtaining of geranylmethoxyphenols 16 and 23 and acetylated derivative 24.
Scheme 6.
Obtaining of geranylmethoxyphenols 16 and 23 and acetylated derivative 24.
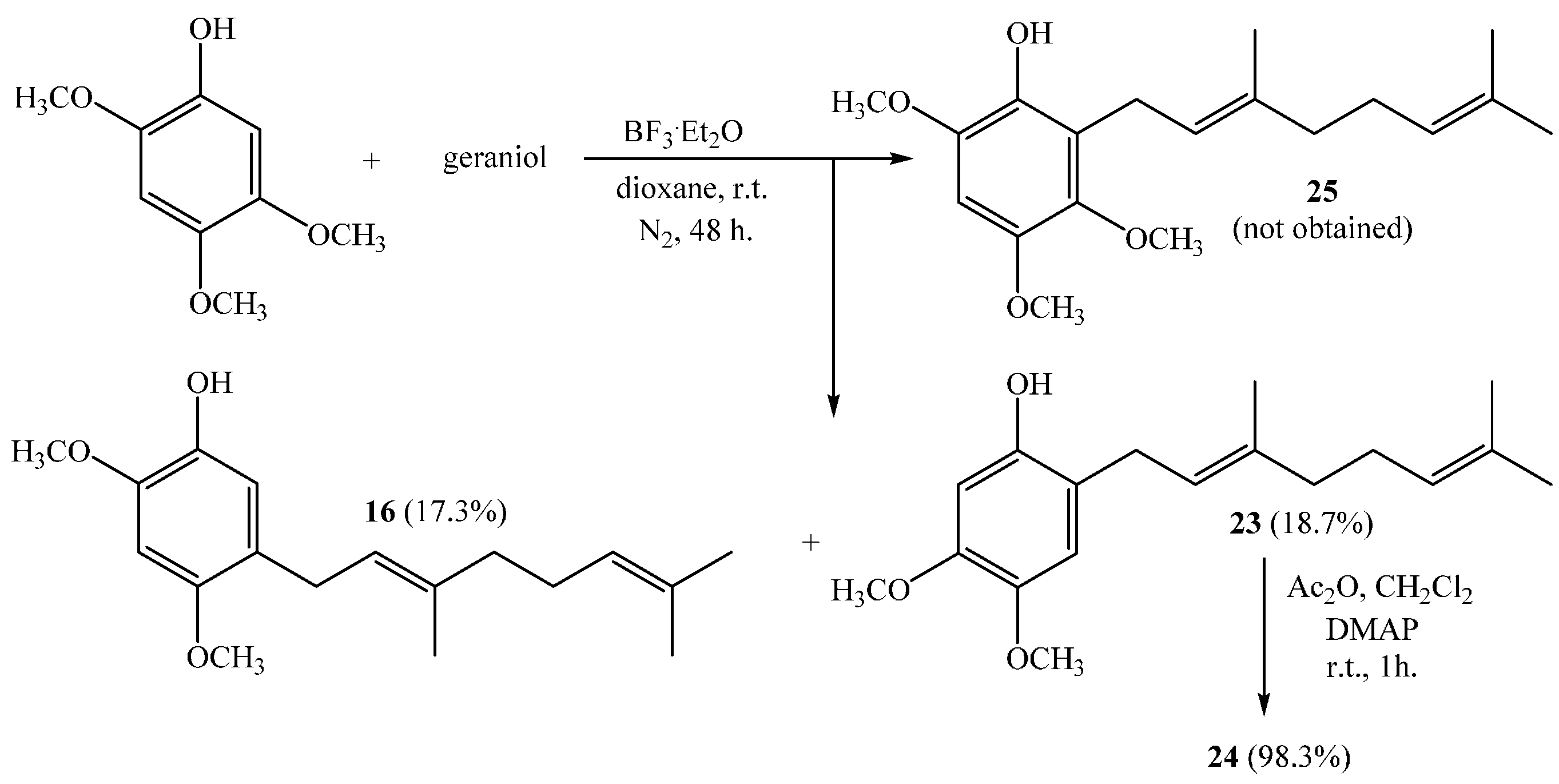
The formation of these compounds has been previously reported by our research group with slightly lower yields. It has been explained in terms of a competing reaction occurring via formation of a geranyl-ether intermediate, a subsequent Claisen rearrangement, and loss of a formaldehyde molecule from the methoxyl group [10]. The NMR data for both (16 and 23) compounds were consistent with those previously reported [10].
Finally, compound 24 was obtained from 23 with a 98.3% yield by standard acetylation.

Table 1.
Synthesis conditions and yield reactions of geranylated phenols and phenyl acetate derivatives.
| Compound | Reactants | Catalyst | Solvent | Yield |
|---|---|---|---|---|
| 6 | 4-methoxyphenol/geraniol | BF3·Et2O/AgNO3 | Acetonitrile | 18.0 |
| 8 | 2-methoxyphenol/geraniol | BF3·Et2O/AgNO3 | Acetonitrile | 4.5 |
| 10 | 2-methoxyphenol/geraniol | BF3·Et2O/AgNO3 | Acetonitrile | 6.3 |
| 11 | 2-methoxyphenol/geraniol | BF3·Et2O/AgNO3 | Acetonitrile | 0.6 |
| 12 | 3-methoxyphenol/geraniol | BF3·Et2O/AgNO3 | Acetonitrile | 7.4 |
| 13 | 3-methoxyphenol/geraniol | BF3·Et2O/AgNO3 | Acetonitrile | 4.2 |
| 14 | 2,4-dimethoxyphenol/geraniol | BF3·Et2O | Dioxane | 1.0 |
| 15 | 2,4-dimethoxyphenol/geraniol | BF3·Et2O | Dioxane | 1.2 |
| 16 | 2,4,5-trimethoxyphenol/geraniol | BF3·Et2O | Dioxane | 17.3 |
| 17 | 2,3,4-trimethoxyphenol/geraniol | BF3·Et2O | Dioxane | 8.6 |
| 18 | 2,3,4-trimethoxyphenol/geraniol | BF3·Et2O | Dioxane | 4.1 |
| 20 | 2,4,6-trimethoxyphenol/geraniol | BF3·Et2O | Dioxane | 16.4 |
| 21 | 2,4,6-trimethoxyphenol/geraniol | BF3·Et2O | Dioxane | 14.0 |
| 23 | 2,4,5-trimethoxyphenol/geraniol | BF3·Et2O | Dioxane | 18.7 |
2.2. Structure Determination
The chemical structure of all new compounds obtained through the synthesis described above was mainly established by NMR spectroscopy. All NMR spectra are given as Supplementary Material (Figure S1). Thus, in this section, the NMR data used to determine the chemical structure of geranylphenol derivatives (12–13, 15, 18, 20–21) and acetylated derivatives (9, 19, 22, 24) is discussed in detail. Compounds 12–13: The 1H NMR spectrum of compound 12 shows a pattern characteristic of aromatic tri-substitution, i.e., doublet signal at 6.99 ppm (J = 8.0 Hz, 1H, H-3); double doublet at 6.44 ppm (J = 2.5 and 8.0 Hz, 1H, H-4); and doublet at 6.42 ppm (J = 2.5 Hz, 1H, H-6). The position of the geranyl chain on the aromatic ring was established by two-dimensional (2D) HMBC correlations. In this spectrum, a 2JH–C coupling of H-1ʹ with C-2 (δC = 118.9 ppm) and C-2ʹ (δC = 122.0 ppm), and a 3JH–C coupling between the signals of C-1, C-3 and C-3ʹ at δC = 155.4, 130.3 and 138.4 ppm, respectively, were observed. In addition, a correlation at 3JH–C between the CH3O group (δH = 3.76 ppm) and C-5 (δC = 159.4 ppm), and correlations between 2JH–C and 3JH–C of the OH group (δH = 5.25 ppm) with C-1 (δC = 155.4 ppm) and C-6 (δC = 102.0 ppm) were also observed (Figure 3a).
Similarly, for compound 13, the 1H NMR spectrum shows a double doublet signal at 7.06 ppm (J = 8.2 and 8.2 Hz, 1H, H-5) and a doublet signal at 6.49 ppm (J = 8.2 Hz, 2H, H-4 and H-6). The aromatic substitution pattern shows unequivocally that the geranyl chain is attached to the ortho position between hydroxyl and methoxyl groups. The position of the geranyl chain on the aromatic ring has been confirmed by 2D HMBC correlations. In this spectrum, the signal at δH = 3.43 ppm assigned to H-1ʹ (d, J = 7.0 Hz, 2H) shows 3JH–C coupling with C-1 (δC = 155.6) and C-3ʹ (δC = 138.1 ppm), and 2JH–C coupling with C-2 and C-2ʹ (δC = 115.2 and 121.9 ppm, respectively), while the signal at δH = 5.24 ppm assigned to H-2ʹ (t, J = 7.0 Hz, 1H) shows 3JH–C coupling with C-2 (δC = 115.2) and C-4ʹ (δC = 39.7 ppm). Additionally, the proton signal of the hydroxyl group (δH = 5.32 ppm) shows 2JH–C coupling with C-1 (δC = 155.6 ppm) and 3JH–C with C-6 (δC = 108.1 ppm). The correlation at 3JH–C between the CH3O group (δH = 3.81 ppm) and C-3 (δC = 157.9 ppm) was also observed (Figure 3b).
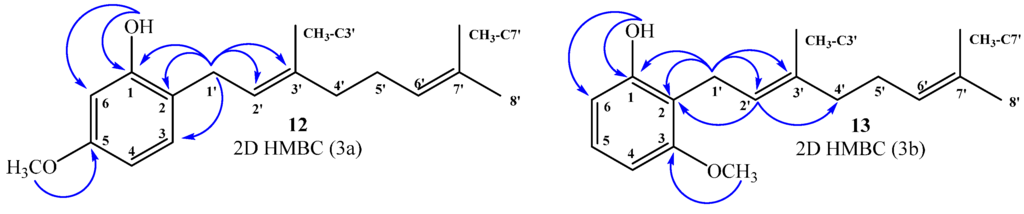
Figure 3.
Most important correlations 2D 1H–13C HMBC, compound 12 (3a) and compound 13 (3b).
Figure 3.
Most important correlations 2D 1H–13C HMBC, compound 12 (3a) and compound 13 (3b).
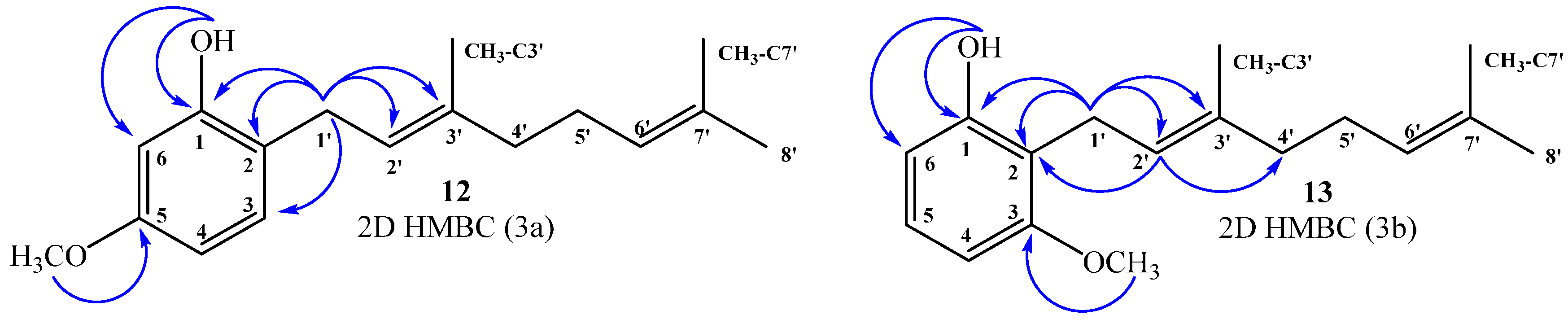
Compound 15: In the 1H NMR spectrum a single hydrogen aromatic signal was observed at δH = 6.42 ppm (s, 1H), confirming the aromatic disubstitution pattern. The positions of geranyl chains on the aromatic ring were established by selective one-dimensional (1D) NOESY experiments, wherein the aromatic signal at δH = 6.42 ppm assigned to H-5 showed spatial correlations with two singlets at 3.87 and 3.77 ppm, assigned to CH3O-C6 and CH3O-C4 (by 2D HSQC and HMBC), respectively. In addition, the signal at δH = 3.39 ppm (d, J = 6.0 Hz, 2H) assigned to hydrogen H-1ʹ showed spatial correlations with the signals at δH = 5.31 (s, 1H) and δH = 1.76 ppm (s, 3H), assigned to OH and CH3-C3ʹ, respectively. The signal at δH = 3.31 ppm (d, J = 6.0 Hz, 2H) assigned to hydrogen H-1ʹʹ showed spatial correlations with CH3O-C4 and with δH = 1.74 ppm (s, 3H) assigned to CH3-C3ʹʹ. Ortho aromatic disubstitution of both geranyl chains was confirmed by the observation of spatial NOE interaction between hydrogen H-1ʹ and H-1ʹʹ (see Figure 4a). Finally, the complete structural determination was established by heteronuclear 2D HSQC and 2D HMBC correlations, where H-1ʹ showed 3JH–C coupling with C-3 (δC = 122.1 ppm), C-3ʹ (δC = 135.2 ppm) and C-1 (δC = 137.9 ppm) and 2JH–C coupling with C-2 and C-2ʹ (δC = 127.3 and 123.8 ppm, respectively). H-1ʹʹ showed 3JH–C coupling with C-4 (δC = 150.7 ppm), C-3ʹʹ (δC = 134.4 ppm) and C-2 (δC = 127.3 ppm) and 2JH–C coupling with C-2ʹʹ and C-3 (δC = 122.8 and 122.1 ppm, respectively). The signal of H-5 at δH = 6.42 ppm (s, 1H-Ar) showed3JH–C coupling with C-3 and C-1 (δC = 122.1 and 137.9 ppm, respectively) and 2JH–C coupling with C-6 and C-4 (δC = 144.4 and 150.7 ppm, respectively). These and other heteronuclear correlations are shown in Figure 4b.
Figure 4.
Main correlations observed for compound 15, selective 1D NOESY (4a), and 2D1H–13C HMBC (4b).
Figure 4.
Main correlations observed for compound 15, selective 1D NOESY (4a), and 2D1H–13C HMBC (4b).
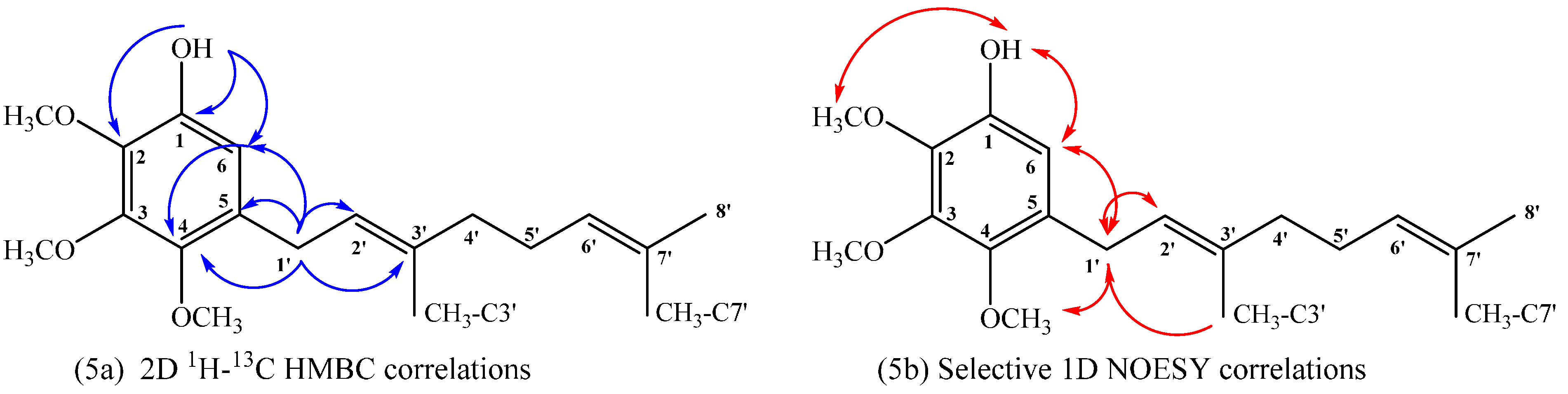
Compound 18: The 1H NMR of compound 18 shows aromatic signal at δH = 6.50 ppm (s, 1H) confirming the aromatic monosubstitution. On the other hand, in the HMBC spectrum, the signal at δH = 3.27 ppm, assigned to H-1ʹ (d, J = 7.2 Hz, 2H), shows 3JH–C coupling with C-6 (δC = 109.5 ppm), C-3ʹ (δC = 136.2 ppm) and C-4 (δC = 144.9 ppm) and 2JH–C coupling with C-2ʹ and C-5 (δC = 122.5 and 130.6 ppm, respectively). While the signal at δH = 6.50 ppm, assigned to H-6 (s, 1H), shows 3JH–C coupling with C-1ʹ (δC = 27.8), C-2 (δC = 138.0 ppm) and C-4 (δC = 144.9 ppm) and the signal at δH = 5.46 ppm, assigned to OH (s, 1H), shows 3JH–C coupling with C-6 (δC = 109.5 ppm) and C-2 (δC = 138.0 ppm) and 2JH–C coupling with C-1 (δC = 144.5 ppm). These HMBC correlations are shown in Figure 5a. Finally, selective 1D NOESY NMR experiments were recorded for compound 18. These correlations are shown in Figure 5b, where the most important ones correspond to the correlations observed between H-6 with OH and H-1ʹ at (5.46 and 3.27 ppm, respectively) and correlations between H-1ʹ with CH3O-C4 and CH3-C3ʹ (3.78 and 1.70 ppm, respectively).
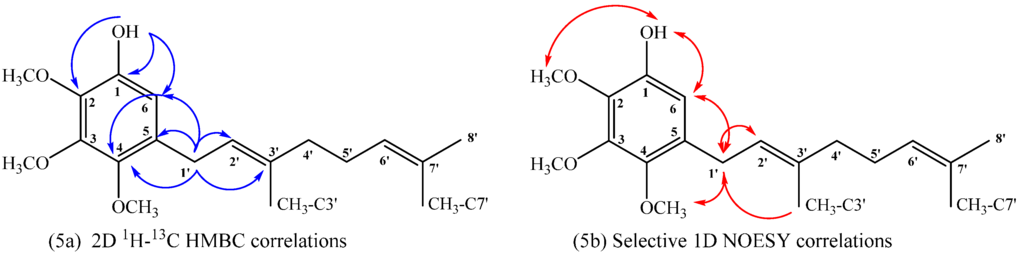
Figure 5.
Main correlations observed for compound 18, 2D 1H–13C HMBC (5a), selective 1D NOESY (5b).
Figure 5.
Main correlations observed for compound 18, 2D 1H–13C HMBC (5a), selective 1D NOESY (5b).
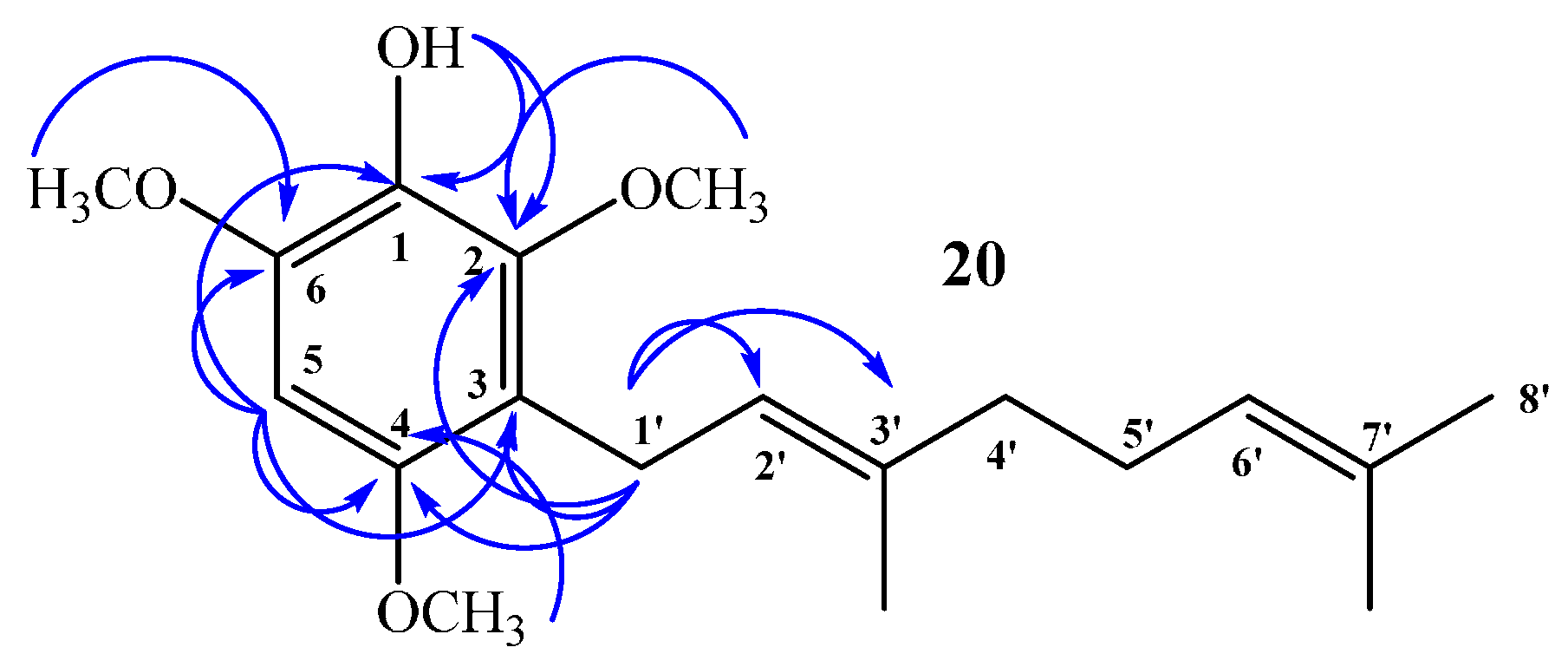
Compound 20: In the 1H NMR spectrum a single signal at δH = 6.32 ppm (s, 1H) confirms aromatic monosubstitution. Unequivocally, there is only one possibility of aromatic monosubstitution and this is reflected in the HMBC spectrum, i.e., the signal at δH = 3.30 ppm, assigned to H-1ʹ (d, J = 6.8 Hz, 2H), correlated with δC = 22.5 ppm (by 2D HSQC) and shows 3JH–C coupling with C-2 (δC = 145.9 ppm), C-4 (δC = 150.6 ppm) and with C-3ʹ (δC = 134.4 ppm) and 2JH–C coupling with C-3 and C-2ʹ (δC = 116.8 and 123.5 ppm, respectively). The signal at δH = 6.32 ppm, assigned to H-5 (s, 1H, by 2D HSQC), shows 3JH–C coupling with C-3 (δC = 116.8 ppm) and C-1 (δC = 133.0 ppm), and 2JH–C coupling with C-2 (δC = 145.9 ppm) and C-4 (δC = 150.6 ppm). In addition, the signal at δH = 5.15 ppm, assigned to OH (s, 1H), shows 3JH–C coupling with C-6 (δC = 145.3 ppm) and C-2 (δC = 145.9 ppm), and 2JH–C coupling with C-1 (δC = 133.0 ppm). These and other HMBC correlations are shown in Figure 6.
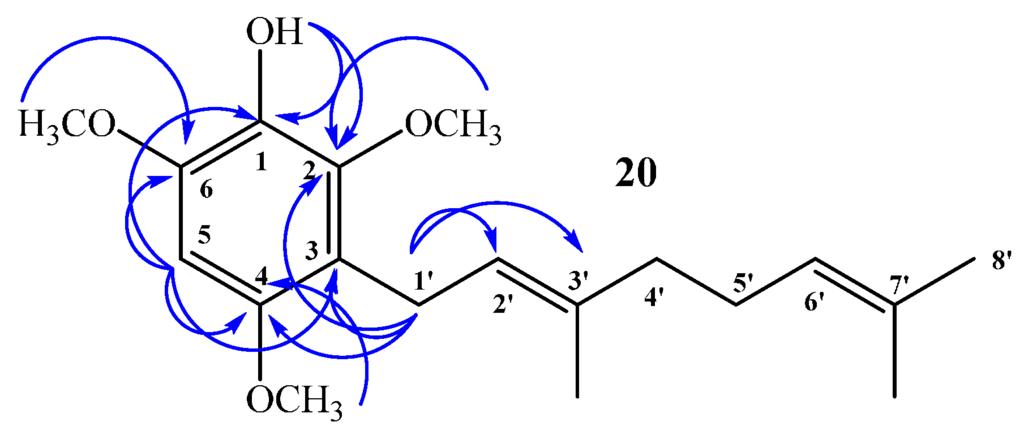
Figure 6.
Main correlations 2D 1H–13C HMBC for compound 20.
Figure 6.
Main correlations 2D 1H–13C HMBC for compound 20.
Compound 21: No aromatic hydrogen signals were detected in the 1H NMR spectrum, whereas a 1:2 ratio of integration areas was observed between the signal at δH = 3.66 ppm, assigned to CH3O-C4 (by 2D HSQC), and the signal at δH = 3.81 ppm, assigned to CH3O-C2 + CH3O-C6 (by 2D HSQC), confirming that this is a symmetrical disubstituted aromatic compound. The complete structure determination of compound 21 was established mainly by 2D HSQC and 2D HMBC experiments. The signal at δH = 3.34 ppm (d, J = 6.6 Hz, 4H), assigned to hydrogen atoms H-1ʹ and H-1ʹʹ, correlated with δC = 23.5 ppm (by 2D HSQC) and shows 3JH–C coupling with C-2 and C-6 (δC = 144.4 ppm, 2 × C), respectively, and with C-4 (δC = 149.5 ppm). In addition, 3JH–C coupling with C-3ʹ and C-3ʹʹ (δC = 134.8 ppm, 2 × C), and 2JH–C coupling with C-3, C-5 (δC = 124.4 ppm, 2 × C), and with C-2ʹ and C-2ʹʹ (δC = 123.8 ppm, 2 × C). The signal at δH = 5.34 ppm, assigned to OH (s, 1H), shows 3JH–C coupling with C-2 and C-6 (δC = 144.4 ppm, 2 × C) and 2JH–C coupling with C-1 (δC = 139.0 ppm). These and other HMBC correlations are shown in Figure 7.
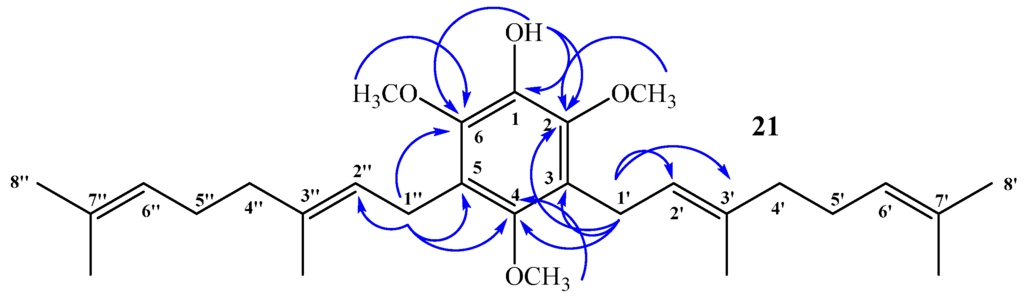
Figure 7.
Most important 2D 1H–13C HMBC correlations observed for compound 21.
Figure 7.
Most important 2D 1H–13C HMBC correlations observed for compound 21.
Phenyl acetate derivative compounds 9, 19, 22 and 24: In the 1H NMR spectrum of these compounds, a signal at δH = 2.32 ppm (s, 3H, CH3CO) is observed, whereas signals at δC = 168.9 ppm (CH3CO) and δC = 20.5 ppm (CH3CO) in the 13C NMR spectrum are found. This confirms the presence of the acetylated derivative.
2.3. Antifungal Activity against B. cinerea in Vitro
The effect of compounds 6–24 (Figure 2) on the mycelial growth of plant pathogen B. cinerea was evaluated in vitro after 48 h of incubation by using the agar-radial test with potato dextrose agar (PDA) as a medium. Figure 8 shows an assay where the mycelium of B. cinerea grows in the presence of Captan (Figure 8a, commercial fungicide widely applied to control diseases of fruits and vegetable crops and used in this study as a positive control) or of different geranylphenol compounds (Figure 8c–Figure 8f). Figure 8b corresponds to the negative control, i.e., no inhibitory compounds. As it can be seen, Captan and compounds 9 and 17 almost completely inhibit the fungus growth, while compounds 15 and 6 are less active at a concentration of 250 mg/L.
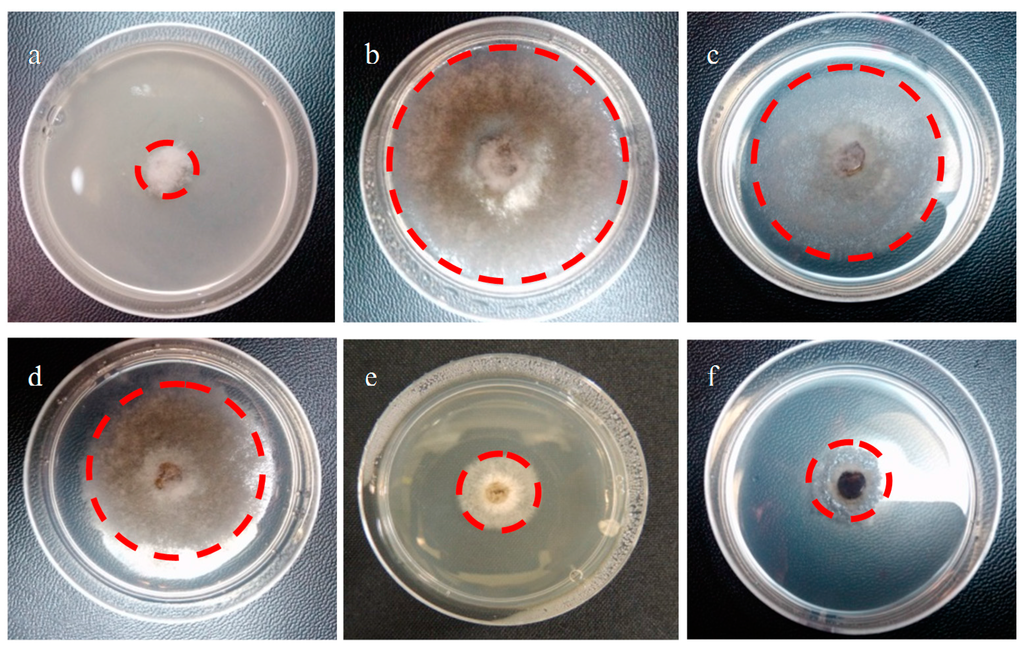
Figure 8.
Effect of geranylated phenol/phenyl acetate derivatives on in vitro mycelial growth of B. cinerea. (a) Positive control, Captan; (b) negative control, the medium contains only PDA and 1% ethanol; (c) compound 15; (d) compound 6; (e) compound 9; (f) compound 17. Concentration of Captan and all tested compounds is 250 mg/L. The red line gives the growth radius of the negative control (b) and in presence of different compounds.
Figure 8.
Effect of geranylated phenol/phenyl acetate derivatives on in vitro mycelial growth of B. cinerea. (a) Positive control, Captan; (b) negative control, the medium contains only PDA and 1% ethanol; (c) compound 15; (d) compound 6; (e) compound 9; (f) compound 17. Concentration of Captan and all tested compounds is 250 mg/L. The red line gives the growth radius of the negative control (b) and in presence of different compounds.

The effect on mycelial growth is evaluated by comparing the growth areas with that observed for the negative control. The results are expressed as a percentage of inhibition, which is calculated as the ratio of the area of B. cinerea in the presence and absence of geranylated phenol/phenyl acetate derivatives, and they are summarized in Table 2.
The data in Table 2 indicates that for most of the studied compounds the percentage of inhibition increases with the increasing compound concentration, reaching maximum values that are between 53% and 98%. For compounds 6, 15, 19 and 20, the effect levels off at 150 mg/L, and compounds 6, 8, 10, 12, 15 and 22 reduce growth in a quantity lower than 50%. On the other hand, the monomethoxygeranyl acetates 7 and 9 and trimethoxygeranylphenols 17 and 18 exhibit an inhibitory effect on mycelial growth similar to that found for Captan, i.e., 86% to 98% at 250 mg/L. In a previous work, we have found that the inhibitory effect of Captan changed more deeply with concentration, i.e., 40%, 60% and 80% at 50, 150 and 250 mg/L, respectively [9]. Both sets of experiments were carried out under the same conditions and, therefore, this remarkable difference is difficult to explain. It could be attributed to seasonal changes in the growing rate of B. cinerea or changes in the concentration of the commercial fungicide. However, as Captan is used only for comparison, the following discussion of the antifungal activity of geranylphenols as a function of chemical structure is not affected by this change.
Table 2.
Effect of geranylated phenol/phenyl acetate derivatives on in vitro mycelial growth of B. cinerea, measured as a percentage of inhibition.
| Geranylated Phenols/Phenyl Acetates Derivatives | Percentage of Inhibition on in Vitro Mycelial Growth of B. cinerea (%) * | ||
|---|---|---|---|
| 50 mg/L | 150 mg/L | 250 mg/L | |
| 6 | 24 ± 4.6 | 42 ± 4.5 | 40 ± 4.8 |
| 7 | 5 ± 1.9 | 56 ± 5.3 | 86 ± 7.2 |
| 8 | 18 ± 3.5 | 31 ± 3.4 | 41 ± 4.6 |
| 9 | 13 ± 2.8 | 79 ± 8.7 | 98 ± 1.4 |
| 10 | 0 | 4 ± 1.4 | 26 ± 5.4 |
| 12 | 5 ± 1.0 | 20 ± 4.8 | 45 ± 4.0 |
| 14 | 9 ± 2.1 | 46 ± 2.8 | 54 ± 4.8 |
| 15 | 0 | 20 ± 2.0 | 21 ± 1.9 |
| 16 | 32 ± 2.2 | 41 ± 4.7 | 71 ± 3.8 |
| 17 | 13 ± 2.6 | 39 ± 4.0 | 91 ± 0.7 |
| 18 | 0 | 66 ± 6.4 | 91 ± 0.7 |
| 19 | 36 ± 6.6 | 55 ± 6.1 | 56 ± 4.6 |
| 20 | 18 ± 2.9 | 67 ± 5.5 | 73 ± 3.1 |
| 21 | 1 ± 0.4 | 20 ± 4.2 | 66 ± 3.9 |
| 22 | 4 ± 1.4 | 18 ± 3.0 | 39 ± 4.5 |
| 23 | 49 ± 3.3 | 59 ± 3.0 | 79 ± 2.1 |
| 24 | 42 ± 4.1 | 57 ± 3.0 | 77 ± 3.3 |
| C− | 0 | 0 | 0 |
| C+ | 89 ± 2.1 | 94 ± 1.1 | 94 ± 1.3 |
* The percentage of inhibition of mycelial growth was based on colony diameter measurements after 72 h of incubation. Each point represents the mean of at least three independent experiments ± standard deviation.
The dependence of antifungal activity on chemical structure is more clearly understood by separating the studied compounds in structurally related groups. In the first place, the results obtained for monogeranyl compounds show that the activity depends clearly on the number of methoxy groups. The inhibitory effect of monomethoxyphenol compounds is so low that they are considered inactive, but when the number of methoxy groups is increased from one to three, the percentage of inhibition increases by a factor of two, reaching values similar to that measured for Captan. The order of ascending activity is monomethoxy (6, 8, 10, 12) < dimethoxy (14, 16, 23) < trimethoxy (17, 18, 22). In the case of acetylated derivatives this effect is the opposite, i.e., the activity decreases with the increasing number of methoxy groups and the order is monomethoxy (7 and 9) > dimethoxy (24) > trimethoxy (19 and 22). Interestingly, the acetylation of the hydroxyl group in monomethoxy compounds increases the activity dramatically, whereas the reverse effect is observed for trimethoxy derivatives. It has been shown that acetylation on geranylphenols with no methoxy groups produces a slight decrease of inhibitory activity [9]. These results confirm that the activity of geranyl compounds against the mycelial growth of B. cinerea is mainly determined by the geranyl chain. However, the activity of digeranyl derivatives is lower than that exhibited by the respective monogeranyl compound (comparison of 20 and 21, 16 and 15 in Table 2). A similar behavior is observed when the activities of 5 and 2,6-digeranylphloroglucinol, a digeranyl derivative obtained from the reaction of phloroglucinol and geraniol, are compared [9]. These results are in line with previous work that established that the efficiency decreases with the increasing number of prenyl moieties [6,9].
It is worth mentioning that the effect of chemical structure on antifungal activity could be associated with differences in water solubility, which finally determines the biodisponibility of these compounds, or with their biochemical mechanism of action. This point is a very important issue that we will intend to clarify in a study where polymer micelles are used to increase the water solubility of these compounds.
3. Experimental Section
3.1. General
Unless otherwise stated, all chemical reagents purchased (Merck, Darmstadt, Germany or Aldrich, St. Louis, MO, USA) were of the highest commercially available purity and were used without previous purification. Fourier transform infrared (FT-IR) spectra were recorded as thin films in a FT-IR Nicolet 6700 spectrometer (Thermo Scientific, San Jose, CA, USA) and frequencies are reported in cm−1. Low resolution mass spectra were recorded on an Agilent 5973 spectrometer (Agilent Technologies, Santa Clara, CA, USA) at 70 eV ionizing voltage coupled with a 6890 N gas chromatograph equipped with a DB-5 column (30 m × 0.25 mm × 0.25 μm), and data are given as percentage of relative intensity m/z (% rel. int.). High resolution mass spectra were recorded on an LTQ Orbitrap XL spectrometer (Thermo Scientific, San Jose, CA, USA) by applying a voltage of 1.8 kV in the positive and 1.9 kV in the negative ionization mode. The spectra were recorded using full scan mode, covering a mass range from m/z 100–1300. The resolution was set to 50,000 and maximum loading time for the ion cyclotron resonance (ICR) cell was set to 250 ms. 1H, 13C, 13C DEPT-135, sel. gs-1D 1H NOESY, gs-2D HSQC and gs-2D HMBC spectra were recorded in CDCl3 solutions and are referenced at the residual peaks of CHCl3 at δ = 7.26 and 77.0 ppm for 1H and 13C, respectively, on a Bruker Avance 400 Digital NMR spectrometer (Bruker, Rheinstetten, Germany), operating at 400.1 MHz for 1H and 100.6 MHz for 13C. Chemical shifts are reported in δ ppm and coupling constants (J) are given in Hz. Silica gel (Merck 200–300 mesh) was used for C.C. and silica gel plates HF254 for thin layer chromatography (TLC). TLC spots were detected by heating after spraying with 25% H2SO4 in H2O.
3.2. Synthesis
3.2.1. Geranylation Reaction
The coupling of geraniol and phenols were carried out using boric trifluoride etherate BF3·OEt2 as catalyst and dioxane as solvent. Alternatively, acetonitrile was used as solvent and AgNO3 as secondary catalyst. In a typical reaction, BF3·OEt2 (0.46 g, 3.2 mmol) is slowly added dropwise to an equimolar solution of a phenol derivative (9.1 mmol) and geraniol (1.4 g, 9.1 mmol) in dioxane (or acetonitrile saturated with AgNO3), with stirring at room temperature and under a N2 atmosphere. After the addition is completed, the stirring is continued for 48 h. The end of the reaction is verified by TLC, and then the mixture is poured onto crushed ice (30 g) and extracted with EtOAc (3 × 20 mL). The organic layer is washed with 5% NaHCO3 (15 mL) and water (2 × 15 mL), dried over Na2SO4, and filtered. The solvent is then evaporated under reduced pressure, and the crude is re-dissolved in CH2Cl2 (5 mL) and chromatographed on silica gel with petroleum ether/EtOAc mixtures of increasing polarity (19.8:0.2→0.2:19.8).
3.2.2. Acetylation of Geranylated Phenols
Geranylated phenols were acetylated by standard acetylation using Ac2O and dimethylaminopyridine (DMAP) in CH2Cl2. In a typical reaction, Ac2O (1.08 g, 10.6 mmol) is added to a solution of a geranylated phenol (0.284 mmol), DMAP (3.0 mg) and pyridine (1.0 mL) in dichloromethane (30 mL). The end of the reaction is verified by TLC (1 h), and then the mixture is extracted with EtOAc (2 × 10 mL). The organic layer is washed with 5% KHSO4 (2 × 10 mL) and water (2 × 10 mL), dried over Na2SO4, and filtered. The solvent is evaporated under reduced pressure.
3.2.3. Synthesis of Geranylated Methoxyphenol/Phenylacetate Derivatives
(E)-2-(3,7-Dimethylocta-2,6-dienyl)-4-methoxyphenol (6)
Compound 6 was obtained as colorless viscous oil (375.5 mg, 18%) by coupling of 4-methoxyphenol (1.00 g, 8.0 mmol) and geraniol (1.2 g, 7.9 mmol) in acetonitrile (15 mL), saturated with AgNO3, and BF3·OEt2 (0.2 g 1.6 mmol) as catalyst. NMR data of 10 was consistent with that reported in literature [15].
(E)-2-(3,7-Dimethylocta-2,6-dienyl)-4-methoxyphenyl Acetate (7)
Compound 7 was obtained by standard acetylation of compound 10 (90 mg, 0.346 mmol) with Ac2O (1.0 mL), DMAP (5.0 mg) and pyridine (2.0 mL) in dichloromethane (30 mL). Compound 7 was obtained as colorless viscous oil (99.3 mg, 95%). NMR data of compound 7 was consistent with that found in literature [15].
(E)-2-(3,7-Dimethylocta-2,6-dienyl)-6-methoxyphenyl Acetate (9)
Compound 9 was obtained by standard acetylation of compound 8 (50 mg, 0.192 mmol) with Ac2O (1.0 mL), DMAP (2.0 mg) and pyridine (1.0 mL) in dichloromethane (20 mL). Compound 13 was obtained as a pale yellow viscous oil (53.9 mg, 93%). Compound 9: IR (cm−1) 2924, 1762, 1496, 1195, 1041; 1H NMR (CDCl3, 400.1 MHz) δ 7.12 (1H, dd, J = 8.0 and 8.0 Hz, H-4), 6.98 (1H, s, H-5), 6.82 (1H, d, J = 8.0 Hz, H-3), 5.23 (1H, t, J = 7.8 Hz, H-2ʹ), 5.09 (1H, t, J = 6.3 Hz, H-6ʹ), 3.81 (3H, s, CH3O), 3.24 (2H, d, J = 7.2 Hz, H-1ʹ), 2.32 (3H, s, CH3CO), 2.10–2.07 (2H, m, H-5ʹ), 2.04–2.03 (2H, m, H-4ʹ), 1.72 (3H, s, CH3-C3ʹ), 1.59 (3H, s, H-8ʹ, 1.56 (3H, s, CH3-C7ʹ); 13C NMR (CDCl3, 100.6 MHz) δ 168.9 (C, CH3CO), 151.5 (C, C-6), 145.4 (C, C-1), 135.8 (C, C-3ʹ), 131.5 (C, C-2), 128.3 (C, C-7ʹ), 126.3 (CH, C-4), 125.5 (CH, C-6ʹ), 124.2 (CH, C-2ʹ), 121.5 (CH, C-3), 109.9 (CH, C-5), 55.9 (CH3, CH3O), 39.7 (CH2, C-4ʹ), 29.7 (CH2, C-1ʹ), 28.5 (CH2, C-5ʹ), 25.7 (CH3, C-8ʹ), 20.5 (CH3, CH3CO), 17.7 (CH3, CH3-C7ʹ), 16.1 (CH3, CH3-C3ʹ).
(E)-2-(3,7-Dimethylocta-2,6-dienyl)-6-methoxyphenol (8), (E)-3-(3,7-Dimethylocta-2,6-dienyl)-2-methoxyphenol (10), (E)-5-(3,7-Dimethylocta-2,6-dien-1-yl)-2-methoxyphenol (11)
Guayacol (1.5 g, 12.1 mmol) and geraniol (0.9 g, 6.0 mmol) were reacted in acetonitrile (15 mL), saturated with AgNO3, using BF3·OEt2 (0.45 g, 3.2 mmol) as catalyst. By following the general procedure described above, three fractions were obtained: Fraction I, 70.6 mg of yellow viscous oil (4.5% yield, compound 8); Fraction II, 98.5 mg of yellow viscous oil (6.3% yield, compound 10); and Fraction III, 9.3 mg of yellow viscous oil (0.6% yield, compound 11). NMR data of compounds 8, 10 and 11 were consistent with those reported in literature [11].
(E)-2-(3,7-Dimethylocta-2,6-dienyl)-5-methoxyphenol (12), (E)-2-(3,7-Dimethylocta-2,6-dienyl)-3-methoxyphenol (13)
Reaction of 3-methoxyphenol (0.8 g, 6.5 mmol) and geraniol (1.0 g, 6.5 mmol) was carried out in acetonitrile (20 mL), saturated with AgNO3, using BF3·OEt2 (0.46 g 3.2 mmol) as catalyst. By following the general procedure described above, two fractions were obtained: Fraction I, 125 mg of reddish viscous oil (7.4% yield, compound 12); and Fraction II, 71.1 mg of reddish viscous oil (4.2% yield, compound 13). Compound 12: 1H NMR (CDCl3, 400.1 MHz) δ 6.99 (1H, d, J = 8.0 Hz, H-3), 6.44 (1H, dd, J = 2.5 and 8.0 Hz, H-4), 6.42 (1H, d, J = 2.5 Hz, H-6), 5.31 (1H, t, J = 7.2 Hz, H-2ʹ), 5.25 (1H, s, OH), 5.08 (1H, t, J = 5.9 Hz, H-6ʹ), 3.76 (3H, s, CH3O), 3.31 (2H, d, J = 7.2 Hz, H-1ʹ), 2.13–2.12 (2H, m, H-5ʹ), 2.09–2.06 (2H, m, H-4ʹ), 4.77 (3H, s, CH3-C3ʹ), 1.69 (3H, s, H-8ʹ), 1.60 (3H, s, CH3-C7ʹ); 13C NMR (CDCl3, 100.6 MHz) δ 159.4 (C, C-5), 155.4 (C, C-1), 138.4 (C, C-3ʹ), 132.0 (C, C-7ʹ), 130.3 (CH, C-3), 123.8 (CH, C-6ʹ), 122.0 (CH, C-2ʹ), 118.9 (C, C-2), 106.1 (CH, C-4), 102.0 (CH, C-6), 55.3 (CH3, CH3O), 39.7 (CH2, C-4ʹ), 29.2 (CH2, C-1ʹ), 26.4 (CH2, C-5ʹ), 25.7 (CH3, C-8ʹ), 17.7 (CH3, CH3-C7ʹ), 16.1 (CH3, CH3-C3ʹ); HRMS m/z 261.1785 (calcd for C17H24O2, 261.1776). Compound 13: 1H NMR (CDCl3, 400.1 MHz) δ 7.06 (1H, dd, J = 8.2 and 8.2 Hz, H-5), 6.49 (2H, d, J = 8.2 Hz, H-4 and H-6), 5.32 (1H, s, OH), 5.24 (1H, t, J = 7.0 Hz, H-2ʹ), 5.06 (1H, t, J = 6.6 Hz, H-6ʹ), 3.81 (3H, s, CH3O), 3.43 (2H, d, J = 7.0 Hz, H-1ʹ), 2.11–2.08 (2H, m, H-5ʹ), 2.06–2.04 (2H, m, H-4ʹ), 1.81 (3H, s, CH3-C3ʹ), 1.68 (3H, s, H-8ʹ), 1.59 (3H, s, CH3-C7ʹ); 13C NMR (CDCl3, 100.6 MHz) δ 157.9 (C, C-3), 155.6 (C, C-1), 138.1 (C, C-3ʹ), 131.8 (C, C-7ʹ), 127.1 (CH, C-5), 123.9 (CH, C-6ʹ), 121.9 (CH, C-2ʹ), 115.2 (C, C-2), 108.1 (CH, C-6), 103.1 (CH, C-4), 55.8 (CH3, CH3O), 39.7 (CH2, C-4ʹ), 26.4 (CH2, C-5ʹ), 25.6 (CH3, C-8ʹ), 22.2 (CH2, C-1ʹ), 17.7 (CH3, CH3-C7ʹ), 16.1 (CH3, CH3-C3ʹ); HRMS m/z 259.17151 (calcd for C17H24O2, 259.1776).
(E)-2-(3,7-Dimethylocta-2,6-dienyl)-4,6-dimethoxyphenol (14), 2,3-bis((E)-3,7-Dimethylocta-2,6-dienyl)-4,6-dimethoxyphenol (15), (E)-5-(3,7-Dimethylocta-2,6-dien-1-yl)-2,4-dimethoxyphenol (16)
Reaction of 2,4-dimethoxyphenol (0.807 g, 5.2 mmol) and geraniol (0.802 g, 5.2 mmol) was carried out in dioxane (20 mL) with BF3·OEt2 (0.31 g, 2.2 mmol) as catalyst. By following the general procedure described above, three fractions were obtained: Fraction I, 15.0 mg of viscous light brown oil (1.0% yield, compound 14); Fraction II, 28.0 mg of viscous light brown oil (1.2% yield, compound 15); Fraction III, 45.0 mg of viscous dark brown oil (3.0% yield, compound 16). Compound 14: IR (cm−1): 3556, 2964, 2919, 2854, 1613, 1497, 1466, 1432, 1376, 1227, 1197, 1148, 1089, 1055, 939, 829; 1H NMR (CDCl3, 400.1 MHz) δ 6.36 (1H, d, J = 2.6 Hz, H-5), 6.30 (1H, d, J = 2.6 Hz, H-3), 5.33 (1H, t, J = 7.2 Hz, H-2ʹ), 5.28 (1H, s, OH), 5.11 (1H, t, J = 7.7 Hz, H-6ʹ), 3.86 (3H, s, CH3O-C6), 3.75 (3H, s, CH3O-C4), 3.35 (2H, d, J = 7.2 Hz, H-1ʹ), 2.11–2.08 (2H, m, H-5ʹ), 2.06–2.03 (2H, m, H-4ʹ), 1.72 (3H, s, CH3 C-3ʹ), 1.67 (3H, s, H-8ʹ), 1.60 (3H, s, CH3 C-7ʹ); 13C NMR (CDCl3, 100.6 MHz) δ 152.9 (C, C-4), 146.7 (C, C-6), 137.4 (C, C-1), 136.5 (C, C-3ʹ), 131.4 (C, C-7ʹ), 127.5 (C, C-2), 124.3 (CH, C-6ʹ), 122.0 (CH, C-2ʹ), 105.5 (CH, C-3), 96.7 (CH, C-5), 56.0 (CH3, CH3O-C6), 55.7 (CH3, CH3O-C4), 39.8 (CH2, C-4ʹ), 28.1 (CH2, C-1ʹ), 26.7 (CH2, C-5ʹ), 25.7 (CH3, C-8ʹ), 17.7 (CH3, CH3-C7ʹ), 16.1 (CH3, CH3-C3ʹ); MS m/z 290 (M+) (100), 205 (24), 189 (19), 168 (65) ((M+ + H − 123), (C9H15·)), 167 (39), 166 (45), 161 (29), 69 (18), 41 (21). NMR data of compound 14 was consistent with that in the literature [6]. Compound 15: IR (cm−1): 3556, 2965, 2925, 2854, 1616, 1485, 1451, 1437, 1376, 1341, 1237, 1202, 1089, 941, 855; 1H NMR (CDCl3, 400.1 MHz) δ 6.42 (1H, s, H-5), 5.31 (1H, s, OH), 5.12–5.06 (4H, m, H-2ʹ, H-2ʹʹ, H-6ʹ and H-6ʹʹ), 3.87 (3H, s, CH3O-C6), 3.77 (3H, s, CH3O-C4), 3.39 (2H, d, J = 6.0 Hz, H-1ʹ), 3.31 (2H, d, J = 6.0 Hz, H-1ʹʹ), 2.07–2.05 (4H, m, H-5ʹ and H-5ʹʹ), 2.03–1.98 (4H, m, H-4ʹ and H-4ʹʹ), 1.76 (3H, s, CH3-C3ʹ), 1.74 (3H, s, CH3-C3ʹ), 1.66 (6H, s, H-8ʹ and H-8ʹʹ), 1.58 (6H, s, CH3-C7ʹ and CH3-C7ʹʹ); 13C NMR (CDCl3, 100.6 MHz) δ 150.7 (C, C-4), 144.4 (C, C-6), 137.9 (C, C-1), 135.2 (C, C-3ʹ), 134.4 (C, C-3ʹʹ), 131.2 (C, C-7ʹʹ and C-7ʹ), 127.3 (C, C-2), 124.4 (CH, C-6ʹ and C-6ʹʹ), 123.8 (CH, C-2ʹ), 122.8 (CH, C-2ʹʹ), 122.1 (C, C-3), 95.3 (C, C-5), 56.9 (CH3, CH3O-C4), 56.1 (CH3, CH3O-C6), 39.7 (CH2, C-4ʹ and C-4ʹʹ), 26.7 (CH2, C-5ʹ and C-5ʹʹ), 25.6 (CH3, C-8ʹ and C-8ʹʹ), 25.3 (CH2, C-1ʹʹ), 24.6 (CH2, C-1ʹ), 17.6 (CH3, CH3-C7ʹ and CH3-C7ʹʹ), 16.2 (CH3, CH3-C3ʹ), 16.1 (CH3, CH3-C3ʹʹ); MS m/z 426 (M+) (74), 302 (16), 259 (32), 220 (40), 219 (100), 201 (41), 187 (63), 167 (19), 69 (67), 41 (48); HRMS m/z 427.3141 (calcd for C28H42O3, 427.3134). Compound 16: IR (cm−1) 2924, 1602, 1512, 1194, 1042; 1H NMR (CDCl3, 400.1 MHz) δ 6.68 (1H, s, H-6), 6.54 (1H, s, H-3), 5.51 (1H, s, OH), 5.28 (1H, t, J = 7.1 Hz, H-2ʹ), 5.11 (1H, t, J = 6.9 Hz, H-6ʹ), 3.82 (3H, s, CH3O-C2), 3.76 (3H, s, CH3O-C4), 3.26 (2H, d, J = 7.2 Hz, H-1ʹ), 2.12–2.08 (2H, m, H-5ʹ), 2.06–2.02 (2H, m, H-4ʹ), 1.70 (3H, s, CH3-C3ʹ), 1.67 (3H, s, H-8ʹ), 1.60 (3H, s, CH3-C7ʹ); 13C NMR (CDCl3, 100.6 MHz) δ 151.7 (C, C-4), 144.1 (C, C-1), 140.1 (C, C-2), 136.0 (C, C-3ʹ), 131.4 (C, C-7ʹ), 124.3 (CH, C-6ʹ), 122.8 (CH, C-2ʹ), 120.9 (C, C-5), 112.8 (CH, C-6), 99.3 (CH, C-3), 56.8 (CH3, CH3O-C2), 56.1 (CH3, CH3O-C4), 39.8 (CH2, C-4ʹ), 27.7 (CH2, C-1ʹ), 26.8 (CH2, C-5ʹ), 25.7 (CH3, C-8ʹ), 17.7 (CH3, CH3-C7ʹ), 16.1 (CH3, CH3-C3ʹ); MS m/z 290 (M+) (100), 221 (15), 207 (12), 189 (39), 178 (22), 167 (84) ((M+ − 123), (C9H15·)), 161 (68), 129 (26), 69 (24), 41 (27).
(E)-6-(3,7-Dimethylocta-2,6-dienyl)-2,3,4-trimethoxyphenol (17) and (E)-5-(3,7-Dimethylocta-2,6-dien-1-yl)-2,3,4-trimethoxyphenol (18)
Coupling of 2,3,4-trimethoxyphenol (1.0 g, 5.5 mmol) and geraniol (0.85 g, 5.5 mmol) was carried out in dioxane (20 mL) with BF3·OEt2 (0.46 g, 3.2 mmol) as catalyst. By following the general procedure described above, two fractions were obtained: Fraction I, 150.5 mg of viscous light brown oil (8.6% yield, compound 17); Fraction II, 71.0 mg of viscous dark brown oil (4.1% yield, compound 18). Compound 17: IR (cm−1) 3446; 2966; 1497; 1464; 1125; 1072; 1H NMR (CDCl3, 400.1 MHz) δ 6.44 (1H, s, H-5), 5.47 (1H, s, OH), 5.32 (1H, t, J = 6.8 Hz, H-2ʹ), 5.11 (1H, t, J = 6.6 Hz, H-6ʹ), 3.95 (3H, s, CH3O-C2), 3.87 (3H, s, CH3O-C3), 3.79 (3H, s, CH3O-C4), 3.31 (2H, d, J = 7.2 Hz, H-1ʹ), 2.11–2.09 (2H, m, H-5ʹ), 2.07–2.04 (2H, m, H-4ʹ), 1.72 (3H, s, CH3 C-3ʹ), 1.67 (3H, s, H-8ʹ), 1.60 (3H, s, CH3 C-7ʹ); 13C NMR (CDCl3, 100.6 MHz) δ 146.1 (C, C-4), 140.8 (C, C-1), 140.0 (C, C-2 and C-3), 136.6 (C, C-3ʹ), 131.4 (C, C-7ʹ), 124.2 (CH, C-6ʹ), 122.0 (CH, C-2ʹ), 121.6 (CH, C-6), 108.2 (C, C-5), 61.2 (CH3, CH3O-C2), 60.9 (CH3, CH3O-C3), 56.6 (CH3, CH3O-C4), 39.7 (CH2, C-4ʹ), 27.9 (CH2, C-1ʹ), 26.7 (CH2, C-5ʹ), 25.7 (CH3, C-8ʹ), 17.7 (CH3, CH3-C7ʹ), 16.1 (CH3, CH3-C3ʹ); MS m/z 320 (M+) (71), 235 (15), 198 (18), 197 (100) ((M+ − 123), (C9H15·)), 181 (12), 159 (12), 69 (9), 41 (12). NMR data of compound 17 was consistent with that reported in the literature [6].
Compound 18: IR (cm−1) 3421, 2966, 1590, 1487, 1465, 1198, 1110; 1H NMR (CDCl3, 400.1 MHz) δ 6.50 (1H, s, H-6), 5.46 (1H, s, OH), 5.25 (1H, t, J = 6.2 Hz, H-2ʹ),5.10 (1H, bt, J = 6.8 Hz, H-6ʹ), 3.93 (3H, s, CH3O-C3), 3.92 (3H, s, CH3O-C2), 3.78 (3H, s, CH3O-C4), 3.27 (2H, d, J = 7.2 Hz, H-1ʹ), 2.11–2.08 (2H, m, H-5ʹ), 2.05–2.02 (2H, m, H-4ʹ), 1.70 (3H, s, CH3-C3ʹ), 1.68 (3H, s, H-8ʹ), 1.60 (3H, s, CH3-C7ʹ); 13C NMR (CDCl3, 100.6 MHz) δ 145.9 (C, C-3), 144.9 (C, C-4), 144.5 (C, C-1), 138.0 (C, C-2), 136.2 (C, C-3ʹ), 131.5 (C, C-7ʹ), 130.6 (C, C-5), 124.2 (CH, C-6ʹ), 122.5 (CH, C-2ʹ), 109.5 (CH, C-6), 61.2 (CH3, CH3O-C3), 61.0 (CH3, CH3O-C2), 60.7 (CH3, CH3O-C4), 39.7 (CH2, C-4ʹ), 27.8 (CH2, C-1ʹ), 26.6 (CH2, C-5ʹ), 25.7 (CH3, C-8ʹ), 17.7 (CH3, CH3-C7ʹ), 16.0 (CH3, CH3-C3ʹ); MS m/z 320 (M+) (100), 219 (33), 206 (26), 197 (75) ((M+ − 123), (C9H15·)), 183 (18), 159 (49), 69 (25), 41 (27); HRMS m/z 321.1994 (calcd for C19H28O4, 321.1988).
(E)-6-(3,7-Dimethylocta-2,6-dienyl)-2,3,4-trimethoxyphenyl Acetate (19)
Standard acetylation of compound 17 (48 mg, 0.150 mmol) with Ac2O (1.08 g, 10.6 mmol), DMAP (3.0 mg) and pyridine (1.0 mL) in dichloromethane (20 mL) gives compound 19 as a viscous light brown oil (54 mg, 99.4% yield). Compound 19: IR (cm−1) 2935, 1766, 1587, 1492, 1463, 1124, 1071; 1H NMR (CDCl3, 400.1 MHz) δ 6.49 (1H, s, H-5), 5.22 (1H, t, J = 7.0 Hz, H-2ʹ), 5.10 (1H, t, J = 6.4 Hz, H-6ʹ), 3.87 (3H, s, CH3O-C3), 3.86 (3H, s, CH3O-C2), 3.83 (3H, s, CH3O-C4), 3.18 (2H, d, J = 7.2 Hz, H-1ʹ), 2.31 (3H, s, CH3CO), 2.11–2.07 (2H, m, H-5ʹ), 2.05–2.01 (2H, m, H-4ʹ), 1.69 (3H, s, CH3-C3ʹ), 1.67 (3H, s, H-8ʹ), 1.61 (3H, s, CH3-C7ʹ); 13C NMR (CDCl3, 100.6 MHz) δ 169.4 (C, COCH3), 151.2 (C, C-4), 145.6 (C, C-2), 140.9 (C, C-3), 136.9 (C, C-1), 135.8 (C, C-6), 131.5 (C, C-7ʹ), 128.9 (C, C-3ʹ), 124.1 (CH, C-6ʹ), 121.4 (CH, C-2ʹ), 107.1 (CH, C-5), 61.0 (CH3, CH3O-C2), 60.9 (CH3, CH3O-C3), 56.1 (CH3, CH3O-C4), 39.7 (CH2, C-4ʹ), 28.6 (CH2, C-1ʹ), 26.7 (CH2, C-5ʹ), 25.7 (CH3, C-8ʹ), 20.4 (CH3, COCH3), 17.7 (CH3, CH3-C7ʹ), 16.2 (CH3, CH3-C3ʹ); MS m/z 362 (M+) (11), 320 (32), 235 (16), 221 (10), 197 (100) ((M+ − 123), (C9H15·)), 181 (9), 159 (10), 69 (10), 41 (13).
(E)-3-(3,7-Dimethylocta-2,6-dien-1-yl)-2,4,6-trimethoxyphenol (20) and 3,5-bis((E)-3,7-Dimethylocta-2,6-dien-1-yl)-2,4,6-trimethoxyphenol (21)
Coupling of 2,4,6-trimethoxyphenol (2.58 g, 14.0 mmol) and geraniol (2.49 g, 16.0 mmol) was carried out in dioxane (20 mL) with BF3·OEt2 (0.90 g, 6.3 mmol) as catalyst. Two fractions were obtained: Fraction I, 735.9 mg of viscous brown oil (16.4% yield, compound 20); Fraction II, 901.8 mg of viscous light brown oil (14.0% yield, compound 21). Compound 20: IR (cm−1) 3447, 2965, 2929, 2838, 1668, 1615, 1500, 1454, 1377, 1345, 1245, 1199, 1105, 910, 872, 801; 1H NMR (CDCl3, 400.1 MHz) δ 6.32 (1H, s, H-5), 5.17 (1H, t, J = 6.2 Hz, H-2ʹ), 5.15 (1H, s, OH), 5.06 (1H, t, J = 6.7 Hz, H-6ʹ), 3.88 (3H, s, CH3O-C6), 3.83 (3H, s, CH3O-C2), 3.78 (3H, s, CH3O-C4), 3.30 (2H, d, J = 6.8 Hz, H-1ʹ), 2.06–2.02 (2H, m, H-5ʹ), 1.98–1.94 (2H, m, H-4ʹ), 1.76 (3H, s, CH3-C3ʹ), 1.64 (3H, s, H-8ʹ), 1.57 (3H, s, CH3-C7ʹ); 13C NMR (CDCl3, 100.6 MHz) δ 150.6 (C, C-4), 145.9 (C, C-2), 145.3 (C, C-6), 134.4 (C, C-3ʹ), 133.0 (C, C-1), 131.1 (C, C-7ʹ), 124.4 (CH, C-6ʹ), 123.5 (CH, C-2ʹ), 116.8 (C, C-3), 93.0 (CH, C-5), 60.8 (CH3, CH3O-C2), 56.5 (CH3, CH3O-C4), 56.3 (CH3, CH3O-C6), 39.7 (CH2, C-4ʹ), 26.7 (CH2, C-5ʹ), 25.6 (CH3, C-8ʹ), 22.5 (CH2, C-1ʹ), 17.6 (CH3, CH3-C7ʹ), 16.0 (CH3, CH3-C3ʹ); MS m/z 320 (M+) (77), 251 (22), 237 (16), 221 (31), 219 (33), 197 (100) ((M+ − 123), (C9H15·)), 186 (27), 183 (24), 159 (17), 69 (18), 41 (23). Compound 21: IR (cm−1) 3447, 3086, 2968, 2926, 2856, 1675, 1642, 1604, 1460, 1422, 1376, 1235, 1106, 1031, 920, 835; 1H NMR (CDCl3, 400.1 MHz) δ 5.34 (1H, s, OH), 5.24–5.20 (2H, m, H-2ʹ and H-2ʹʹ), 5.08 (2H, m, H-6ʹ and H-6ʹʹ), 3.81 (6H, s, CH3O-C6 and CH3O-C2), 3.66 (3H, s, CH3O-C4), 3.34 (4H, d, J = 6.6 Hz, H-1ʹ and H-1ʹʹ), 2.07–2.04 (4H, m, H-5ʹ and H-5ʹʹ), 2.01–1.97 (4H, m, H-4ʹ and H-4ʹʹ), 1.77 (6H, s, CH3-C3ʹ and CH3-C3ʹʹ), 1.64 (6H, s, H-8ʹ and H-8ʹʹ), 1.57 (6H, s, CH3-C7ʹ and CH3-C7ʹʹ); 13C NMR (CDCl3, 100.6 MHz) δ 149.5 (C, C-4), 144.4 (C, C-2 and C-6), 139.0 (C, C-1), 134.8 (C, C-3ʹ and C-3ʹʹ), 131.3 (C, C-7ʹ and C-7ʹʹ), 124.4 (C, C-3 and C-5), 124.3 (C, C-6ʹ and C-6ʹʹ), 123.8 (CH, C-2ʹ and C-2ʹʹ), 61.7 (CH3, CH3O-C4), 60.9 (CH3, CH3O-C2 and CH3O-C6), 39.7 (CH2, C-4ʹ and C-4ʹʹ), 26.6 (CH2, C-5ʹ and C-5ʹʹ), 25.6 (CH3, C-8ʹ and C-8ʹʹ), 23.5 (CH2, C-1ʹ and C-1ʹʹ), 17.6 (CH3, CH3-C7ʹ and CH3-C7ʹʹ), 16.2 (CH3, CH3-C3ʹ and CH3-C3ʹʹ); MS m/z 456 (M+) (33), 333 (22) ((M+ − 123), (C9H15·)), 301 (11), 263 (100), 211 (34), 69 (54), 41 (38); HRMS m/z 457.3247 (calcd for C29H44O4, 457.3240).
(E)-3-(3,7-Dimethylocta-2,6-dienyl)-2,4,6-trimethoxyphenyl Acetate (22)
Standard acetylation of compound 20 (65 mg, 0.18 mmol) with Ac2O (0.5 mL, 5.3 mmol), DMAP (5.0 mg) and pyridine (1.0 mL) in dichloromethane (30 mL) gives compound 22 as a viscous yellow oil (72 mg, 99% yield). Compound 22: IR (cm−1) 3402, 2961, 2970, 2843, 1767, 1607, 1496, 1458, 1375, 1201, 1108, 1024, 927; 1H NMR (CDCl3, 400.1 MHz) δ 6.32 (1H, s, H-5), 5.16 (1H, t, J = 6.4 Hz, H-2ʹ), 5.06 (1H, t, J = 6.8 Hz, H-6ʹ), 3.81 (6H, s, CH3O-C4 and CH3O-C6), 3.74 (3H, s, CH3O-C2), 3.29 (2H, d, J = 6.7 Hz, H-1ʹ), 2.33 (3H, s, COCH3), 2.05–2.02 (2H, m, H-5ʹ), 1.98–1.94 (2H, m, H-4ʹ), 1.75 (3H, s, CH3-C3ʹ), 1.64 (3H, s, H-8ʹ), 1.57 (3H, s, CH3-C7ʹ); 13C NMR (CDCl3, 100.6 MHz) δ 169.1 (C, COCH3), 155.8 (C, C-4), 151.3 (C, C-2), 150.2 (C, C-6), 134.5 (C, C-3ʹ), 131.1 (C, C-7ʹ), 127.0 (C, C-1), 124.4 (CH, C-6ʹ), 123.2 (CH, C-2ʹ), 116.4 (C, C-3), 92.5 (CH, C-5), 61.3 (CH3, CH3O-C2), 56.1 (CH3, CH3O-C4), 55.8 (CH3, CH3O-C6), 39.7 (CH2, C-4ʹ), 26.6 (CH2, C-5ʹ), 25.6 (CH3, C-8ʹ), 22.5 (CH2, C-1ʹ), 20.4 (CH3, COCH3), 17.6 (CH3, CH3-C7ʹ), 15.9 (CH3, CH3-C3ʹ).
(E)-5-(3,7-Dimethylocta-2,6-dienyl)-2,4-dimethoxyphenol (16), (E)-2-(3,7-Dimethylocta-2,6-dienyl)-4,5-dimethoxyphenol (23)
Reaction of 2,4,5-trimethoxyphenol (2.46 g, 13.3 mmol) and geraniol (2.07 g, 13.3 mmol) was carried out in dioxane (20 mL) with BF3·OEt2 (0.62g, 4.4 mmol) as catalyst. By following the described general procedure of geranylation reaction, two fractions were obtained: Fraction I, 719.6 mg of viscous brown oil (18.7% yield, compound 23); Fraction II, 677.5 mg of viscous light brown oil (17.3% yield, compound 16). Spectroscopic data of compound 16 was consistent with those found above. Compound 23: IR (cm−1) 2924, 1619, 1512, 1451, 1195; 1H NMR (CDCl3, 400.1 MHz) δ 6.62 (1H, s, H-3), 6.45 (1H, s, H-6), 5.30 (1H, t, J = 7.1 Hz, H-2ʹ), 5.07 (1H, t, J = 7.1 Hz, H-6ʹ), 4.93 (1H, s, OH), 3.82 (6H, s, OCH3 × 2), 3.30 (2H, d, J = 7.0 Hz, H-1ʹ), 2.09 (4H, m, H-5ʹ and H-4ʹ), 1.78 (3H, s, CH3-C3ʹ), 1.68 (3H, s, H-8ʹ), 1.60 (3H, s, CH3-C7ʹ); 13C NMR (CDCl3, 100.6 MHz) δ 148.4 (C, C-1), 148.3 (C, C-5), 142.8 (C, C-4), 138.6 (C, C-3ʹ), 132.0 (C, C-7ʹ), 123.7 (CH, C-6ʹ), 121.9 (CH, C-2ʹ), 117.3 (C, C-2), 113.7 (CH, C-3), 101.2 (CH, C-6), 56.6 (CH3, CH3O-C5), 55.9 (CH3, CH3O-C4), 39.6 (CH2, C-4ʹ), 29.6 (CH2, C-1ʹ), 26.4 (CH2, C-5ʹ), 25.6 (CH3, C-8ʹ), 17.7 (CH3, CH3-C7ʹ), 16.2 (CH3, CH3-C3ʹ); MS m/z 290 (M+) (45), 205 (19), 167 (100) ((M+ − 123), (C9H15·)), 69 (11), 41 (13). NMR data of compound 27 was consistent with those in the literature [6,10].
(E)-2-(3,7-Dimethylocta-2,6-dienyl)-4,5-dimethoxyphenyl Acetate (24)
Standard acetylation of compound 23 (50.8 mg, 0.175 mmol) with Ac2O (100 µL), DMAP (3.0 mg) and pyridine (1.0 mL) in dichloromethane (20 mL) gives compound 24 as a dark brown oil (57.2 mg, 98.3% yield). Compound 24: IR (cm−1) 2973, 2929, 2854, 1765, 1616, 1514, 1448, 1369, 1206, 1101, 1015, 907, 855; 1H NMR (CDCl3, 400.1 MHz) δ 6.70 (1H, s, H-3), 6.56 (1H, s, H-6), 5.23 (1H, t, J = 6.9 Hz, H-2ʹ), 5.10 (1H, t, J = 6.4 Hz, H-6ʹ), 3.84 (3H, s, CH3O-C4), 3.83 (3H, s, CH3O-C5), 3.17 (2H, d, J = 7.0 Hz, H-1ʹ), 2.29 (3H, s, COCH3), 2.09–2.05 (2H, m, H-5ʹ), 2.05–2.04 (2H, m, H-4ʹ), 1.69 (3H, s, CH3-C3ʹ), 1.67 (3H, s, H-8ʹ), 1.60 (3H, s, CH3-C7ʹ); 13C NMR (CDCl3, 100.6 MHz) δ 169.8 (C, COCH3), 147.5 (C, C-5), 146.9 (C, C-4), 141.8 (C, C-1), 136.8 (C, C-3ʹ), 131.5 (C, C-7ʹ), 124.8 (C, C-2), 124.1 (CH, C-6ʹ), 121.8 (CH, C-2ʹ), 112.3 (CH, C-3), 106.1 (CH, C-6), 56.1 (CH3, CH3O-C4), 56.0 (CH3, CH3O-C5), 39.7 (CH2, C-4ʹ), 28.2 (CH2, C-1ʹ), 26.7 (CH2, C-5ʹ), 25.7 (CH3, C-8ʹ), 20.8 (CH3, COCH3), 17.7 (CH3, CH3-C7ʹ), 16.2 (CH3, CH3-C3ʹ); MS m/z 332 (M+) (39), 290 (34), 221 (16), 205 (25), 167 (100) ((M+ − CH3CO − 123), (C9H15·)), 69 (13), 41 (13).
3.2.4. Synthesis of Trimethoxyphenols
In a typical reaction, m-chloroperoxybenzoic acid (mCPBA) is added to a stirred solution of trimethoxybenzaldehyde and sodium bicarbonate in dichloromethane (70 mL). The mixture is stirred at room temperature until the end of the reaction is reached (3 h). Then, the reaction mixture is filtered in a vacuum; the organic phase is washed with NaHCO3 (3 × 50 mL) and water (2 × 50 mL), and dried over anhydrous Na2SO4. The solvent is evaporated under reduced pressure, whereas the crude is re-dissolved in CH2Cl2 (5 mL) and chromatographed on silica gel with petroleum ether/EtOAc of increasing polarity mixtures (50:0→30:20). Trimethoxyphenyl formate is obtained as a dark brown solid. Then, triethylamine (2 mL) is added to a solution of trimethoxyphenyl formate in methanol (50 mL), and the mixture is stirred at room temperature until the end of reaction is verified by TLC (3 h). The solvent is evaporated under reduced pressure; the crude is diluted with ethyl acetate (20 mL) and the organic phase is washed with 5% HCl (2 × 30 mL) and water (2 × 15 mL), and dried over anhydrous Na2SO4. The solvent is evaporated under reduced pressure; the crude is re-dissolved in CH2Cl2 (5 mL) and chromatographed on silica gel with petroleum ether/EtOAc of increasing polarity mixtures (50:0→30:20).
2,3,4-Trimethoxyphenol. mCPBA (3.30 g, 19 mmol) was added to a solution of 2,3,4-trimethoxybenzaldehyde (2.0 g, 10 mmol) and sodium bicarbonate (1.82 g, 21 mmol) in dichloromethane (70 mL). 2,3,4-trimethoxyphenyl formate was obtained as a dark brown solid (1.81 g, 89.2% yield). Triethylamine (2 mL; 1.45 g; 14.0 mmol) was added to a solution of 2,3,4-trimethoxyphenyl formate (1.7 g, 8.0 mmol) in methanol (50 mL). 2,3,4-trimethoxyphenol was obtained as a dark brown oil (1.80 g, 99.6% yield). IR (cm−1) 3421, 2995, 2940, 1601, 1479, 1427, 1360, 1052, 955, 797; 1H NMR (CDCl3, 400.1 MHz) δ 6.62 (1H, d, J = 9.0 Hz, H-6), 6.55 (1H, d, J = 9.0 Hz, H-5), 3.95 (3H, s, CH3O), 3.89 (3H, s, CH3O), 3.80 (3H, s, CH3O); 13C NMR (CDCl3, 100.6 MHz) δ 146.9 (C, C-1), 143.3 (C, C-2), 142.2 (C, C-3), 140.4(C, C-4), 108.5 (CH, C-6), 107.6 (CH, C-5), 61.2 (CH3, CH3O), 60.9 (CH3, CH3O), 55.5 (CH3, CH3O).
2,4,6-Trimethoxyphenol. mCPBA (4.45 g, 25.8 mmol) was added to a solution of 2,4,6-trimethoxybenzaldehyde (3.03 g, 15.4 mmol) and sodium bicarbonate (2.70 g, 32.1 mmol) in dichloromethane (70 mL). 2,4,6-trimethoxyphenyl formate was obtained as a dark brown solid (2.95 g, 90.2% yield). Triethylamine (6 mL, 42.0 mmol) was added to a solution of 2,4,6-trimethoxyphenyl formate (2.95 g, 13.9 mmol) in methanol (75 mL). 2,4,6-trimethoxyphenol was obtained as a dark brown solid (2.44 g, 95.0% yield). mp 60.5–62.9 °C; IR (cm−1) 3422, 3010, 2964, 1626, 1469, 1439, 1361, 1236, 1053, 942, 815; 1H NMR (CDCl3, 400.1 MHz) δ 6.16 (2H, s, H-3 and H-5), 3.83 (6H, s, 2 × CH3O), 3.74 (3H, s, CH3O); 13C NMR (CDCl3, 100.6 MHz) δ 153.0 (C, C-1), 147.2 (C, C-2 and C-6), 128.9 (C, C-4), 91.7 (CH, C-3 and C-5), 56.1 (CH3, 2 × CH3O), 55.6 (CH3, CH3O).
2,4,5-Trimethoxyphenol. mCPBA (5.0 g, 29.0 mmol) was added to a solution of 2,4,5-trimethoxybenzaldehyde (3.01 g, 15.0 mmol) and sodium bicarbonate (1.8 g, 32.0 mmol) in dichloromethane (70 mL). 2,4,5-trimethoxyphenyl formate was obtained as a dark brown solid (2.99 g, 91.6% yield). Triethylamine (4 mL, 28.0 mmol) was added to a solution of 2,4,5-trimethoxyphenyl formate (2.99 g, 14.0 mmol) in methanol (75 mL). 2,4,5-trimethoxyphenol was obtained as a dark brown solid (2.46 g, 94.3% yield). mp 83.5–87.0 °C; IR (cm−1) 3480, 3067, 2996, 1621, 1479, 1441, 1375, 1209, 1027, 823M; 1H NMR (CDCl3, 400.1 MHz) δ 6.61 (1H, s, H-6), 6.57 (1H, s, H-3), 5.29 (1H, s, OH), 3.87 (3H, s, CH3O), 3.84 (6H, s, 2 × CH3O); 13C NMR (CDCl3, 100.6 MHz) δ 144.2 (C, C-2), 143.6 (C, C-5), 143.1 (C, C-1), 141.9 (C, C-4), 104.8 (CH, C-3), 100.1 (CH, C-6).
3.3. Fungal Isolate and Culture Condition
In this study, the strain UK of B. cinerea was used in all experiments. This strain was isolated from a naturally infected grape (Vitis vinifera) and was maintained on potato dextrose agar medium (PDA; Difco, Detroit, MI, USA) at 4 °C. The inoculum of the pathogen was grown on PDA in photoperiod of 16 h light/8 h dark at 23 °C for five days.
3.4. Effect of the Compounds on the Mycelial Growth of B. cinerea in Vitro
The anti-phytophatogenic activities of compounds, the negative control (C−), and the positive control (C+, commercial fungicide Captan) were assessed using the radial growth test technique on PDA medium [18]. All tested compounds were dissolved in ethanol (1%) and water, added at different amounts to obtain final concentrations of 50, 150 and 250 mg/L in PDA medium. Negative and positive control experimental conditions for the growth of mycelia of B. cinerea were included. Negative control conditions means PDA medium containing 1% ethanol, whereas positive control indicates PDA medium including the commercial fungicide Captan at the same concentration specified for the compounds of interest.
A plug (4 mm) of PDA medium with five-day-old mycelium colonies of the pathogen was placed at the center of a Petri dish with PDA medium with or without studied compounds. Subsequently, they were incubated under controlled conditions of temperature to 23 °C and photoperiod 16 h light/8 h for 48 h. The percentage of inhibition was determined for each compound by expressing the area of B. cinerea as a percentage of the negative control. The evaluation was conducted through measuring diameters of mycelial growth after 72 h of incubation. The inhibition percentages of mycelial growth were calculated for each compound and compared with the negative control according to Hou et al. [19]. All treatments were performed independently three times in triplicate.
4. Conclusions
The evaluation of the inhibitory effect of a series of geranylphenols on the mycelial growth of the plant pathogen B. cinerea was made in vitro after 72 h of incubation using the radial growth test technique on PDA medium, and included a commercial fungicide as a positive control. The results show that most of the compounds exhibit activities against the mycelial growth of B. cinerea in the range of 51% and 98% between 150 and 250 ppm, whereas compounds 6, 8, 10, 12, 15 and 22 are less active with a percentage of inhibition lower than 50%. The analysis of the relation between the activity and the structure of compounds indicates that the biological activity depends strongly on the number of methoxy, hydroxyl, acetate, and prenyl groups. For geranylphenols, the activity improves from 40% to 90% by increasing the number of methoxy groups in the aromatic ring. The position of the methoxy relative to the hydroxyl group has also a slight effect on the activity. On the other hand, for geranylacetates, the opposite effect is observed, i.e., the activity decreases with an increasing number of methoxy groups. Finally, the activity of digeranyl derivatives is lower than that exhibited by the respective monogeranyl compounds. These results confirm that the activity of geranyl compounds against the mycelial growth of B. cinerea is mainly determined by the geranyl chain.
These structural effects may be related to differences in the response of the plant pathogen by activating or inhibiting different mechanisms (collapse of the hyphae, degradation of fungus cellular wall, or loss of turgidity) which may lead to growth arrest of the pathogenic hyphae. An alternative explanation could be the different biodisponibility of these compounds due to differences in water solubility. Thus, additional studies are required to elucidate what is the reason for the observed effects of these compounds that affect the fungus growth.
Supplementary Materials
Supplementary materials can be found at http://www.mdpi.com/1422-0067/16/08/19130/s1.
Acknowledgments
The authors thank to FONDECYT (grant No. 1120996) and the Dirección General de Investigación y Postgrado (DGIP-USM grant No. 131305) of Universidad Técnica Federico Santa María.
Author Contributions
Luis Espinoza supervised the whole work. María I. Chávez and Mauricio Soto performed the synthesis of all compounds. Lautaro Taborga collaborated in the synthesis and structure determination of geranylphenols by spectroscopic methods. Camila Bay carried out the study of mycelial growth of B. cinerea under Katy Diaz’s supervision. Hugo Peña-Cortés, Katy Diaz, Lautaro Taborga, Luis Espinoza and, Andrés F. Olea collaborated in the discussion and interpretation of the results. Andrés F. Olea wrote the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- De Rosa, S.; de Giulio, A.; Iodice, C. Biological effects of prenylated hydroquinones—Structure–activity relationship studies in antimicrobial, brine shrimp, and fish lethality assays. J. Nat. Prod. 1994, 57, 1711–1716. [Google Scholar] [CrossRef] [PubMed]
- Danelutte, A.P.; Lago, J.H.G.; Young, M.C.M.; Kato, M.J. Antifungal flavanones and prenylated hydroquinones from Piper crassinervium Kunth. Phytochemistry 2003, 64, 555–559. [Google Scholar] [CrossRef]
- Malami, I. Prenylated benzoic acid derivates from Piper species as source of anti-infective agents. Int. J. Pharm. Sci. Res. 2012, 3, 1554–1559. [Google Scholar]
- Manners, G.D. The hydroquinone terpenoids of Cordia alliodora. J. Chem. Soc. Perkin Trans. I 1977, 1977, 405–410. [Google Scholar] [CrossRef]
- Tziveleka, L.-A.; Kourounakis, A.P.; Kourounakis, P.N.; Roussis, V.; Vagias, V. Antioxidant potential of natural and synthesised polyprenylated hydroquinones. Bioorg. Med. Chem. 2002, 10, 935–939. [Google Scholar] [CrossRef]
- Fedorov, S.N.; Radchenko, O.S.; Shubina, L.K.; Balaneva, N.N.; Bode, A.M.; Stonik, V.A.; Dong, Z.G. Evaluation of cancer-preventive activity and structure–activity relationships of 3-demethylubiquinone Q2, isolated from the ascidian Aplidium glabrum, and its synthetic analogs. Pharm. Res. 2006, 23, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, L.F.; Lago, J.H.G.; Tanizaki, T.M.; di Mascio, P.; Kato, M.J. Antioxidant activity of prenylated hydroquinone and benzoic acid derivatives from Piper crassinervium Kunth. Phytochemistry 2006, 67, 1838–1843. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, L.; Baeza, E.; Catalan, K.; Osorio, M.; Carrasco, H.; Cuellar, M.; Villena, J. Synthesis and antitumor activity of diterpenylhydroquinone derivatives of natural Ent-Labdanes. Molecules 2010, 15, 6502–6511. [Google Scholar]
- Espinoza, L.; Taborga, L.; Diaz, K.; Olea, A.F.; Peña-Cortes, H. Synthesis of linear geranylphenols and their effect on mycelial growth of plant pathogen Botrytis cinerea. Molecules 2014, 19, 1512–1526. [Google Scholar] [CrossRef] [PubMed]
- Baeza, E.; Catalan, K.; Villena, J.; Carrasco, H.; Cuellar, M.; Espinoza, L. Synthesis and cytotoxic activity of geranylmethoxyhydroquinone derivatives. J. Chil. Chem. Soc. 2012, 57, 1219–1223. [Google Scholar] [CrossRef]
- Taborga, L.; Vergara, A.; Osorio, M.; Carvajal, M.; Madrid, A.; Marilaf, F.; Carrasco, H.; Espinoza, L. Synthesis and NMR structure determination of new linear geranylphenols by direct geranylation of activated phenols. J. Chil. Chem. Soc. 2013, 58, 1790–1796. [Google Scholar] [CrossRef]
- Kumano, T.; Richard, S.B.; Noel, J.P.; Nishiyama, M.; Kuzuyama, T. Chemoenzymatic syntheses of prenylated aromatic small molecules using streptomyces prenyltransferases with relaxed substrate specificities. Bioorg. Med. Chem. 2008, 16, 8117–8126. [Google Scholar] [CrossRef] [PubMed]
- Lopez, S.N.; Lopes, A.A.; Batista, J.M., Jr.; Flausino, O., Jr.; Bolzani, V.S.; Kato Massuo, J.; Furlan, M. Geranylation of benzoic acid derivatives by enzymatic extracts from Piper crassinervium (Piperaceae). Bioresour. Technol. 2010, 101, 4251–4260. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yan, Z.; Lei, Y.; Sheng, K.; Yao, Q.; Lu, K.; Yu, P. Concise synthesis of prenylated and geranylated chalcone natural products by regiospecific iodination and Suzuki coupling reactions. Tetrahedron Lett. 2014, 55, 897–899. [Google Scholar] [CrossRef]
- Baeza, E.; Catalan, K.; Pena-Cortes, H.; Espinoza, L.; Villena, J.; Carrasco, H. Synthesis of geranylhydroquinone derivatives with potential cytotoxic activity. Quim. Nova 2012, 35, 523–526. [Google Scholar] [CrossRef]
- Khalil, I.M.; Barker, D.; Copp, B.R. Biomimetic synthesis of thiaplidiaquinones A and B. J. Nat. Prod. 2012, 75, 2256–2260. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.T.; Pearce, A.; Januario, A.H.; Page, M.J.; Kaiser, M.; McLaughlin, R.J.; Harper, J.L.; Webb, V.L.; Barker, D.; Copp, B.R. Anti-inflammatory and antimalarial meroterpenoids from the New Zealand ascidian aplidium scabellum. J. Org. Chem. 2011, 76, 9151–9156. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, L.; Espinoza, P.; Urzua, A.; Vivanco, M.; Cotoras, M. In vitro antifungal activity of the diterpenoid 7á-hydroxy-8(17)-labden-15-oic acid and its derivatives against Botrytis cinerea. Molecules 2009, 14, 1966–1979. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.; Yang, R.; Zhang, C.; Zhu, L.; Miao, F.; Yang, X.; Zhou, L. 2-(Substituted phenyl)-3,4-dihydroisoquinolin-2-iums as novel antifungal lead compounds: Biological evaluation and structure–activity relationships. Molecules 2013, 18, 10413–10424. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).