
Cellular Pathways in Response to Ionizing Radiation and Their Targetability for Tumor Radiosensitization


Abstract
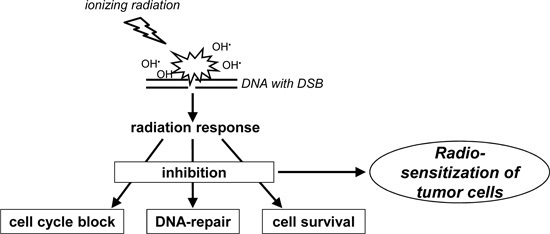
:
1. Introduction
2. Properties and Biological Effects of Ionizing Radiation
3. Signaling Pathways of the Cellular Radiation Response
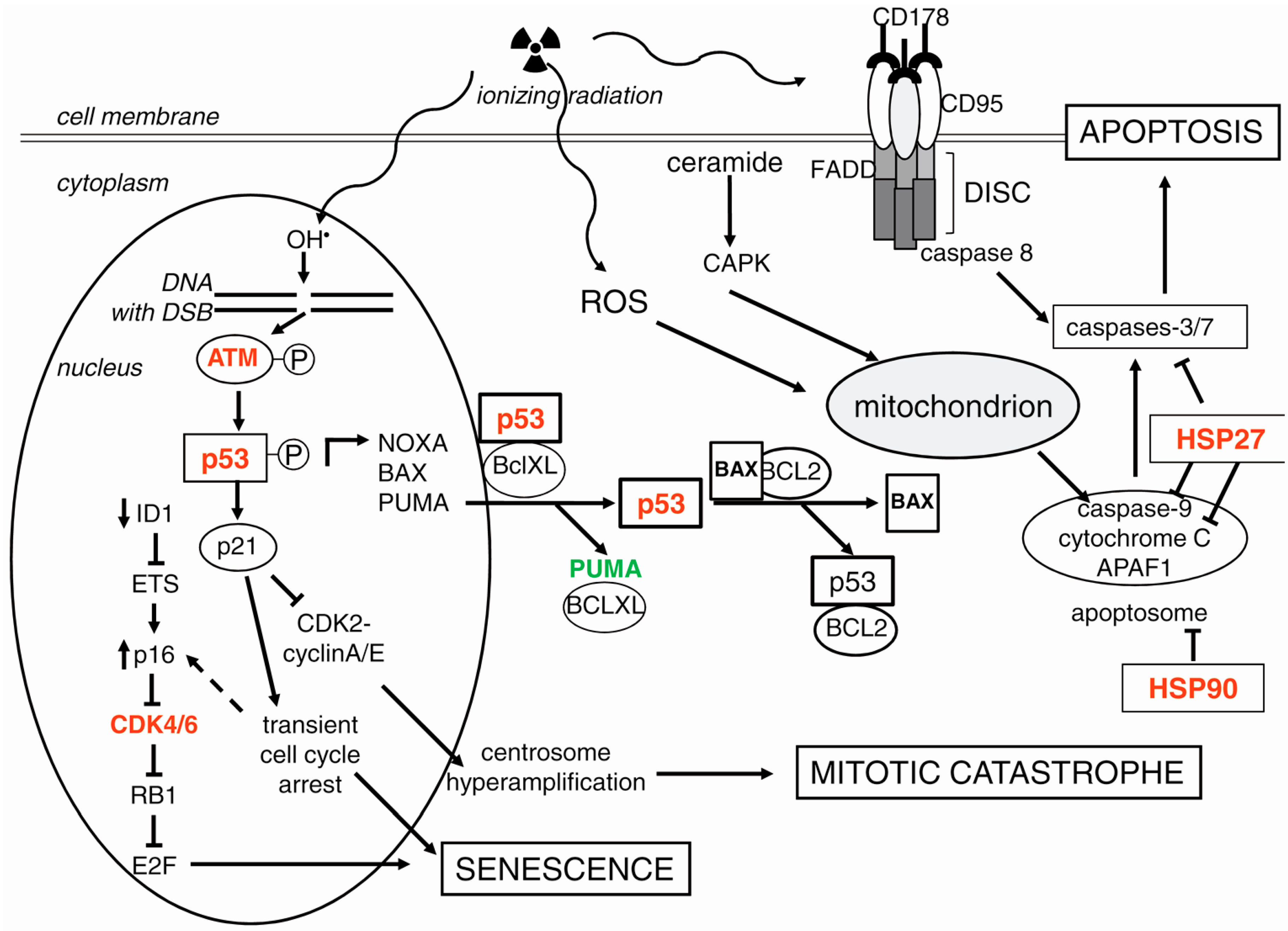
3.1. p53 as a Guardian of Genomic Stability: Repair or Cell Death

3.2. Signaling for Double-Strand Break Repair
3.3. Apoptosis

3.3.1. The Intrinsic Apoptotic Pathway
3.3.2. The Extrinsic Apoptotic Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Substance | Radiosensitization of Cell Line/Tumor Entity | Comments | Reference |
|---|---|---|---|---|
| ATM | CP466722 | HeLa (cervix carcinoma) | only in vitro results | [69] |
| ATM | KU-55933 | various tumor cell lines HeLa, MCF-7, ovary cancer cells, bladder cancer cell, etc. | up to now no clinical trial | [70,71,72,73] |
| ATM | KU-60019 | glioblastoma and glioblastoma-initiating cells | successor of KU-55933 | [74,75,76] |
| increased radiosensitivity in p53-deficient cells | [75,77] | |||
| ATR | NU6027 | MCF-7 (breast carcinoma) | increased effects in combination with various chemotherapeutic drugs | [78] |
| BCR-ABL | imatinib | RT112 (transitional bladder cell carcinoma), H1299 (lung carcinoma), PANC1 (pancreatic adenocarcinoma), PC3 (prostate adenocarcinoma) | no increased radiation gut toxicity in an animal model with xenotransplantation of PC3 | [79] |
| CDK1, 2, 4 | flavopiridol (alvocidib) | various cancer cell lines and xenografts | successful clinical studies in combination with standard chemotherapeutic regimens | [80,81,82] |
| CDK1, 2, 9 | AZD5438 | A549, H1299, and H460 (non-small cell lung cancer) | discontinued clinical development due to low tolerability in phase II studies | [83] |
| CDK4/6 | palbociclib (PD0332991) | human glioblastoma U87 intracranial xenografts and brainstem glioma mouse model | FDA approval for potential treatment of breast cancer | [84,85] |
| CHK1 | UCN-01 | A549 (lung carcinoma), NCI-H460 (large-cell lung carcinoma), K562 (erythroblastoid leukemia cell line), glioblastoma stem-like cells in vitro and in xenografts | no effect on BEAS-2B (immortalized normal bronchial epithelial cell line) enhanced radiosensitivity of lung cancer cell lines in combination with celecoxib and of head and neck squamous cell carcinoma by combination with ATRA (8 all-trans retinoic acid) | [86,87,88,89] |
| CHK2 | PV1019 | MCF-7 (breast carcinoma), U251 (glioblastoma) | radioprotective in mouse thymocytes | [90] |
| CHK2 | XL-844 | HT-29 (colon carcinoma) | only one in vitro study with irradiation | [91] |
| EGFR | cetuximab | several clinical trials combined with standard chemoradiotherapy | FDA approval only for treatment of locally advanced head and neck cancer in combination with radiation | [92,93] |
| HDAC | LBH589 (panobinostat) | prostate cancer and glioblastoma cells | obatoclax, inhibitor of BCL-2, for increased radiosensitization of glioblastoma cells resistant to LBH589 and SAHA | [94,95,96] |
| HDAC | PCI-24781 (abexinostat) | cervical and colon carcinoma cells, nasopharyngeal carcinoma cells in vitro and in xenografts | two phase I studies as mono- or combination (with doxorubicin) therapy in patients with metastatic carcinoma, lymphomas | [97,98] |
| [99,100] | ||||
| HDAC | SAHA (vorinostat) | LN18 and U251 (glioblastoma cells), osteosarcoma (OS) and rhabdomyosarcoma cell lines and OS xenografts | two finished phase I trials to determine the maximum well-tolerated dose | [101,102,103,104,105,106] |
| HSP90 | 17-AAG (geldanamycin) | DU145 (prostate carcinoma), SQ-5 (lung squamous carcinoma), T98G and U87-MG (glioblastoma), esophageal cancer cells | enhanced radiosensitization in combination with the PARP inhibitor olaparib; no radiosensitizing effect in normal tissue cells | [107,108,109] |
| HSP90 | 17-DMAG | MiaPaCa (pancreatic carcinoma), NSCLC cell lines | no radiosensitizing effect in normal tissue cells; radioprotective in PBMC | [110,111] |
| HSP90 | NVP-AUY922, NVP-BEP800, NVP-HSP990 | various tumor cell lines: A549, GaMG, HT 1080, SNB19, MIA PaCa-2 and U251 | no clinical trial | [112,113] |
| HSP90 | STA-9090 (ganetespib) | oropharyngeal squamous cell carcinoma (SCC) tissue samples HCT 116 (colorectal cancer cell line) | effective also in combination with cisplatin and in xenografts combined with capecitabine two ongoing clinical trials in combination with chemoradiation | [114,115] |
| MDM2 | nutlin-3a | prostate cancer cell lines, NSCLC cells | activation of p53 resulted in increased senescence | [116,117,118] |
| MDM2 | PXN727 | HCT116 (colon cancer cell line) | upregulation of secretion of HSP70 | [118] |
| MRN-complex | telomelysin (OBP-301) | orthotopic human esophageal cancer xenograft model | ongoing analysis of the safety and efficacy of telomelysin in patients with hepatocellular carcinoma | [119] |
| p53 | PRIMA-1MET MIRA-1 | SCLC cell lines with mutant p53 in vitro and as xenografts in mouse experiments | reactivation of p53 and radiosensitization | [30] |
| PRKDC | NU7441 | C4-2 and PC3 (prostate carcinoma), MCF-7 SW620 (colon carcinoma) cell culture and xenografts | increased radiosensitization of MCF-7 cells in combination with K55933 no effect in PRKDC-deficient V3 cells | [120,121,122] |
3.3.3. The Membrane Stress Apoptotic Pathway
3.4. Mitotic Catastrophe
3.5. Senescence
3.6. Autophagy
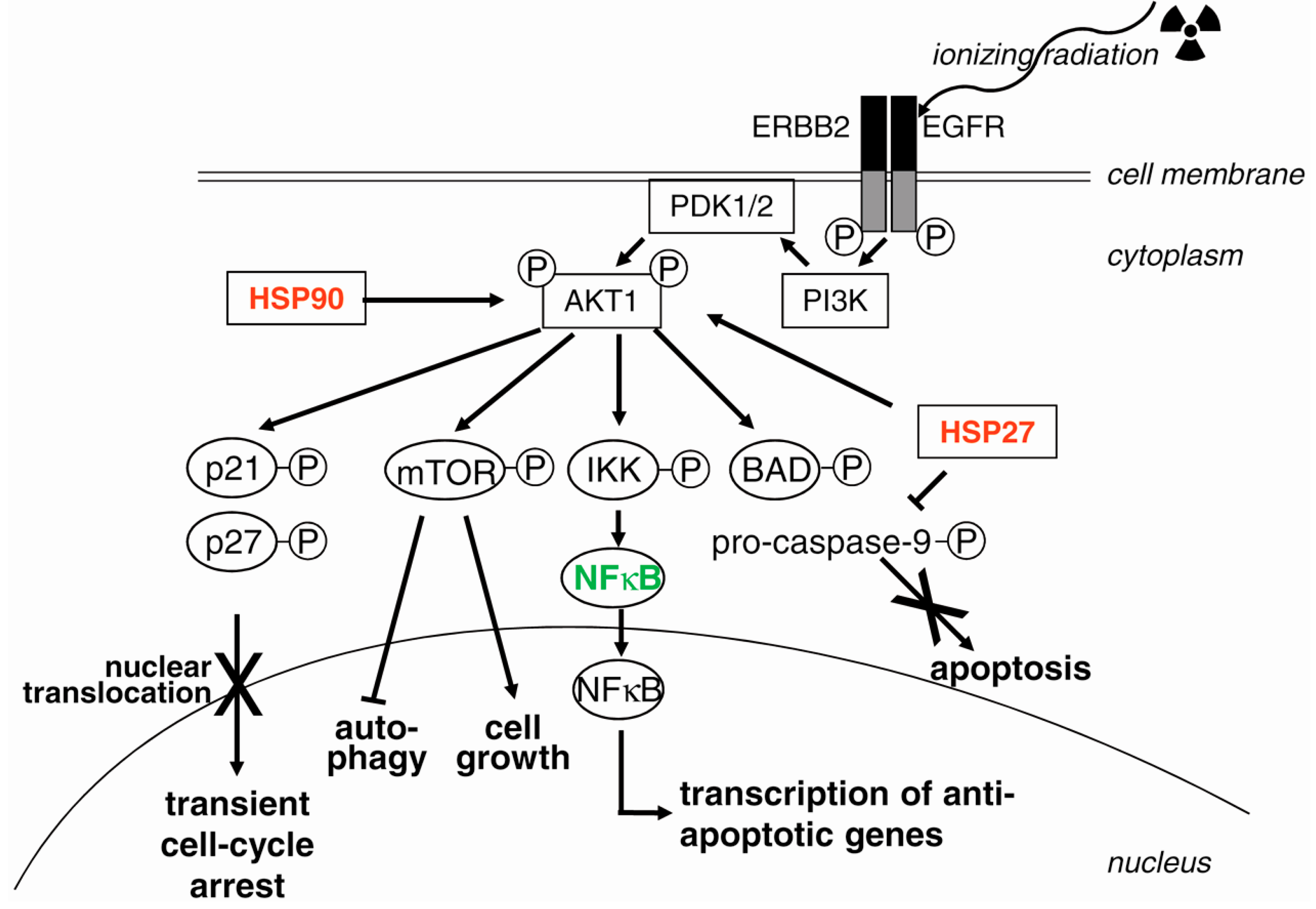
3.7. EGFR (Epidermal Growth Factor Receptor) and PI3K (Phosphatidylinositol 3-Kinase)
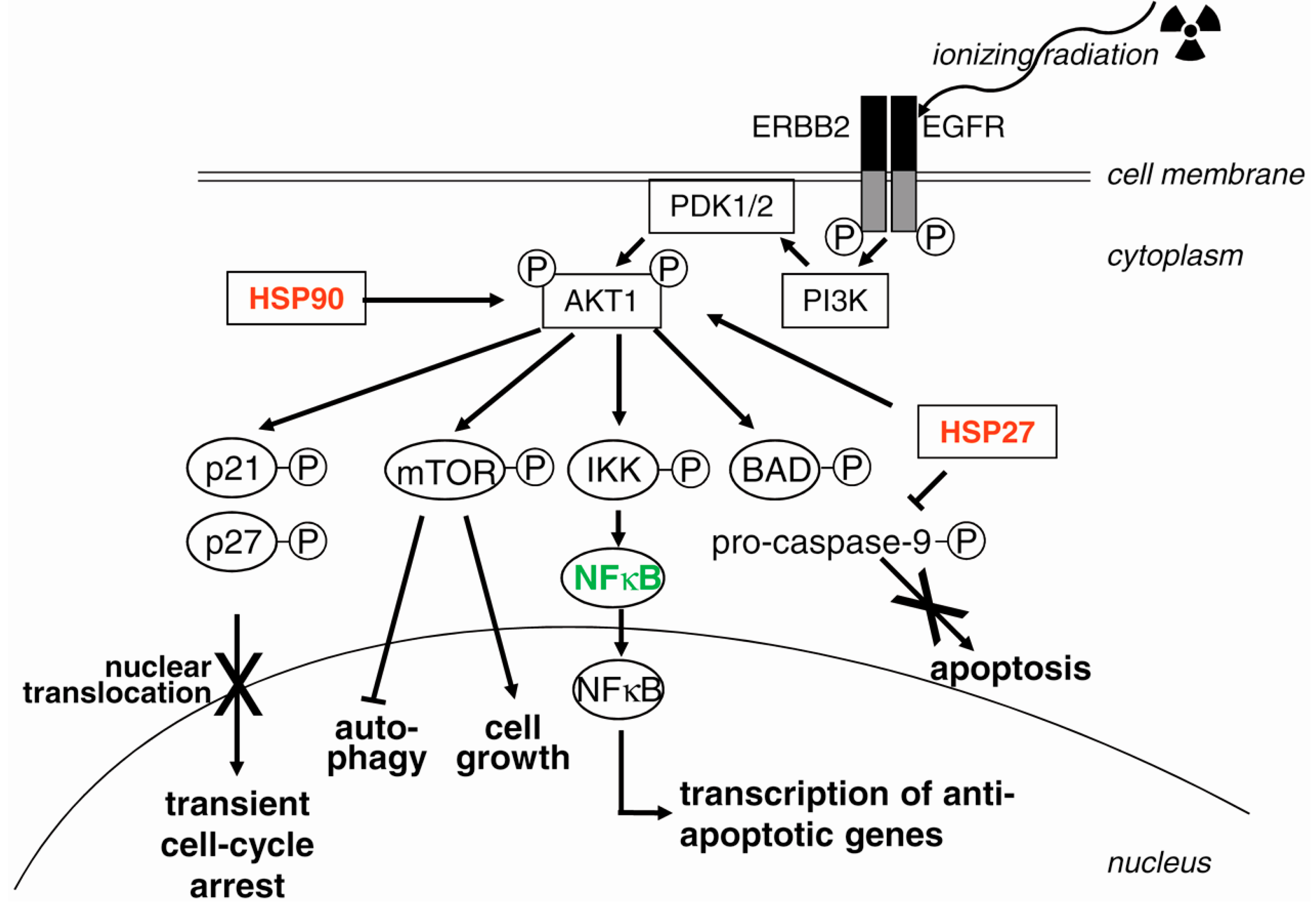

4. Targets for Radiosensitization
4.1. Inhibition of Cell Cycle Control
4.1.1. Inhibition of CHK1 or CHK2 (Checkpoint Kinase-1 and -2)
4.1.2. Inhibition of Cyclin-Dependent Kinases
4.2. Inhibition of DNA Repair
4.2.1. Targeting the MRN Complex
4.2.2. Targeting ATM (Ataxia-Telangiectasia Mutated) or ATR (Ataxia-Telangiectasia and RAD3-Related)
4.2.3. Inhibition of Homologous Recombination (HR)
4.2.4. Inhibition of Non-Homologous End Joining (NHEJ)
4.2.5. Histone Deacetylase (HDAC) Inhibitors
4.3. Suppression of Survival Pathways
4.3.1. Targeting EGFR
4.3.2. Induction of Senescence
4.3.3. Inhibition of Autophagy
4.4. Reactivation of p53
5. Perspectives
6. Conclusions
Conflicts of Interest
References
- Delaney, G.; Jacob, S.; Featherstone, C.; Barton, M. The role of radiotherapy in cancer treatment: Estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer 2005, 104, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.S.; Duke, S.; Jena, R.; Williams, M.V.; Burnet, N.G. Advances in radiotherapy. BMJ 2012, 345, e7765. [Google Scholar] [CrossRef] [PubMed]
- Bentzen, S.M.; Heeren, G.; Cottier, B.; Slotman, B.; Glimelius, B.; Lievens, Y.; van den Bogaert, W. Towards evidence-based guidelines for radiotherapy infrastructure and staffing needs in Europe: The ESTRO QUARTS project. Radiother. Oncol. 2005, 75, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Peters, L.J.; Withers, H.R.; Thames, H.D., Jr.; Fletcher, G.H. Tumor radioresistance in clinical radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 1982, 8, 101–108. [Google Scholar] [CrossRef]
- West, C.M.; Davidson, S.E.; Hendry, J.H.; Hunter, R.D. Prediction of cervical carcinoma response to radiotherapy. Lancet 1991, 338, 818. [Google Scholar] [CrossRef]
- Holthusen, H. Erfahrungen über die verträglichkeit von röntgenstrahlen und deren nutzanwendung zur verhütung von schaden. Strahlenther. Onkol. 1936, 57, 30–36. [Google Scholar]
- Bhide, S.A.; Nutting, C.M. Recent advances in radiotherapy. BMC Med. 2010, 8. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.S.; Fakiris, A.J.; Chang, E.L.; Mayr, N.A.; Wang, J.Z.; Papiez, L.; Teh, B.S.; McGarry, R.C.; Cardenes, H.R.; Timmerman, R.D. Stereotactic body radiation therapy: A novel treatment modality. Nat. Rev. Clin. Oncol. 2010, 7, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Tipton, K.; Launders, J.H.; Inamdar, R.; Miyamoto, C.; Schoelles, K. Stereotactic body radiation therapy: Scope of the literature. Ann. Intern. Med. 2011, 154, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.W. Cancer and radiation therapy: Current advances and future directions. Int. J. Med. Sci. 2012, 9, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Purdy, J.A. Dose to normal tissues outside the radiation therapy patient’s treated volume: A review of different radiation therapy techniques. Health Phys. 2008, 95, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Wenz, F.; Tiefenbacher, U.; Willeke, F.; Weber, K.J. The search for therapeutic gain in radiation oncology. Onkologie 2001, 24 (Suppl. 5), 51–55. [Google Scholar] [CrossRef] [PubMed]
- Verellen, D.; Ridder, M.D.; Linthout, N.; Tournel, K.; Soete, G.; Storme, G. Innovations in image-guided radiotherapy. Nat. Rev. Cancer 2007, 7, 949–960. [Google Scholar] [CrossRef] [PubMed]
- Bentzen, S.M.; Constine, L.S.; Deasy, J.O.; Eisbruch, A.; Jackson, A.; Marks, L.B.; Ten Haken, R.K.; Yorke, E.D. Quantitative Analyses of Normal Tissue Effects in the Clinic (QUANTEC): An introduction to the scientific issues. Int. J. Radiat. Oncol. Biol. Phys. 2010, 76, S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Emami, B.; Lyman, J.; Brown, A.; Coia, L.; Goitein, M.; Munzenrider, J.E.; Shank, B.; Solin, L.J.; Wesson, M. Tolerance of normal tissue to therapeutic irradiation. Int. J. Radiat. Oncol. Biol. Phys. 1991, 21, 109–122. [Google Scholar] [CrossRef]
- Maier, P.; Wenz, F.; Herskind, C. Radioprotection of normal tissue cells. Strahlenther. Onkol. 2014, 190, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Herskind, C.; Wenz, F. Radiobiological aspects of intraoperative tumour-bed irradiation with low-energy X-rays (LEX-IORT). Transl. Cancer Res. 2014, 3, 3–17. [Google Scholar]
- Herskind, C.; Westergaard, O. Inactivation of a single eucaryotic gene irradiated in vitro in transcriptionally active chromatin form. Radiat. Res. 1986, 106, 331–344. [Google Scholar] [CrossRef]
- Herskind, C.; Westergaard, O. Variable protection by OH scavengers against radiation-induced inactivation of isolated transcriptionally active chromatin: The influence of secondary radicals. Radiat. Res. 1988, 114, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.J.; Giaccia, A.J. Radiobiology for the Radiologist, 7th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2012; p. 576. [Google Scholar]
- Prasad, R.; Beard, W.A.; Batra, V.K.; Liu, Y.; Shock, D.D.; Wilson, S.H. A review of recent experiments on step-to-step “hand-off” of the DNA intermediates in mammalian base excision repair pathways. Mol. Biol. 2011, 45, 586–600. [Google Scholar] [CrossRef]
- Shiloh, Y. ATM and related protein kinases: Safeguarding genome integrity. Nat. Rev. Cancer 2003, 3, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196. [Google Scholar] [CrossRef] [PubMed]
- Stracker, T.H.; Roig, I.; Knobel, P.A.; Marjanovic, M. The ATM signaling network in development and disease. Front. Genet. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Stokes, M.P.; Rush, J.; Macneill, J.; Ren, J.M.; Sprott, K.; Nardone, J.; Yang, V.; Beausoleil, S.A.; Gygi, S.P.; Livingstone, M.; et al. Profiling of UV-induced ATM/ATR signaling pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 19855–19860. [Google Scholar] [CrossRef] [PubMed]
- Lukas, J.; Lukas, C.; Bartek, J. Mammalian cell cycle checkpoints: Signalling pathways and their organization in space and time. DNA Repair 2004, 3, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.B.; Bartek, J. Targeting the checkpoint kinases: Chemosensitization versus chemoprotection. Nat. Rev. Cancer 2004, 4, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, H.C.; Schumacher, B. The p53 network: Cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 2012, 28, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Scott, A.; Murray, D. New insights into p53 signaling and cancer cell response to DNA damage: Implications for cancer therapy. J. Biomed. Biotechnol. 2012, 2012. [Google Scholar] [CrossRef]
- Pflaum, J.; Schlosser, S.; Muller, M. p53 Family and cellular stress responses in cancer. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef]
- Zhang, X.P.; Liu, F.; Wang, W. Two-phase dynamics of p53 in the DNA damage response. Proc. Natl. Acad. Sci. USA 2011, 108, 8990–8995. [Google Scholar] [CrossRef] [PubMed]
- Gudkov, A.V.; Komarova, E.A. The role of p53 in determining sensitivity to radiotherapy. Nat. Rev. Cancer 2003, 3, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P. Cancer p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, W.C.; Denning, M.F. P21Waf1 control of epithelial cell cycle and cell fate. Crit. Rev. Oral Biol. Med. 2002, 13, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Smits, V.A.; Klompmaker, R.; Vallenius, T.; Rijksen, G.; Makela, T.P.; Medema, R.H. p21 inhibits Thr161 phosphorylation of CDC2 to enforce the G2 DNA damage checkpoint. J. Biol. Chem. 2000, 275, 30638–30643. [Google Scholar] [CrossRef] [PubMed]
- Boutros, R.; Lobjois, V.; Ducommun, B. CDC25 phosphatases in cancer cells: Key players? Good targets? Nat. Rev. Cancer 2007, 7, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Pannicke, U.; Schwarz, K.; Lieber, M.R. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell 2002, 108, 781–794. [Google Scholar] [CrossRef]
- Bartek, J.; Mistrik, M.; Bartkova, J. Androgen receptor signaling fuels DNA repair and radioresistance in prostate cancer. Cancer Discov. 2013, 3, 1222–1224. [Google Scholar] [CrossRef] [PubMed]
- Polkinghorn, W.R.; Parker, J.S.; Lee, M.X.; Kass, E.M.; Spratt, D.E.; Iaquinta, P.J.; Arora, V.K.; Yen, W.F.; Cai, L.; Zheng, D.; et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 2013, 3, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.F.; Schiewer, M.J.; Dean, J.L.; Schrecengost, R.S.; de Leeuw, R.; Han, S.; Ma, T.; Den, R.B.; Dicker, A.P.; Feng, F.Y.; et al. A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discov. 2013, 3, 1254–1271. [Google Scholar] [CrossRef] [PubMed]
- Spratt, D.E.; Evans, M.J.; Davis, B.J.; Doran, M.G.; Lee, M.X.; Shah, N.; Wongvipat, J.; Carnazza, K.E.; Klee, G.G.; Polkinghorn, W.; et al. Androgen receptor upregulation mediates radioresistance after ionizing radiation. Cancer Res. 2015, 75, 4688–4696. [Google Scholar] [CrossRef] [PubMed]
- Chernikova, S.B.; Game, J.C.; Brown, J.M. Inhibiting homologous recombination for cancer therapy. Cancer Biol. Ther. 2012, 13, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Shivji, M.K.; Davies, O.R.; Savill, J.M.; Bates, D.L.; Pellegrini, L.; Venkitaraman, A.R. A region of human BRCA2 containing multiple BRC repeats promotes RAD51-mediated strand exchange. Nucleic Acids Res. 2006, 34, 4000–4011. [Google Scholar] [CrossRef] [PubMed]
- Buis, J.; Stoneham, T.; Spehalski, E.; Ferguson, D.O. MRE11 regulates CTIP-dependent double-strand break repair by interaction with CDK2. Nat. Struct. Mol. Biol. 2012, 19, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Bunting, S.F.; Callen, E.; Wong, N.; Chen, H.T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 inhibits homologous recombination in BRCA1-deficient cells by blocking resection of DNA breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; van der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010, 17, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.D.; Yu, V.K.; Georgakilas, A.G.; Koumenis, C.; Park, J.H.; Carlson, D.J. Effects of radiation quality and oxygen on clustered DNA lesions and cell death. Radiat. Res. 2011, 176, 587–602. [Google Scholar] [CrossRef] [PubMed]
- Takasawa, R.; Nakamura, H.; Mori, T.; Tanuma, S. Differential apoptotic pathways in human keratinocyte HaCaT cells exposed to UVB and UVC. Apoptosis 2005, 10, 1121–1130. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H. p53: Death star. Cell 2000, 103, 691–694. [Google Scholar] [CrossRef]
- Cory, S.; Adams, J.M. The BCL2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Dogu, Y.; Diaz, J. Mathematical model of a network of interaction between p53 and BCL-2 during genotoxic-induced apoptosis. Biophys. Chem. 2009, 143, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Kuribayashi, K.; Finnberg, N.; Jeffers, J.R.; Zambetti, G.P.; El-Deiry, W.S. The relative contribution of pro-apoptotic p53-target genes in the triggering of apoptosis following DNA damage in vitro and in vivo. Cell Cycle 2011, 10, 2380–2389. [Google Scholar] [CrossRef] [PubMed]
- Oda, E.; Ohki, R.; Murasawa, H.; Nemoto, J.; Shibue, T.; Yamashita, T.; Tokino, T.; Taniguchi, T.; Tanaka, N. Noxa, a BH3-only member of the BCL-2 family and candidate mediator of p53-induced apoptosis. Science 2000, 288, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Dejean, L.M.; Martinez-Caballero, S.; Manon, S.; Kinnally, K.W. Regulation of the mitochondrial apoptosis-induced channel, MAC, by BCL-2 family proteins. Biochim. Biophys. Acta 2006, 1762, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Ogura, A.; Oowada, S.; Kon, Y.; Hirayama, A.; Yasui, H.; Meike, S.; Kobayashi, S.; Kuwabara, M.; Inanami, O. Redox regulation in radiation-induced cytochrome c release from mitochondria of human lung carcinoma A549 cells. Cancer Lett. 2009, 277, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Richter, C.; Frei, B. Ca2+ release from mitochondria induced by prooxidants. Free Radic. Biol. Med. 1988, 4, 365–375. [Google Scholar] [CrossRef]
- Richter, C.; Kass, G.E. Oxidative stress in mitochondria: Its relationship to cellular Ca2+ homeostasis, cell death, proliferation, and differentiation. Chem. Biol. Interact. 1991, 77, 1–23. [Google Scholar] [CrossRef]
- Richter, C.; Schlegel, J. Mitochondrial calcium release induced by prooxidants. Toxicol. Lett. 1993, 67, 119–127. [Google Scholar] [CrossRef]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Grijalba, M.T.; Vercesi, A.E.; Schreier, S. Ca2+-induced increased lipid packing and domain formation in submitochondrial particles. A possible early step in the mechanism of Ca2+-stimulated generation of reactive oxygen species by the respiratory chain. Biochemistry 1999, 38, 13279–13287. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.G.; Pathan, N.; Ethell, I.M.; Krajewski, S.; Yamaguchi, Y.; Shibasaki, F.; McKeon, F.; Bobo, T.; Franke, T.F.; Reed, J.C. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science 1999, 284, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Cain, K.; Bratton, S.B.; Langlais, C.; Walker, G.; Brown, D.G.; Sun, X.M.; Cohen, G.M. APAF-1 oligomerizes into biologically active approximately 700-kDa and inactive approximately 1.4-MDa apoptosome complexes. J. Biol. Chem. 2000, 275, 6067–6070. [Google Scholar] [CrossRef] [PubMed]
- Cain, K.; Brown, D.G.; Langlais, C.; Cohen, G.M. Caspase activation involves the formation of the aposome, a large (approximately 700 kDa) caspase-activating complex. J. Biol. Chem. 1999, 274, 22686–22692. [Google Scholar] [CrossRef] [PubMed]
- Bruey, J.M.; Ducasse, C.; Bonniaud, P.; Ravagnan, L.; Susin, S.A.; Diaz-Latoud, C.; Gurbuxani, S.; Arrigo, A.P.; Kroemer, G.; Solary, E.; et al. HSP27 negatively regulates cell death by interacting with cytochrome c. Nat. Cell Biol. 2000, 2, 645–652. [Google Scholar] [PubMed]
- Saleh, A.; Srinivasula, S.M.; Balkir, L.; Robbins, P.D.; Alnemri, E.S. Negative regulation of the APAF-1 apoptosome by HSP70. Nat. Cell Biol. 2000, 2, 476–483. [Google Scholar] [PubMed]
- Pandey, P.; Saleh, A.; Nakazawa, A.; Kumar, S.; Srinivasula, S.M.; Kumar, V.; Weichselbaum, R.; Nalin, C.; Alnemri, E.S.; Kufe, D.; et al. Negative regulation of cytochrome c-mediated oligomerization of APAF-1 and activation of procaspase-9 by heat shock protein 90. EMBO J. 2000, 19, 4310–4322. [Google Scholar] [CrossRef] [PubMed]
- Rainey, M.D.; Charlton, M.E.; Stanton, R.V.; Kastan, M.B. Transient inhibition of ATM kinase is sufficient to enhance cellular sensitivity to ionizing radiation. Cancer Res. 2008, 68, 7466–7474. [Google Scholar] [CrossRef] [PubMed]
- Teng, P.N.; Bateman, N.W.; Darcy, K.M.; Hamilton, C.A.; Maxwell, G.L.; Bakkenist, C.J.; Conrads, T.P. Pharmacologic inhibition of ATR and ATM offers clinically important distinctions to enhancing platinum or radiation response in ovarian, endometrial, and cervical cancer cells. Gynecol. Oncol. 2015, 136, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Carruthers, R.; Ahmed, S.U.; Strathdee, K.; Gomez-Roman, N.; Amoah-Buahin, E.; Watts, C.; Chalmers, A.J. Abrogation of radioresistance in glioblastoma stem-like cells by inhibition of ATM kinase. Mol. Oncol. 2015, 9, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Mihatsch, J.; Holler, M.; Chaachouay, H.; Rodemann, H.P. Cisplatin-mediated radiosensitization of non-small cell lung cancer cells is stimulated by ATM inhibition. Radiother. Oncol. 2014, 111, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Shen, Y.; Chen, Y.; Hsieh, J.T.; Kong, Z. The ATM inhibitor KU55933 sensitizes radioresistant bladder cancer cells with DAB2IP gene defect. Int. J. Radiat. Biol. 2015, 91, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Vecchio, D.; Daga, A.; Carra, E.; Marubbi, D.; Baio, G.; Neumaier, C.E.; Vagge, S.; Corvo, R.; Pia Brisigotti, M.; Louis Ravetti, J.; et al. Predictability, efficacy and safety of radiosensitization of glioblastoma-initiating cells by the ATM inhibitor KU-60019. Int. J. Cancer 2014, 135, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Biddlestone-Thorpe, L.; Sajjad, M.; Rosenberg, E.; Beckta, J.M.; Valerie, N.C.; Tokarz, M.; Adams, B.R.; Wagner, A.F.; Khalil, A.; Gilfor, D.; et al. ATM kinase inhibition preferentially sensitizes p53-mutant glioma to ionizing radiation. Clin. Cancer Res. 2013, 19, 3189–3200. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Reinhardt, H.C.; Bartkova, J.; Tommiska, J.; Blomqvist, C.; Nevanlinna, H.; Bartek, J.; Yaffe, M.B.; Hemann, M.T. The combined status of ATM and p53 link tumor development with therapeutic response. Genes Dev. 2009, 23, 1895–1909. [Google Scholar] [CrossRef] [PubMed]
- Peasland, A.; Wang, L.Z.; Rowling, E.; Kyle, S.; Chen, T.; Hopkins, A.; Cliby, W.A.; Sarkaria, J.; Beale, G.; Edmondson, R.J.; et al. Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br. J. Cancer 2011, 105, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.; Zhao, H.; Jalali, F.; Al Rashid, S.; Ran, J.; Supiot, S.; Kiltie, A.E.; Bristow, R.G. Targeting homologous recombination using imatinib results in enhanced tumor cell chemosensitivity and radiosensitivity. Mol. Cancer Ther. 2009, 8, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Omura-Minamisawa, M.; Kang, Y.; Cheng, C.; Inoue, T. Flavopiridol potentiates the cytotoxic effects of radiation in radioresistant tumor cells in which p53 is mutated or BCL-2 is overexpressed. Int. J. Radiat. Oncol. Biol. Phys. 2008, 71, 1485–1495. [Google Scholar] [CrossRef] [PubMed]
- Newcomb, E.W.; Lymberis, S.C.; Lukyanov, Y.; Shao, Y.; Schnee, T.; Devitt, M.; Rosenstein, B.S.; Zagzag, D.; Formenti, S.C. Radiation sensitivity of GL261 murine glioma model and enhanced radiation response by flavopiridol. Cell Cycle 2006, 5, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Raju, U.; Ariga, H.; Koto, M.; Lu, X.; Pickett, J.; Valdecanas, D.; Mason, K.A.; Milas, L. Improvement of esophageal adenocarcinoma cell and xenograft responses to radiation by targeting cyclin-dependent kinases. Radiother. Oncol. 2006, 80, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, P.; Tumati, V.; Yu, L.; Chan, N.; Tomimatsu, N.; Burma, S.; Bristow, R.G.; Saha, D. AZD5438, an inhibitor of CDK1, 2, and 9, enhances the radiosensitivity of non-small cell lung carcinoma cells. Int. J. Radiat. Oncol. Biol. Phys. 2012, 84, e507–e514. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Dering, J.; Conklin, D.; Kalous, O.; Cohen, D.J.; Desai, A.J.; Ginther, C.; Atefi, M.; Chen, I.; Fowst, C.; et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009, 11. [Google Scholar] [CrossRef] [PubMed]
- Gelbert, L.M.; Cai, S.; Lin, X.; Sanchez-Martinez, C.; del Prado, M.; Lallena, M.J.; Torres, R.; Ajamie, R.T.; Wishart, G.N.; Flack, R.S.; et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: In vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Investig. New Drugs 2014, 32, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Busby, E.C.; Leistritz, D.F.; Abraham, R.T.; Karnitz, L.M.; Sarkaria, J.N. The radiosensitizing agent 7-hydroxystaurosporine (UCN-01) inhibits the DNA damage checkpoint kinase hChk1. Cancer Res. 2000, 60, 2108–2112. [Google Scholar] [PubMed]
- Kim, Y.M.; Jeong, I.H.; Pyo, H. Celecoxib enhances the radiosensitizing effect of 7-hydroxystaurosporine (UCN-01) in human lung cancer cell lines. Int. J. Radiat. Oncol. Biol. Phys. 2012, 83, e399–e407. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, G.; Maalouf, M.; Boivin, A.; Battiston-Montagne, P.; Beuve, M.; Levy, A.; Jalade, P.; Fournier, C.; Ardail, D.; Magne, N.; et al. Targeting head and neck cancer stem cells to overcome resistance to photon and carbon ion radiation. Stem Cell Rev. 2014, 10, 114–126. [Google Scholar] [CrossRef]
- Signore, M.; Pelacchi, F.; di Martino, S.; Runci, D.; Biffoni, M.; Giannetti, S.; Morgante, L.; de Majo, M.; Petricoin, E.F.; Stancato, L.; et al. Combined PDK1 and CHK1 inhibition is required to kill glioblastoma stem-like cells in vitro and in vivo. Cell Death Dis. 2014, 5, e1223. [Google Scholar] [CrossRef] [PubMed]
- Jobson, A.G.; Lountos, G.T.; Lorenzi, P.L.; Llamas, J.; Connelly, J.; Cerna, D.; Tropea, J.E.; Onda, A.; Zoppoli, G.; Kondapaka, S.; et al. Cellular inhibition of checkpoint kinase 2 (Chk2) and potentiation of camptothecins and radiation by the novel Chk2 inhibitor PV1019 (7-nitro-1H-indole-2-carboxylic acid (4-(1-(guanidinohydrazone)-ethyl)-phenyl)-amide). J. Pharmacol. Exp. Ther. 2009, 331, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Riesterer, O.; Matsumoto, F.; Wang, L.; Pickett, J.; Molkentine, D.; Giri, U.; Milas, L.; Raju, U. A novel Chk inhibitor, XL-844, increases human cancer cell radiosensitivity through promotion of mitotic catastrophe. Investig. New Drugs 2011, 29, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Cuneo, K.C.; Nyati, M.K.; Ray, D.; Lawrence, T.S. EGFR targeted therapies and radiation: Optimizing efficacy by appropriate drug scheduling and patient selection. Pharmacol. Ther. 2015, 154, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Vincenzi, B.; Zoccoli, A.; Pantano, F.; Venditti, O.; Galluzzo, S. Cetuximab: From bench to bedside. Curr. Cancer Drug Targets 2010, 10, 80–95. [Google Scholar] [CrossRef] [PubMed]
- Berghauser Pont, L.M.; Spoor, J.K.; Venkatesan, S.; Swagemakers, S.; Kloezeman, J.J.; Dirven, C.M.; van der Spek, P.J.; Lamfers, M.L.; Leenstra, S. The BCL-2 inhibitor Obatoclax overcomes resistance to histone deacetylase inhibitors SAHA and LBH589 as radiosensitizers in patient-derived glioblastoma stem-like cells. Genes Cancer 2014, 5, 445–459. [Google Scholar] [PubMed]
- Pont, L.M.; Naipal, K.; Kloezeman, J.J.; Venkatesan, S.; van den Bent, M.; van Gent, D.C.; Dirven, C.M.; Kanaar, R.; Lamfers, M.L.; Leenstra, S. DNA damage response and anti-apoptotic proteins predict radiosensitization efficacy of HDAC inhibitors SAHA and LBH589 in patient-derived glioblastoma cells. Cancer Lett. 2015, 356, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Graham, P.H.; Hao, J.; Chang, L.; Ni, J.; Power, C.A.; Dong, Q.; Kearsley, J.H.; Li, Y. Combination therapy with the histone deacetylase inhibitor LBH589 and radiation is an effective regimen for prostate cancer cells. PLoS ONE 2013, 8, e74253. [Google Scholar] [CrossRef] [PubMed]
- Gressette, M.; Verillaud, B.; Jimenez-Pailhes, A.S.; Lelievre, H.; Lo, K.W.; Ferrand, F.R.; Gattolliat, C.H.; Jacquet-Bescond, A.; Kraus-Berthier, L.; Depil, S.; et al. Treatment of nasopharyngeal carcinoma cells with the histone-deacetylase inhibitor abexinostat: Cooperative effects with cis-platin and radiotherapy on patient-derived xenografts. PLoS ONE 2014, 9, e91325. [Google Scholar] [CrossRef] [PubMed]
- Banuelos, C.A.; Banath, J.P.; MacPhail, S.H.; Zhao, J.; Reitsema, T.; Olive, P.L. Radiosensitization by the histone deacetylase inhibitor PCI-24781. Clin. Cancer Res. 2007, 13, 6816–6826. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.; Flamand, Y.; Balasubramanian, S.; Butrynski, J.E.; Harmon, D.C.; George, S.; Cote, G.M.; Wagner, A.J.; Morgan, J.A.; Sirisawad, M.; et al. Phase 1 study of oral abexinostat, a histone deacetylase inhibitor, in combination with doxorubicin in patients with metastatic sarcoma. Cancer 2015, 121, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Morschhauser, F.; Terriou, L.; Coiffier, B.; Bachy, E.; Varga, A.; Kloos, I.; Lelievre, H.; Sarry, A.L.; Depil, S.; Ribrag, V. Phase 1 study of the oral histone deacetylase inhibitor abexinostat in patients with Hodgkin lymphoma, non-Hodgkin lymphoma, or chronic lymphocytic leukaemia. Investig. New Drugs 2015, 33, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Purrucker, J.C.; Fricke, A.; Ong, M.F.; Rube, C.; Rube, C.E.; Mahlknecht, U. HDAC inhibition radiosensitizes human normal tissue cells and reduces DNA Double-Strand Break repair capacity. Oncol. Rep. 2010, 23, 263–269. [Google Scholar] [PubMed]
- Barazzuol, L.; Jeynes, J.C.; Merchant, M.J.; Wera, A.C.; Barry, M.A.; Kirkby, K.J.; Suzuki, M. Radiosensitization of glioblastoma cells using a histone deacetylase inhibitor (SAHA) comparing carbon ions with X-rays. Int. J. Radiat. Biol. 2015, 91, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Blattmann, C.; Thiemann, M.; Stenzinger, A.; Christmann, A.; Roth, E.; Ehemann, V.; Debus, J.; Kulozik, A.E.; Weichert, W.; Huber, P.E.; et al. Radiosensitization by histone deacetylase inhibition in an osteosarcoma mouse model. Strahlenther. Onkol. 2013, 189, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Blattmann, C.; Oertel, S.; Ehemann, V.; Thiemann, M.; Huber, P.E.; Bischof, M.; Witt, O.; Deubzer, H.E.; Kulozik, A.E.; Debus, J.; et al. Enhancement of radiation response in osteosarcoma and rhabdomyosarcoma cell lines by histone deacetylase inhibition. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Ree, A.H.; Dueland, S.; Folkvord, S.; Hole, K.H.; Seierstad, T.; Johansen, M.; Abrahamsen, T.W.; Flatmark, K. Vorinostat, a histone deacetylase inhibitor, combined with pelvic palliative radiotherapy for gastrointestinal carcinoma: The Pelvic Radiation and Vorinostat (PRAVO) phase 1 study. Lancet Oncol. 2010, 11, 459–464. [Google Scholar] [CrossRef]
- Shi, W.; Lawrence, Y.R.; Choy, H.; Werner-Wasik, M.; Andrews, D.W.; Evans, J.J.; Judy, K.D.; Farrell, C.J.; Moshel, Y.; Berger, A.C.; et al. Vorinostat as a radiosensitizer for brain metastasis: A phase I clinical trial. J. Neurooncol. 2014, 118, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, M.; Yu, D.; Hirayama, R.; Ninomiya, Y.; Sekine, E.; Kubota, N.; Ando, K.; Okayasu, R. Inhibition of homologous recombination repair in irradiated tumor cells pretreated with HSP90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Biochem. Biophys. Res. Commun. 2006, 351, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Dungey, F.A.; Caldecott, K.W.; Chalmers, A.J. Enhanced radiosensitization of human glioma cells by combining inhibition of poly(ADP-ribose) polymerase with inhibition of heat shock protein 90. Mol. Cancer Ther. 2009, 8, 2243–2254. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wanders, A.; Wardega, P.; Tinge, B.; Gedda, L.; Bergstrom, S.; Sooman, L.; Gullbo, J.; Bergqvist, M.; Hesselius, P.; et al. HSP90 is expressed and represents a therapeutic target in human oesophageal cancer using the inhibitor 17-allylamino-17-demethoxygeldanamycin. Br. J. Cancer 2009, 100, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Dote, H.; Burgan, W.E.; Camphausen, K.; Tofilon, P.J. Inhibition of HSP90 compromises the DNA damage response to radiation. Cancer Res. 2006, 66, 9211–9220. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, R.; Kiang, J.G. Geldanamycin analog 17-DMAG limits apoptosis in human peripheral blood cells by inhibition of p53 activation and its interaction with heat-shock protein 90 kDa after exposure to ionizing radiation. Radiat. Res. 2011, 176, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Milanovic, D.; Firat, E.; Grosu, A.L.; Niedermann, G. Increased radiosensitivity and radiothermosensitivity of human pancreatic MIA PaCa-2 and U251 glioblastoma cell lines treated with the novel HSP90 inhibitor NVP-HSP990. Radiat. Oncol. 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Stingl, L.; Stuhmer, T.; Chatterjee, M.; Jensen, M.R.; Flentje, M.; Djuzenova, C.S. Novel HSP90 inhibitors, NVP-AUY922 and NVP-BEP800, radiosensitise tumour cells through cell-cycle impairment, increased DNA damage and repair protraction. Br. J. Cancer 2010, 102, 1578–1591. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Wen, J.; Magliocca, K.; Muller, S.; Liu, Y.; Chen, Z.G.; Saba, N.; Diaz, R. Heat shock protein 90 (HSP90) is overexpressed in p16-negative oropharyngeal squamous cell carcinoma, and its inhibition in vitro potentiates the effects of chemoradiation. Cancer Chemother. Pharmacol. 2014, 74, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Smith, D.L.; Sequeira, M.; Sang, J.; Bates, R.C.; Proia, D.A. The HSP90 inhibitor ganetespib has chemosensitizer and radiosensitizer activity in colorectal cancer. Investig. New Drugs 2014, 32, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; McCubrey, J.A.; Jefferson, H.S.; Paine, M.S.; Chappell, W.H.; Terrian, D.M. A dominant role for p53-dependent cellular senescence in radiosensitization of human prostate cancer cells. Cell Cycle 2007, 6, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Yount, C.; Lang, H.; Yang, A.; Riemer, E.C.; Lyons, K.; Vanek, K.N.; Silvestri, G.A.; Schulte, B.A.; Wang, G.Y. Activation of p53 with Nutlin-3a radiosensitizes lung cancer cells via enhancing radiation-induced premature senescence. Lung Cancer 2013, 81, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Schilling, D.; Duwel, M.; Molls, M.; Multhoff, G. Radiosensitization of wildtype p53 cancer cells by the MDM2-inhibitor PXN727 is associated with altered heat shock protein 70 (HSP70) levels. Cell. Stress Chaperones 2013, 18, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, S.; Fujiwara, T.; Shirakawa, Y.; Yamasaki, Y.; Yano, S.; Uno, F.; Tazawa, H.; Hashimoto, Y.; Watanabe, Y.; Noma, K.; et al. Telomerase-dependent oncolytic adenovirus sensitizes human cancer cells to ionizing radiation via inhibition of DNA repair machinery. Cancer Res. 2010, 70, 9339–9348. [Google Scholar] [CrossRef] [PubMed]
- Cmielova, J.; Havelek, R.; Vavrova, J.; Rezacova, M. Changes in the response of MCF-7 cells to ionizing radiation after the combination of ATM and DNA-PK inhibition. Med. Oncol. 2015, 32. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Tumati, V.; Tseng, S.F.; Hsu, F.M.; Kim, D.N.; Hong, D.; Hsieh, J.T.; Jacobs, C.; Kapur, P.; Saha, D. DAB2IP regulates autophagy in prostate cancer in response to combined treatment of radiation and a DNA-PKcs inhibitor. Neoplasia 2012, 14, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Thomas, H.D.; Batey, M.A.; Cowell, I.G.; Richardson, C.J.; Griffin, R.J.; Calvert, A.H.; Newell, D.R.; Smith, G.C.; Curtin, N.J. Preclinical evaluation of a potent novel DNA-dependent protein kinase inhibitor NU7441. Cancer Res. 2006, 66, 5354–5362. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M. On the TRAIL from p53 to apoptosis? Nat. Genet. 1997, 17, 130–131. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Strand, S.; Hug, H.; Heinemann, E.M.; Walczak, H.; Hofmann, W.J.; Stremmel, W.; Krammer, P.H.; Galle, P.R. Drug-induced apoptosis in hepatoma cells is mediated by the CD95 (APO-1/FAS) receptor/ligand system and involves activation of wild-type p53. J. Clin. Investig. 1997, 99, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Harms, K.; Nozell, S.; Chen, X. The common and distinct target genes of the p53 family transcription factors. Cell. Mol. Life Sci. 2004, 61, 822–842. [Google Scholar] [CrossRef] [PubMed]
- Sheard, M.A. Ionizing radiation as a response-enhancing agent for CD95-mediated apoptosis. Int. J. Cancer 2001, 96, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, C.; Fulda, S.; Srinivasan, A.; Friesen, C.; Li, F.; Tomaselli, K.J.; Debatin, K.M.; Krammer, P.H.; Peter, M.E. Two CD95 (APO-1/FAS) signaling pathways. EMBO J. 1998, 17, 1675–1687. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.R.; Meyer, C.F.; Tan, T.H. Persistent activation of c-Jun N-terminal kinase 1 (JNK1) in γ radiation-induced apoptosis. J. Biol. Chem. 1996, 271, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Verheij, M.; Bose, R.; Lin, X.H.; Yao, B.; Jarvis, W.D.; Grant, S.; Birrer, M.J.; Szabo, E.; Zon, L.I.; Kyriakis, J.M.; et al. Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis. Nature 1996, 380, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Verheij, M.; Ruiter, G.A.; Zerp, S.F.; van Blitterswijk, W.J.; Fuks, Z.; Haimovitz-Friedman, A.; Bartelink, H. The role of the stress-activated protein kinase (SAPK/JNK) signaling pathway in radiation-induced apoptosis. Radiother. Oncol. 1998, 47, 225–232. [Google Scholar] [CrossRef]
- Westwick, J.K.; Bielawska, A.E.; Dbaibo, G.; Hannun, Y.A.; Brenner, D.A. Ceramide activates the stress-activated protein kinases. J. Biol. Chem. 1995, 270, 22689–22692. [Google Scholar] [CrossRef] [PubMed]
- Charette, S.J.; Lavoie, J.N.; Lambert, H.; Landry, J. Inhibition of Daxx-mediated apoptosis by heat shock protein 27. Mol. Cell. Biol. 2000, 20, 7602–7612. [Google Scholar] [CrossRef] [PubMed]
- Frisch, S.M.; Vuori, K.; Kelaita, D.; Sicks, S. A role for Jun-N-terminal kinase in anoikis; suppression by BCL-2 and crmA. J. Cell Biol. 1996, 135, 1377–1382. [Google Scholar] [CrossRef] [PubMed]
- Bossy-Wetzel, E.; Bakiri, L.; Yaniv, M. Induction of apoptosis by the transcription factor c-Jun. EMBO J. 1997, 16, 1695–1709. [Google Scholar] [CrossRef] [PubMed]
- Shiah, S.G.; Chuang, S.E.; Chau, Y.P.; Shen, S.C.; Kuo, M.L. Activation of c-Jun NH2-terminal kinase and subsequent CPP32/Yama during topoisomerase inhibitor β-lapachone-induced apoptosis through an oxidation-dependent pathway. Cancer Res. 1999, 59, 391–398. [Google Scholar] [PubMed]
- Giusti, A.M.; Raimondi, M.; Ravagnan, G.; Sapora, O.; Parasassi, T. Human cell membrane oxidative damage induced by single and fractionated doses of ionizing radiation: A fluorescence spectroscopy study. Int. J. Radiat. Biol. 1998, 74, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Kolesnick, R. The therapeutic potential of modulating the ceramide/sphingomyelin pathway. J. Clin. Investig. 2002, 110, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Kolesnick, R.N. 1,2-Diacylglycerols but not phorbol esters stimulate sphingomyelin hydrolysis in GH3 pituitary cells. J. Biol. Chem. 1987, 262, 16759–16762. [Google Scholar] [PubMed]
- Okazaki, T.; Bell, R.M.; Hannun, Y.A. Sphingomyelin turnover induced by vitamin D3 in HL-60 cells. Role in cell differentiation. J. Biol. Chem. 1989, 264, 19076–19080. [Google Scholar] [PubMed]
- Lin, T.; Genestier, L.; Pinkoski, M.J.; Castro, A.; Nicholas, S.; Mogil, R.; Paris, F.; Fuks, Z.; Schuchman, E.H.; Kolesnick, R.N.; et al. Role of acidic sphingomyelinase in FAS/CD95-mediated cell death. J. Biol. Chem. 2000, 275, 8657–8663. [Google Scholar] [CrossRef] [PubMed]
- Ch’ang, H.J.; Maj, J.G.; Paris, F.; Xing, H.R.; Zhang, J.; Truman, J.P.; Cardon-Cardo, C.; Haimovitz-Friedman, A.; Kolesnick, R.; Fuks, Z. ATM regulates target switching to escalating doses of radiation in the intestines. Nat. Med. 2005, 11, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.C.; Haimovitz-Friedman, A.; Persaud, R.S.; McLoughlin, M.; Ehleiter, D.; Zhang, N.; Gatei, M.; Lavin, M.; Kolesnick, R.; Fuks, Z. Ataxia telangiectasia-mutated gene product inhibits DNA damage-induced apoptosis via ceramide synthase. J. Biol. Chem. 1999, 274, 17908–17917. [Google Scholar] [CrossRef] [PubMed]
- Nehs, M.A.; Lin, C.I.; Kozono, D.E.; Whang, E.E.; Cho, N.L.; Zhu, K.; Moalem, J.; Moore, F.D., Jr.; Ruan, D.T. Necroptosis is a novel mechanism of radiation-induced cell death in anaplastic thyroid and adrenocortical cancers. Surgery 2011, 150, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Yuan, J. Expansion and evolution of cell death programmes. Nat. Rev. Mol. Cell Biol. 2008, 9, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Jonathan, E.C.; Bernhard, E.J.; McKenna, W.G. How does radiation kill cells? Curr. Opin. Chem. Biol. 1999, 3, 77–83. [Google Scholar] [CrossRef]
- Galluzzi, L.; Maiuri, M.C.; Vitale, I.; Zischka, H.; Castedo, M.; Zitvogel, L.; Kroemer, G. Cell death modalities: Classification and pathophysiological implications. Cell Death Differ. 2007, 14, 1237–1243. [Google Scholar] [CrossRef] [PubMed]
- Castedo, M.; Perfettini, J.L.; Roumier, T.; Andreau, K.; Medema, R.; Kroemer, G. Cell death by mitotic catastrophe: A molecular definition. Oncogene 2004, 23, 2825–2837. [Google Scholar] [CrossRef] [PubMed]
- Hirose, Y.; Katayama, M.; Mirzoeva, O.K.; Berger, M.S.; Pieper, R.O. AKT activation suppresses Chk2-mediated, methylating agent-induced G2 arrest and protects from temozolomide-induced mitotic catastrophe and cellular senescence. Cancer Res. 2005, 65, 4861–4869. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Hager, C.; Bastians, H. Mechanisms of mitotic cell death induced by chemotherapy-mediated G2 checkpoint abrogation. Cancer Res. 2007, 67, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Heald, R.; McLoughlin, M.; McKeon, F. Human wee1 maintains mitotic timing by protecting the nucleus from cytoplasmically activated CDC2 kinase. Cell 1993, 74, 463–474. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Kalejs, M.; Cragg, M.S. Mitotic catastrophe and endomitosis in tumour cells: An evolutionary key to a molecular solution. Cell Biol. Int. 2005, 29, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, D.; Lofroth, P.O.; Johansson, L.; Riklund, K.A.; Stigbrand, T. Cell cycle disturbances and mitotic catastrophes in HeLa Hep2 cells following 2.5 to 10 Gy of ionizing radiation. Clin. Cancer Res. 2007, 13, 5501s–5508s. [Google Scholar] [CrossRef] [PubMed]
- Bourke, E.; Dodson, H.; Merdes, A.; Cuffe, L.; Zachos, G.; Walker, M.; Gillespie, D.; Morrison, C.G. DNA damage induces Chk1-dependent centrosome amplification. EMBO Rep. 2007, 8, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Dodson, H.; Wheatley, S.P.; Morrison, C.G. Involvement of centrosome amplification in radiation-induced mitotic catastrophe. Cell Cycle 2007, 6, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, K.; Morita, N.; Domiki, C.; Fujikawa-Yamamoto, K.; Hashimoto, M.; Iwabuchi, K.; Suzuki, K. Induction of centrosome amplification in p53 siRNA-treated human fibroblast cells by radiation exposure. Cancer Sci. 2006, 97, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ji, P.; Liu, J.; Broaddus, R.R.; Xue, F.; Zhang, W. Centrosome-associated regulators of the G2/M checkpoint as targets for cancer therapy. Mol. Cancer 2009, 8. [Google Scholar] [CrossRef] [PubMed]
- Hanashiro, K.; Kanai, M.; Geng, Y.; Sicinski, P.; Fukasawa, K. Roles of cyclins A and E in induction of centrosome amplification in p53-compromised cells. Oncogene 2008, 27, 5288–5302. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, D.; Stigbrand, T. Radiation-induced cell death mechanisms. Tumour Biol. 2010, 31, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Galluzzi, L.; Castedo, M.; Kroemer, G. Mitotic catastrophe: A mechanism for avoiding genomic instability. Nat. Rev. Mol. Cell Biol. 2011, 12, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Castedo, M.; Perfettini, J.L.; Roumier, T.; Valent, A.; Raslova, H.; Yakushijin, K.; Horne, D.; Feunteun, J.; Lenoir, G.; Medema, R.; et al. Mitotic catastrophe constitutes a special case of apoptosis whose suppression entails aneuploidy. Oncogene 2004, 23, 4362–4370. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, D.; Lofroth, P.O.; Johansson, L.; Riklund, K.; Stigbrand, T. Apoptotic signalling in HeLa Hep2 cells following 5 Gy of cobalt-60 γ radiation. Anticancer Res. 2009, 29, 4361–4366. [Google Scholar] [PubMed]
- Ruth, A.C.; Roninson, I.B. Effects of the multidrug transporter p-glycoprotein on cellular responses to ionizing radiation. Cancer Res. 2000, 60, 2576–2578. [Google Scholar] [PubMed]
- Weaver, B.A.; Cleveland, D.W. Decoding the links between mitosis, cancer, and chemotherapy: The mitotic checkpoint, adaptation, and cell death. Cancer Cell 2005, 8, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Castedo, M.; Coquelle, A.; Vivet, S.; Vitale, I.; Kauffmann, A.; Dessen, P.; Pequignot, M.O.; Casares, N.; Valent, A.; Mouhamad, S.; et al. Apoptosis regulation in tetraploid cancer cells. EMBO J. 2006, 25, 2584–2595. [Google Scholar] [CrossRef] [PubMed]
- Dimitrijevic-Bussod, M.; Balzaretti-Maggi, V.S.; Gadbois, D.M. Extracellular matrix and radiation G1 cell cycle arrest in human fibroblasts. Cancer Res. 1999, 59, 4843–4847. [Google Scholar] [PubMed]
- Campisi, J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol 2001, 11, S27–S31. [Google Scholar] [CrossRef]
- Dikomey, E.; Borgmann, K.; Brammer, I.; Kasten-Pisula, U. Molecular mechanisms of individual radiosensitivity studied in normal diploid human fibroblasts. Toxicology 2003, 193, 125–135. [Google Scholar] [CrossRef]
- Borgmann, K.; Dede, M.; Wrona, A.; Brammer, I.; Overgaard, J.; Dikomey, E. For X-irradiated normal human fibroblasts, only half of cell inactivation results from chromosomal damage. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Quick, Q.A.; Gewirtz, D.A. An accelerated senescence response to radiation in wild-type p53 glioblastoma multiforme cells. J. Neurosurg. 2006, 105, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Klein, L.E.; Freeze, B.S.; Smith, A.B., 3rd; Horwitz, S.B. The microtubule stabilizing agent discodermolide is a potent inducer of accelerated cell senescence. Cell Cycle 2005, 4, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Roninson, I.B.; Broude, E.V.; Chang, B.D. If not apoptosis, then what? Treatment-induced senescence and mitotic catastrophe in tumor cells. Drug Resist. Updat. 2001, 4, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Uetake, Y.; Sluder, G. Cell cycle progression after cleavage failure: Mammalian somatic cells do not possess a “tetraploidy checkpoint”. J. Cell Biol. 2004, 165, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Roninson, I.B. Tumor cell senescence in cancer treatment. Cancer Res. 2003, 63, 2705–2715. [Google Scholar] [PubMed]
- Rodemann, H.P.; Peterson, H.P.; Schwenke, K.; von Wangenheim, K.H. Terminal differentiation of human fibroblasts is induced by radiation. Scanning Microsc. 1991, 5, 1135–1142. [Google Scholar] [PubMed]
- Herskind, C.; Johansen, J.; Bentzen, S.M.; Overgaard, M.; Overgaard, J.; Bamberg, M.; Rodemann, H.P. Fibroblast differentiation in subcutaneous fibrosis after postmastectomy radiotherapy. Acta Oncol. 2000, 39, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Herskind, C.; Rodemann, H.P. Spontaneous and radiation-induced differentiationof fibroblasts. Exp. Gerontol. 2000, 35, 747–755. [Google Scholar] [CrossRef]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-induced senescence in cancer. J. Natl. Cancer Inst. 2010, 102, 1536–1546. [Google Scholar] [CrossRef] [PubMed]
- Zahnreich, S.; Melnikova, L.; Winter, M.; Nasonova, E.; Durante, M.; Ritter, S.; Fournier, C. Radiation-induced premature senescence is associated with specific cytogenetic changes. Mutat. Res. 2010, 701, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.A. The cat and mouse games that genes, viruses, and cells play. Cell 1997, 88, 573–575. [Google Scholar] [CrossRef]
- Gewirtz, D.A. Autophagy and senescence in cancer therapy. J. Cell. Physiol. 2014, 229, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Gewirtz, D.A. Autophagy, senescence and tumor dormancy in cancer therapy. Autophagy 2009, 5, 1232–1234. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Hansen, G.; Murray, D. Role of p16(INK4A) in replicative senescence and DNA damage-induced premature senescence in p53-deficient human cells. Biochem. Res. Int. 2012, 2012, 951574. [Google Scholar] [CrossRef] [PubMed]
- Alcorta, D.A.; Xiong, Y.; Phelps, D.; Hannon, G.; Beach, D.; Barrett, J.C. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc. Natl. Acad. Sci. USA 1996, 93, 13742–13747. [Google Scholar] [CrossRef] [PubMed]
- Stein, G.H.; Drullinger, L.F.; Soulard, A.; Dulic, V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol. Cell. Biol. 1999, 19, 2109–2117. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.R.; Elmore, L.W.; Jackson-Cook, C.; Demasters, G.; Povirk, L.F.; Holt, S.E.; Gewirtz, D.A. p53-Dependent accelerated senescence induced by ionizing radiation in breast tumour cells. Int. J. Radiat. Biol. 2005, 81, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, Y.; Yang, X.; Zhu, H.; Guo, Q.; Chen, X.; Zhang, H.; Cheng, H.; Sun, X. Autophagy and its function in radiosensitivity. Tumour Biol. 2015, 36, 4079–4087. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Levine, B. BCL-2 inhibition of autophagy: A new route to cancer? Cancer Res. 2006, 66, 2885–2888. [Google Scholar] [CrossRef] [PubMed]
- Honscheid, P.; Datta, K.; Muders, M.H. Autophagy: Detection, regulation and its role in cancer and therapy response. Int. J. Radiat. Biol. 2014, 90, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Goehe, R.W.; Di, X.; Sharma, K.; Bristol, M.L.; Henderson, S.C.; Valerie, K.; Rodier, F.; Davalos, A.R.; Gewirtz, D.A. The autophagy-senescence connection in chemotherapy: Must tumor cells (self) eat before they sleep? J. Pharmacol. Exp. Ther. 2012, 343, 763–778. [Google Scholar] [CrossRef] [PubMed]
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavare, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Dittmann, K.; Mayer, C.; Fehrenbacher, B.; Schaller, M.; Raju, U.; Milas, L.; Chen, D.J.; Kehlbach, R.; Rodemann, H.P. Radiation-induced epidermal growth factor receptor nuclear import is linked to activation of DNA-dependent protein kinase. J. Biol. Chem. 2005, 280, 31182–31189. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhu, F.; Wang, Z. Identification of EGF receptor C-terminal sequences 1005–1017 and di-leucine motif 1010LL1011 as essential in EGF receptor endocytosis. Exp. Cell Res. 2007, 313, 3349–3363. [Google Scholar] [CrossRef] [PubMed]
- Hanada, N.; Lo, H.W.; Day, C.P.; Pan, Y.; Nakajima, Y.; Hung, M.C. Co-regulation of B-Myb expression by E2F1 and EGF receptor. Mol. Carcinog. 2006, 45, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.W.; Xia, W.; Wei, Y.; Ali-Seyed, M.; Huang, S.F.; Hung, M.C. Novel prognostic value of nuclear epidermal growth factor receptor in breast cancer. Cancer Res. 2005, 65, 338–348. [Google Scholar] [PubMed]
- Toulany, M.; Rodemann, H.P. Membrane receptor signaling and control of DNA repair after exposure to ionizing radiation. Nuklearmedizin 2010, 49 (Suppl. 1), S26–S30. [Google Scholar] [PubMed]
- Karni, R.; Jove, R.; Levitzki, A. Inhibition of pp60c-Src reduces BCL-XL expression and reverses the transformed phenotype of cells overexpressing EGF and HER-2 receptors. Oncogene 1999, 18, 4654–4662. [Google Scholar] [CrossRef] [PubMed]
- Lerdrup, M.; Bruun, S.; Grandal, M.V.; Roepstorff, K.; Kristensen, M.M.; Hommelgaard, A.M.; van Deurs, B. Endocytic down-regulation of ErbB2 is stimulated by cleavage of its C-terminus. Mol. Biol. Cell 2007, 18, 3656–3666. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Minjgee, M.; Kehlbach, R.; Chen, J.; Baumann, M.; Rodemann, H.P. ErbB2 expression through heterodimerization with ERBB1 is necessary for ionizing radiation- but not EGF-induced activation of AKT survival pathway. Radiother. Oncol. 2010, 97, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Dittmann, K.; Mayer, C.; Kehlbach, R.; Rodemann, H.P. Radiation-induced caveolin-1 associated EGFR internalization is linked with nuclear EGFR transport and activation of DNA-PK. Mol. Cancer 2008, 7. [Google Scholar] [CrossRef] [PubMed]
- Dittmann, K.; Mayer, C.; Rodemann, H.P. Nuclear EGFR as novel therapeutic target: Insights into nuclear translocation and function. Strahlenther. Onkol. 2010, 186, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Liccardi, G.; Hartley, J.A.; Hochhauser, D. EGFR nuclear translocation modulates DNA repair following cisplatin and ionizing radiation treatment. Cancer Res. 2011, 71, 1103–1114. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Dittmann, K.; Fehrenbacher, B.; Schaller, M.; Baumann, M.; Rodemann, H.P. PI3K-AKT signaling regulates basal, but MAP-kinase signaling regulates radiation-induced XRCC1 expression in human tumor cells in vitro. DNA Repair 2008, 7, 1746–1756. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.W.; Chen, B.P.; Prithivirajsingh, S.; Kurimasa, A.; Story, M.D.; Qin, J.; Chen, D.J. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev. 2002, 16, 2333–2338. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Lee, K.J.; Fattah, K.R.; Lin, Y.F.; Fehrenbacher, B.; Schaller, M.; Chen, B.P.; Chen, D.J.; Rodemann, H.P. AKT promotes post-irradiation survival of human tumor cells through initiation, progression, and termination of DNA-PKcs-dependent DNA double-strand break repair. Mol. Cancer Res. 2012, 10, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Viniegra, J.G.; Martinez, N.; Modirassari, P.; Hernandez Losa, J.; Parada Cobo, C.; Sanchez-Arevalo Lobo, V.J.; Aceves Luquero, C.I.; Alvarez-Vallina, L.; Ramon y Cajal, S.; Rojas, J.M.; et al. Full activation of PKB/AKT in response to insulin or ionizing radiation is mediated through ATM. J. Biol. Chem. 2005, 280, 4029–4036. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Kasten-Pisula, U.; Brammer, I.; Wang, S.; Chen, J.; Dittmann, K.; Baumann, M.; Dikomey, E.; Rodemann, H.P. Blockage of epidermal growth factor receptor-phosphatidylinositol 3-kinase-AKT signaling increases radiosensitivity of K-RAS mutated human tumor cells in vitro by affecting DNA repair. Clin. Cancer Res. 2006, 12, 4119–4126. [Google Scholar] [CrossRef] [PubMed]
- Yacoub, A.; McKinstry, R.; Hinman, D.; Chung, T.; Dent, P.; Hagan, M.P. Epidermal growth factor and ionizing radiation up-regulate the DNA repair genes XRCC1 and ERCC1 in DU145 and LNCaP prostate carcinoma through MAPK signaling. Radiat. Res. 2003, 159, 439–452. [Google Scholar] [CrossRef]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, E.J.; Stanbridge, E.J.; Gupta, S.; Gupta, A.K.; Soto, D.; Bakanauskas, V.J.; Cerniglia, G.J.; Muschel, R.J.; McKenna, W.G. Direct evidence for the contribution of activated N-RAS and K-RAS oncogenes to increased intrinsic radiation resistance in human tumor cell lines. Cancer Res. 2000, 60, 6597–6600. [Google Scholar] [PubMed]
- Gupta, A.K.; Bakanauskas, V.J.; Cerniglia, G.J.; Cheng, Y.; Bernhard, E.J.; Muschel, R.J.; McKenna, W.G. The RAS radiation resistance pathway. Cancer Res. 2001, 61, 4278–4282. [Google Scholar] [PubMed]
- Cengel, K.A.; Voong, K.R.; Chandrasekaran, S.; Maggiorella, L.; Brunner, T.B.; Stanbridge, E.; Kao, G.D.; McKenna, W.G.; Bernhard, E.J. Oncogenic K-RAS signals through epidermal growth factor receptor and wild-type H-RAS to promote radiation survival in pancreatic and colorectal carcinoma cells. Neoplasia 2007, 9, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Baumann, M.; Rodemann, H.P. Stimulated PI3K-AKT signaling mediated through ligand or radiation-induced EGFR depends indirectly, but not directly, on constitutive K-RAS activity. Mol. Cancer Res. 2007, 5, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Dittmann, K.; Kruger, M.; Baumann, M.; Rodemann, H.P. Radioresistance of K-RAS mutated human tumor cells is mediated through EGFR-dependent activation of PI3K-AKT pathway. Radiother. Oncol. 2005, 76, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Kehlbach, R.; Florczak, U.; Sak, A.; Wang, S.; Chen, J.; Lobrich, M.; Rodemann, H.P. Targeting of AKT1 enhances radiation toxicity of human tumor cells by inhibiting DNA-PKcs-dependent DNA double-strand break repair. Mol. Cancer Ther. 2008, 7, 1772–1781. [Google Scholar] [CrossRef] [PubMed]
- Wahl, D.R.; Lawrence, T.S. Integrating chemoradiation and molecularly targeted therapy. Adv. Drug Deliv. Rev. 2015. [Google Scholar] [CrossRef] [PubMed]
- Paglin, S.; Lee, N.Y.; Nakar, C.; Fitzgerald, M.; Plotkin, J.; Deuel, B.; Hackett, N.; McMahill, M.; Sphicas, E.; Lampen, N.; et al. Rapamycin-sensitive pathway regulates mitochondrial membrane potential, autophagy, and survival in irradiated MCF-7 cells. Cancer Res. 2005, 65, 11061–11070. [Google Scholar] [CrossRef] [PubMed]
- Dingli, D.; Michor, F. Successful therapy must eradicate cancer stem cells. Stem Cells 2006, 24, 2603–2610. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [PubMed]
- Phillips, T.M.; McBride, W.H.; Pajonk, F. The response of CD24−/low/CD44+ breast cancer-initiating cells to radiation. J. Natl. Cancer Inst. 2006, 98, 1777–1785. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.K.; Hur, B.I.; Ko, M.H.; Kim, C.H.; Cha, S.H.; Kang, S.K. Potential identity of multi-potential cancer stem-like subpopulation after radiation of cultured brain glioma. BMC Neurosci. 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.; Webb, B.; Gerson, S.L. CD133+ cells contribute to radioresistance via altered regulation of DNA repair genes in human lung cancer cells. Radiother. Oncol. 2014, 110, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, I.; Kobayashi, E.; Asano, K.; Yoshida, M.; Nakano, H. UCN-01, a selective inhibitor of protein kinase C from Streptomyces. J. Antibiot. 1987, 40, 1782–1784. [Google Scholar] [CrossRef] [PubMed]
- Cicenas, J.; Kalyan, K.; Sorokinas, A.; Jatulyte, A.; Valiunas, D.; Kaupinis, A.; Valius, M. Highlights of the Latest Advances in Research on CDK Inhibitors. Cancers 2014, 6, 2224–2242. [Google Scholar] [CrossRef] [PubMed]
- Boss, D.S.; Schwartz, G.K.; Middleton, M.R.; Amakye, D.D.; Swaisland, H.; Midgley, R.S.; Ranson, M.; Danson, S.; Calvert, H.; Plummer, R.; et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of the oral cyclin-dependent kinase inhibitor AZD5438 when administered at intermittent and continuous dosing schedules in patients with advanced solid tumours. Ann. Oncol. 2010, 21, 884–894. [Google Scholar] [CrossRef] [PubMed]
- Blachly, J.S.; Byrd, J.C. Emerging drug profile: Cyclin-dependent kinase inhibitors. Leuk. Lymphoma 2013, 54, 2133–2143. [Google Scholar] [CrossRef] [PubMed]
- Dickson, M.A. Molecular pathways: CDK4 inhibitors for cancer therapy. Clin. Cancer Res. 2014, 20, 3379–3383. [Google Scholar] [CrossRef] [PubMed]
- Aleem, E.; Arceci, R.J. Targeting cell cycle regulators in hematologic malignancies. Front. Cell Dev. Biol. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Michaud, K.; Solomon, D.A.; Oermann, E.; Kim, J.S.; Zhong, W.Z.; Prados, M.D.; Ozawa, T.; James, C.D.; Waldman, T. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res. 2010, 70, 3228–3238. [Google Scholar] [CrossRef] [PubMed]
- Cen, L.; Carlson, B.L.; Schroeder, M.A.; Ostrem, J.L.; Kitange, G.J.; Mladek, A.C.; Fink, S.R.; Decker, P.A.; Wu, W.; Kim, J.S.; et al. p16-CDK4-RB axis controls sensitivity to a cyclin-dependent kinase inhibitor PD0332991 in glioblastoma xenograft cells. Neuro Oncol. 2012, 14, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Barton, K.L.; Misuraca, K.; Cordero, F.; Dobrikova, E.; Min, H.D.; Gromeier, M.; Kirsch, D.G.; Becher, O.J. PD-0332991, a CDK4/6 inhibitor, significantly prolongs survival in a genetically engineered mouse model of brainstem glioma. PLoS ONE 2013, 8, e77639. [Google Scholar] [CrossRef] [PubMed]
- Dupre, A.; Boyer-Chatenet, L.; Sattler, R.M.; Modi, A.P.; Lee, J.H.; Nicolette, M.L.; Kopelovich, L.; Jasin, M.; Baer, R.; Paull, T.T.; et al. A forward chemical genetic screen reveals an inhibitor of the MRE11-RAD50-NBS1 complex. Nat. Chem. Biol. 2008, 4, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Hickson, I.; Zhao, Y.; Richardson, C.J.; Green, S.J.; Martin, N.M.; Orr, A.I.; Reaper, P.M.; Jackson, S.P.; Curtin, N.J.; Smith, G.C. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004, 64, 9152–9159. [Google Scholar] [CrossRef] [PubMed]
- Golding, S.E.; Rosenberg, E.; Valerie, N.; Hussaini, I.; Frigerio, M.; Cockcroft, X.F.; Chong, W.Y.; Hummersone, M.; Rigoreau, L.; Menear, K.A.; et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol. Cancer Ther. 2009, 8, 2894–2902. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, M.; Allen, C.; Nickoloff, J.A.; Hromas, R. Synthetic lethality: Exploiting the addiction of cancer to DNA repair. Blood 2011, 117, 6074–6082. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, A. A synthetic lethal therapeutic approach: Poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J. Clin. Oncol. 2008, 26, 3785–3790. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Mansour, W.Y.; Rhein, T.; Dahm-Daphi, J. The alternative end-joining pathway for repair of DNA double-strand breaks requires PARP1 but is not dependent upon microhomologies. Nucleic Acids Res. 2010, 38, 6065–6077. [Google Scholar] [CrossRef] [PubMed]
- Sinha, G. Downfall of iniparib: A PARP inhibitor that doesn’t inhibit PARP after all. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.; Cao, C.; Geng, X.; Tian, B.; Wu, Y.; Zhang, C.; Chen, Z.; Li, W.; Shen, H.; Ying, S. Effectiveness and safety of poly (ADP-ribose) polymerase inhibitors in cancer therapy: A systematic review and meta-analysis. Oncotarget 2015, in press. [Google Scholar]
- Morgan, M.A.; Lawrence, T.S. Molecular pathways: Overcoming radiation resistance by targeting DNA damage response pathways. Clin. Cancer Res. 2015, 21, 2898–2904. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, M.; Kastan, M.B. The DNA damage response: Implications for tumor responses to radiation and chemotherapy. Annu. Rev. Med. 2015, 66, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Foedermayr, M.; Sebesta, M.; Rudas, M.; Berghoff, A.S.; Promberger, R.; Preusser, M.; Dubsky, P.; Fitzal, F.; Gnant, M.; Steger, G.G.; et al. BRCA-1 methylation and TP53 mutation in triple-negative breast cancer patients without pathological complete response to taxane-based neoadjuvant chemotherapy. Cancer Chemother. Pharmacol. 2014, 73, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Stachelek, G.C.; Peterson-Roth, E.; Liu, Y.; Fernandez, R.J., 3rd; Pike, L.R.; Qian, J.M.; Abriola, L.; Hoyer, D.; Hungerford, W.; Merkel, J.; et al. YU238259 Is a Novel Inhibitor of homology-dependent DNA repair that exhibits synthetic lethality and radiosensitization in repair-deficient tumors. Mol. Cancer Res. 2015, 13, 1389–1397. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, A.; Bastian, P.J.; Bellmunt, J.; Bolla, M.; Joniau, S.; van der Kwast, T.; Mason, M.; Matveev, V.; Wiegel, T.; Zattoni, F.; et al. EAU guidelines on prostate cancer. Part 1: Screening, diagnosis, and local treatment with curative intent-update 2013. Eur. Urol. 2014, 65, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Bolla, M.; van Tienhoven, G.; Warde, P.; Dubois, J.B.; Mirimanoff, R.O.; Storme, G.; Bernier, J.; Kuten, A.; Sternberg, C.; Billiet, I.; et al. External irradiation with or without long-term androgen suppression for prostate cancer with high metastatic risk: 10-Year results of an EORTC randomised study. Lancet Oncol. 2010, 11, 1066–1073. [Google Scholar] [CrossRef]
- Ciszewski, W.M.; Tavecchio, M.; Dastych, J.; Curtin, N.J. DNA-PK inhibition by NU7441 sensitizes breast cancer cells to ionizing radiation and doxorubicin. Breast Cancer Res. Treat. 2014, 143, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Shang, Z.F.; Hsu, F.M.; Zhang, Z.; Tumati, V.; Lin, Y.F.; Chen, B.P.; Saha, D. NSCLC cells demonstrate differential mode of cell death in response to the combined treatment of radiation and a DNA-PKcs inhibitor. Oncotarget 2015, 6, 3848–3860. [Google Scholar] [CrossRef] [PubMed]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Chu, Y.; Ma, H.; Zhang, Y.; Zhang, X.; Zhao, D.; Li, Z.; Wang, J.; Gao, Y.E.; Xiao, L.; et al. Epigenetic interventions increase the radiation sensitivity of cancer cells. Curr. Pharm. Des. 2014, 20, 1857–1865. [Google Scholar] [CrossRef] [PubMed]
- Cerna, D.; Camphausen, K.; Tofilon, P.J. Histone deacetylation as a target for radiosensitization. Curr. Top. Dev. Biol. 2006, 73, 173–204. [Google Scholar] [PubMed]
- Brand, T.M.; Iida, M.; Luthar, N.; Starr, M.M.; Huppert, E.J.; Wheeler, D.L. Nuclear EGFR as a molecular target in cancer. Radiother. Oncol. 2013, 108, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Chen, Y.J.; Li, L.Y.; Wei, Y.L.; Hsu, S.C.; Tsai, S.L.; Chiu, P.C.; Huang, W.P.; Wang, Y.N.; Chen, C.H.; et al. Nuclear translocation of epidermal growth factor receptor by AKT-dependent phosphorylation enhances breast cancer-resistant protein expression in gefitinib-resistant cells. J. Biol. Chem. 2011, 286, 20558–20568. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Iida, M.; Dunn, E.F.; Ghia, A.J.; Wheeler, D.L. Nuclear EGFR contributes to acquired resistance to cetuximab. Oncogene 2009, 28, 3801–3813. [Google Scholar] [CrossRef] [PubMed]
- Poklepovic, A.; Gewirtz, D.A. Outcome of early clinical trials of the combination of hydroxychloroquine with chemotherapy in cancer. Autophagy 2014, 10, 1478–1480. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.R.; Ye, X.; Supko, J.G.; Desideri, S.; Grossman, S.A.; Brem, S.; Mikkelson, T.; Wang, D.; Chang, Y.C.; Hu, J.; et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014, 10, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Puentes, L.L.; Gonzalez-Pinedo, M.; Crismatt, A.; Ortega-Gomez, A.; Gamboa-Vignolle, C.; Nunez-Gomez, R.; Dorantes-Gallareta, Y.; Arce-Salinas, C.; Arrieta, O. Phase II randomized, double-blind, placebo-controlled study of whole-brain irradiation with concomitant chloroquine for brain metastases. Radiat. Oncol. 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.; Issaeva, N.; Zache, N.; Shilov, A.; Hultcrantz, M.; Bergman, J.; Selivanova, G.; Wiman, K.G. Reactivation of mutant p53 and induction of apoptosis in human tumor cells by maleimide analogs. J. Biol. Chem. 2005, 280, 30384–30391. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.; Bykov, V.J.; Ali, D.; Andren, O.; Cherif, H.; Tidefelt, U.; Uggla, B.; Yachnin, J.; Juliusson, G.; Moshfegh, A.; et al. Targeting p53 in vivo: A first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J. Clin. Oncol. 2012, 30, 3633–3639. [Google Scholar] [CrossRef] [PubMed]
- Supiot, S.; Zhao, H.; Wiman, K.; Hill, R.P.; Bristow, R.G. PRIMA-1(MET) radiosensitizes prostate cancer cells independent of their MTp53-status. Radiother. Oncol. 2008, 86, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.J.; Galanis, E.; Anderson, S.K.; Schiff, D.; Kaufmann, T.J.; Peller, P.J.; Giannini, C.; Brown, P.D.; Uhm, J.H.; McGraw, S.; et al. A phase II trial of everolimus, temozolomide, and radiotherapy in patients with newly diagnosed glioblastoma: NCCTG N057K. Neuro Oncol. 2015, 17, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Higgins, G.S.; O’Cathail, S.M.; Muschel, R.J.; McKenna, W.G. Drug radiotherapy combinations: Review of previous failures and reasons for future optimism. Cancer Treat. Rev. 2015, 41, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Morris, Z.S.; Harari, P.M. Interaction of radiation therapy with molecular targeted agents. J. Clin. Oncol. 2014, 32, 2886–2893. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, A.; Loeffler, J.S.; Dyson, N.J. Insulin-like growth factor receptor I mediates resistance to anti-epidermal growth factor receptor therapy in primary human glioblastoma cells through continued activation of phosphoinositide 3-kinase signaling. Cancer Res. 2002, 62, 200–207. [Google Scholar] [PubMed]
- Johnke, R.M.; Sattler, J.A.; Allison, R.R. Radioprotective agents for radiation therapy: Future trends. Future Oncol. 2014, 10, 2345–2357. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maier, P.; Hartmann, L.; Wenz, F.; Herskind, C. Cellular Pathways in Response to Ionizing Radiation and Their Targetability for Tumor Radiosensitization. Int. J. Mol. Sci. 2016, 17, 102. https://doi.org/10.3390/ijms17010102
Maier P, Hartmann L, Wenz F, Herskind C. Cellular Pathways in Response to Ionizing Radiation and Their Targetability for Tumor Radiosensitization. International Journal of Molecular Sciences. 2016; 17(1):102. https://doi.org/10.3390/ijms17010102
Chicago/Turabian StyleMaier, Patrick, Linda Hartmann, Frederik Wenz, and Carsten Herskind. 2016. "Cellular Pathways in Response to Ionizing Radiation and Their Targetability for Tumor Radiosensitization" International Journal of Molecular Sciences 17, no. 1: 102. https://doi.org/10.3390/ijms17010102
APA StyleMaier, P., Hartmann, L., Wenz, F., & Herskind, C. (2016). Cellular Pathways in Response to Ionizing Radiation and Their Targetability for Tumor Radiosensitization. International Journal of Molecular Sciences, 17(1), 102. https://doi.org/10.3390/ijms17010102