
Insights into Mechanisms of Chronic Neurodegeneration


 ,
,
Abstract
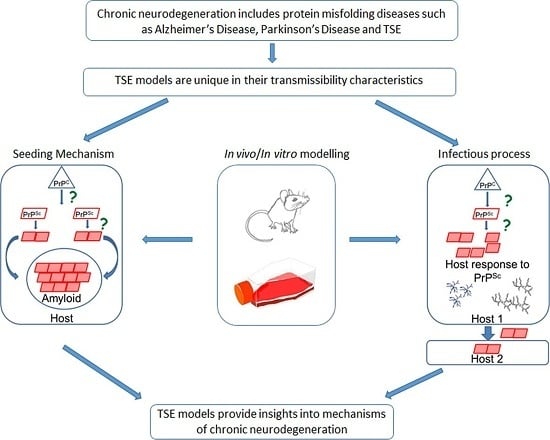
:
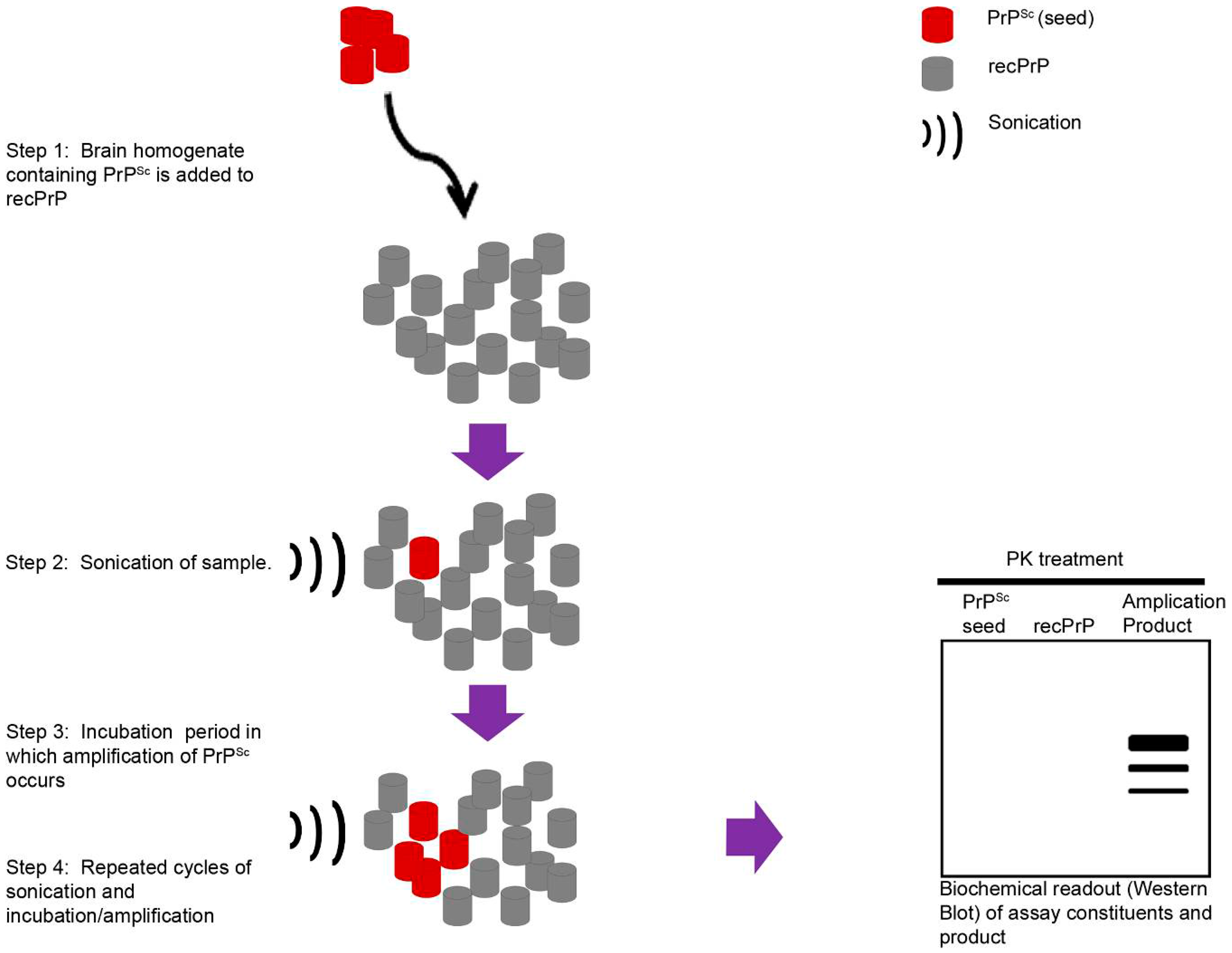
{kind=link}
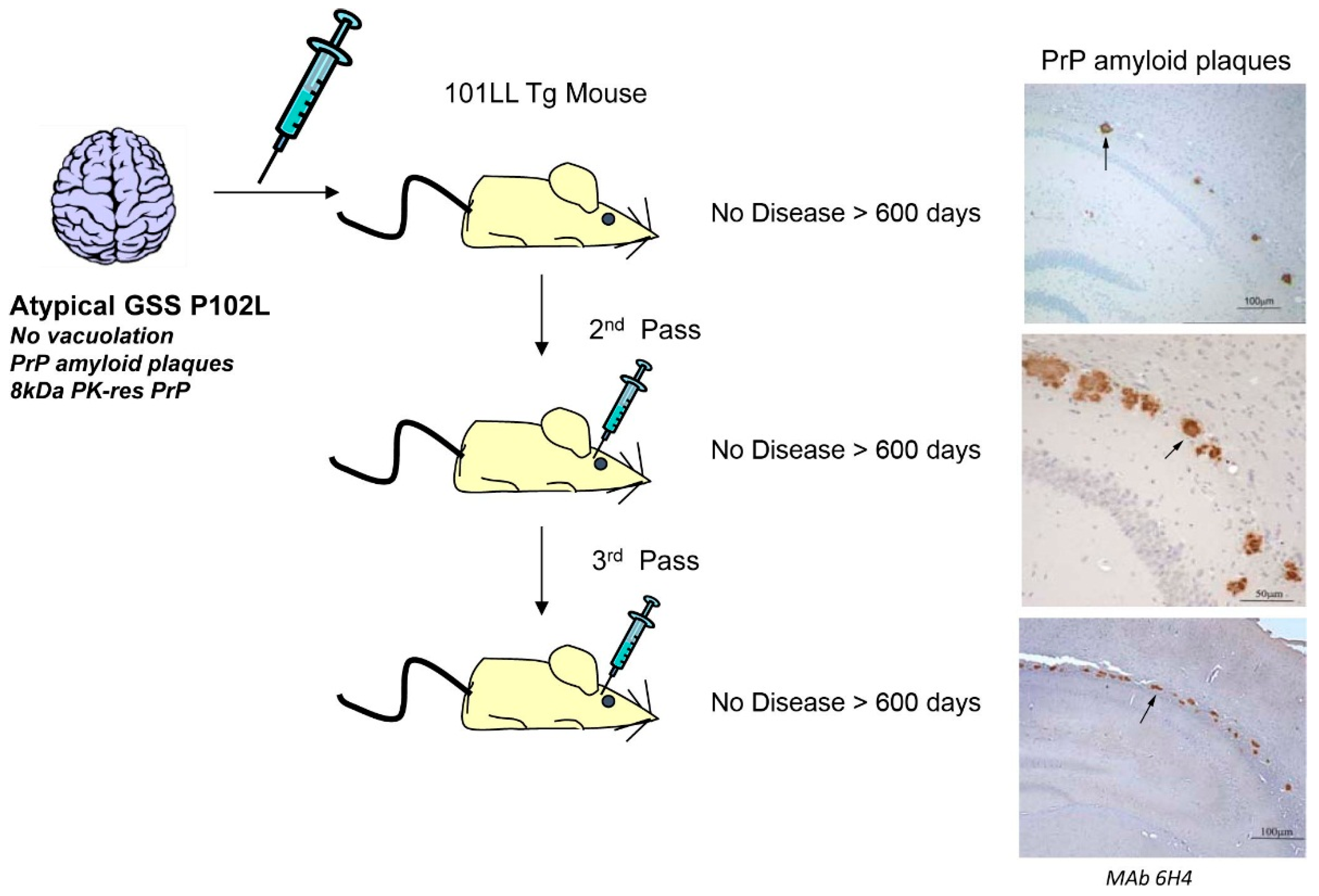
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. In Vitro Modelling of Protein Misfolding

3. In Vivo TSE Models of Chronic Neurodegeneration
4. Time Course Studies of TSE

5. Neurodegeneration and Protein Misfolding
6. Glial Cells and Neurodegeneration
7. Protein Misfolding and Infection

8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kayed, R.; Glabe, C.G. Conformation-dependent anti-amyloid oligomer antibodies. Methods Enzymol. 2006, 413, 326–344. [Google Scholar] [PubMed]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Head, E.; Sarsoza, F.; Saing, T.; Cotman, C.W.; Necula, M.; Margol, L.; Wu, J.; Breydo, L.; Thompson, J.L.; et al. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener. 2007, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Roberts, G.W.; Lofthouse, R.; Allsop, D.; Landon, M.; Kidd, M.; Prusiner, S.B.; Crow, T.J. CNS amyloid proteins in neurodegenerative diseases. Neurology 1988, 38, 1534–1540. [Google Scholar] [CrossRef] [PubMed]
- Piccardo, P.; Manson, J.C.; King, D.; Ghetti, B.; Barron, R.M. Accumulation of prion protein in the brain that is not associated with transmissible disease. Proc. Natl. Acad. Sci. USA 2007, 104, 4712–4717. [Google Scholar] [CrossRef] [PubMed]
- Klein, W.L.; Krafft, G.A.; Finch, C.E. Targeting small abeta oligomers: The solution to an alzheimer’s disease conundrum? Trends Neurosci. 2001, 24, 219–224. [Google Scholar] [CrossRef]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.; Lansbury, P.T. Protofibrils, pores, fibrils, and neurodegeneration: Separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 2003, 26, 267–298. [Google Scholar] [CrossRef] [PubMed]
- Novitskaya, V.; Bocharova, O.V.; Bronstein, I.; Baskakov, I.V. Amyloid fibrils of mammalian prion protein are highly toxic to cultured cells and primary neurons. J. Biol. Chem. 2006, 281, 13828–13836. [Google Scholar] [CrossRef] [PubMed]
- Simoneau, S.; Rezaei, H.; Sales, N.; Kaiser-Schulz, G.; Lefebvre-Roque, M.; Vidal, C.; Fournier, J.G.; Comte, J.; Wopfner, F.; Grosclaude, J.; et al. In vitro and in vivo neurotoxicity of prion protein oligomers. PLoS Pathog. 2007, 3, e125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Jackson, A.P.; Zhang, Z.R.; Han, Y.; Yu, S.; He, R.Q.; Perrett, S. Amyloid-like aggregates of the yeast prion protein ure2 enter vertebrate cells by specific endocytotic pathways and induce apoptosis. PLoS ONE 2010, e12529. [Google Scholar] [CrossRef] [PubMed]
- Sanghera, N.; Wall, M.; Venien-Bryan, C.; Pinheiro, T.J. Globular and pre-fibrillar prion aggregates are toxic to neuronal cells and perturb their electrophysiology. Biochim. Biophys. Acta 2008, 1784, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Sokolov, Y.; Edmonds, B.; McIntire, T.M.; Milton, S.C.; Hall, J.E.; Glabe, C.G. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 2004, 279, 46363–46366. [Google Scholar] [CrossRef] [PubMed]
- Quist, A.; Doudevski, I.; Lin, H.; Azimova, R.; Ng, D.; Frangione, B.; Kagan, B.; Ghiso, J.; Lal, R. Amyloid ion channels: A common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA 2005, 102, 10427–10432. [Google Scholar] [CrossRef] [PubMed]
- Price, J.L.; Morris, J.C. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann. Neurol. 1999, 45, 358–368. [Google Scholar] [CrossRef]
- Ehret, M.J.; Chamberlin, K.W. Current practices in the treatment of alzheimer disease: Where is the evidence after the phase iii trials? Clin. Ther. 2015, 37, 1604–1616. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Thomas, R.G.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; Raman, R.; Sun, X.; Aisen, P.S.; et al. Phase 3 trials of solanezumab for mild-to-moderate alzheimer’s disease. N. Engl. J. Med. 2014, 370, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Salloway, S.; Sperling, R.; Fox, N.C.; Blennow, K.; Klunk, W.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Porsteinsson, A.P.; Ferris, S.; et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Luehmann, M.; Coomaraswamy, J.; Bolmont, T.; Kaeser, S.; Schaefer, C.; Kilger, E.; Neuenschwander, A.; Abramowski, D.; Frey, P.; Jaton, A.L.; et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 2006, 313, 1781–1784. [Google Scholar] [CrossRef] [PubMed]
- Eisele, Y.S.; Bolmont, T.; Heikenwalder, M.; Langer, F.; Jacobson, L.H.; Yan, Z.X.; Roth, K.; Aguzzi, A.; Staufenbiel, M.; Walker, L.C.; et al. Induction of cerebral β-amyloidosis: Intracerebral versus systemic Aβ inoculation. Proc. Natl. Acad. Sci. USA 2009, 106, 12926–12931. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef] [PubMed]
- De Calignon, A.; Polydoro, M.; Suarez-Calvet, M.; William, C.; Adamowicz, D.H.; Kopeikina, K.J.; Pitstick, R.; Sahara, N.; Ashe, K.H.; Carlson, G.A.; et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 2012, 73, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Drouet, V.; Wu, J.W.; Witter, M.P.; Small, S.A.; Clelland, C.; Duff, K. Trans-synaptic spread of tau pathology in vivo. PLoS ONE 2012, 7, e31302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Guerrero-Munoz, M.J.; Kiritoshi, T.; Neugebauer, V.; Jackson, G.R.; Kayed, R. Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci. Rep. 2012, 2, 700. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological alpha-synuclein transmission initiates parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Kehm, V.M.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J. Exp. Med. 2012, 209, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Jaunmuktane, Z.; Mead, S.; Ellis, M.; Wadsworth, J.D.; Nicoll, A.J.; Kenny, J.; Launchbury, F.; Linehan, J.; Richard-Loendt, A.; Walker, A.S.; et al. Evidence for human transmission of amyloid-beta pathology and cerebral amyloid angiopathy. Nature 2015, 525, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Freeman, T.B.; Olanow, C.W. Lewy body-like pathology in long-term embryonic nigral transplants in parkinson’s disease. Nat. Med. 2008, 14, 504–506. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.J.; Abrams, J.Y.; Schonberger, L.B.; Leschek, E.W.; Mills, J.L.; Lee, V.M.; Trojanowski, J.Q. Evaluation of potential infectivity of alzheimer and parkinson disease proteins in recipients of cadaver-derived human growth hormone. JAMA Neurol. 2013, 70, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Kocisko, D.A.; Come, J.H.; Priola, S.A.; Chesebro, B.; Raymond, G.J.; Lansbury, P.T.; Caughey, B. Cell-free formation of protease-resistant prion protein. Nature 1994, 370, 471–474. [Google Scholar] [CrossRef] [PubMed]
- Saborio, G.P.; Permanne, B.; Soto, C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 2001, 411, 810–813. [Google Scholar] [CrossRef] [PubMed]
- Edgeworth, J.A.; Gros, N.; Alden, J.; Joiner, S.; Wadsworth, J.D.; Linehan, J.; Brandner, S.; Jackson, G.S.; Weissmann, C.; Collinge, J. Spontaneous generation of mammalian prions. Proc. Natl. Acad. Sci. USA 2010, 107, 14402–14406. [Google Scholar] [CrossRef] [PubMed]
- Deleault, N.R.; Harris, B.T.; Rees, J.R.; Supattapone, S. Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. USA 2007, 104, 9741–9746. [Google Scholar] [CrossRef] [PubMed]
- Weber, P.; Giese, A.; Piening, N.; Mitteregger, G.; Thomzig, A.; Beekes, M.; Kretzschmar, H.A. Generation of genuine prion infectivity by serial pmca. Vet. Microbiol. 2007, 123, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, X.; Yuan, C.G.; Ma, J. Generating a prion with bacterially expressed recombinant prion protein. Science 2010, 327, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- Timmes, A.G.; Moore, R.A.; Fischer, E.R.; Priola, S.A. Recombinant prion protein refolded with lipid and rna has the biochemical hallmarks of a prion but lacks in vivo infectivity. PLoS ONE 2013, 8, e71081. [Google Scholar] [CrossRef] [PubMed]
- Roostaee, A.; Beaudoin, S.; Staskevicius, A.; Roucou, X. Aggregation and neurotoxicity of recombinant alpha-synuclein aggregates initiated by dimerization. Mol. Neurodegener. 2013, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Salvadores, N.; Shahnawaz, M.; Scarpini, E.; Tagliavini, F.; Soto, C. Detection of misfolded abeta oligomers for sensitive biochemical diagnosis of alzheimer’s disease. Cell Rep. 2014, 7, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Jucker, M.; Walker, L.C. Pathogenic protein seeding in alzheimer disease and other neurodegenerative disorders. Ann. Neurol. 2011, 70, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Groschup, M.H.; Buschmann, A. Rodent models for prion diseases. Vet. Res. 2008, 39, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Barron, R.M.; Manson, J.C. A gene-targeted mouse model of p102l gerstmann-straussler-scheinker syndrome. Clin. Lab. Med. 2003, 23, 161–173. [Google Scholar] [CrossRef]
- Cancellotti, E.; Wiseman, F.; Tuzi, N.L.; Baybutt, H.; Monaghan, P.; Aitchison, L.; Simpson, J.; Manson, J.C. Altered glycosylated prp proteins can have different neuronal trafficking in brain but do not acquire scrapie-like properties. J. Biol. Chem. 2005, 280, 42909–42918. [Google Scholar] [CrossRef] [PubMed]
- Browning, S.R.; Mason, G.L.; Seward, T.; Green, M.; Eliason, G.A.; Mathiason, C.; Miller, M.W.; Williams, E.S.; Hoover, E.; Telling, G.C. Transmission of prions from mule deer and elk with chronic wasting disease to transgenic mice expressing cervid PrP. J. Virol. 2004, 78, 13345–13350. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.; Hart, P.; Piccardo, P.; Hunter, N.; Casalone, C.; Baron, T.; Barron, R.M. Bovine PrP expression levels in transgenic mice influence transmission characteristics of atypical bovine spongiform encephalopathy. J. Gen. Virol. 2012, 93, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.C.; Clarke, A.R.; Hooper, M.L.; Aitchison, L.; McConnell, I.; Hope, J. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol. Neurobiol. 1994, 8, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Bueler, H.; Aguzzi, A.; Sailer, A.; Greiner, R.A.; Autenried, P.; Aguet, M.; Weissmann, C. Mice devoid of PrP are resistant to scrapie. Cell 1993, 73, 1339–1347. [Google Scholar] [CrossRef]
- Brandner, S.; Isenmann, S.; Raeber, A.; Fischer, M.; Sailer, A.; Kobayashi, Y.; Marino, S.; Weissmann, C.; Aguzzi, A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature 1996, 379, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Barron, R.M.; Baybutt, H.; Tuzi, N.L.; McCormack, J.; King, D.; Moore, R.C.; Melton, D.W.; Manson, J.C. Polymorphisms at codons 108 and 189 in murine PrP play distinct roles in the control of scrapie incubation time. J. Gen. Virol. 2005, 86, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Barron, R.M.; Thomson, V.; Jamieson, E.; Melton, D.W.; Ironside, J.; Will, R.; Manson, J.C. Changing a single amino acid in the N-terminus of murine PrP alters TSE incubation time across three species barriers. EMBO J. 2001, 20, 5070–5078. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.C.; Jamieson, E.; Baybutt, H.; Tuzi, N.L.; Barron, R.; McConnell, I.; Somerville, R.; Ironside, J.; Will, R.; Sy, M.S.; et al. A single amino acid alteration (101L) introduced into murine PrP dramatically alters incubation time of transmissible spongiform encephalopathy. EMBO J. 1999, 18, 6855–6864. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.; Foster, D.; Mirenda, C.; Serban, D.; Coufal, F.; Walchli, M.; Torchia, M.; Groth, D.; Carlson, G.; DeArmond, S.J.; et al. Transgenic mice expressing hamster prion protein produce species-specific scrapie infectivity and amyloid plaques. Cell 1989, 59, 847–857. [Google Scholar] [CrossRef]
- Asante, E.A.; Linehan, J.M.; Desbruslais, M.; Joiner, S.; Gowland, I.; Wood, A.L.; Welch, J.; Hill, A.F.; Lloyd, S.E.; Wadsworth, J.D.; et al. BSE prions propagate as either variant CJD-like or sporadic CJD-like prion strains in transgenic mice expressing human prion protein. EMBO J. 2002, 21, 6358–6366. [Google Scholar] [CrossRef] [PubMed]
- Brun, A.; Gutierrez-Adan, A.; Castilla, J.; Pintado, B.; Diaz-San Segundo, F.; Cano, M.J.; Alamillo, E.; Espinosa, J.C.; Torres, J.M. Reduced susceptibility to bovine spongiform encephalopathy prions in transgenic mice expressing a bovine PrP with five octapeptide repeats. J. Gen. Virol. 2007, 88, 1842–1849. [Google Scholar] [CrossRef] [PubMed]
- Vilotte, J.L.; Soulier, S.; Essalmani, R.; Stinnakre, M.G.; Vaiman, D.; Lepourry, L.; Da Silva, J.C.; Besnard, N.; Dawson, M.; Buschmann, A.; et al. Markedly increased susceptibility to natural sheep scrapie of transgenic mice expressing ovine PrP. J. Virol. 2001, 75, 5977–5984. [Google Scholar] [CrossRef] [PubMed]
- Tuzi, N.L.; Cancellotti, E.; Baybutt, H.; Blackford, L.; Bradford, B.; Plinston, C.; Coghill, A.; Hart, P.; Piccardo, P.; Barron, R.M.; et al. Host PrP glycosylation: A major factor determining the outcome of prion infection. PLoS Biol. 2008, 6, e100. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, F.K.; Cancellotti, E.; Piccardo, P.; Iremonger, K.; Boyle, A.; Brown, D.; Ironside, J.W.; Manson, J.C.; Diack, A.B. The glycosylation status of PrPc is a key factor in determining transmissible spongiform encephalopathy transmission between species. J. Virol. 2015, 89, 4738–4747. [Google Scholar] [CrossRef] [PubMed]
- Neuendorf, E.; Weber, A.; Saalmueller, A.; Schatzl, H.M.; Reifenberg, K.; Pfaff, E.; Groschup, M.H. Glycosylation deficiency at either one of the two glycan attachment sites of cellular prion protein preserves susceptibility to bovine spongiform encephalopathy and scrapie infections. J. Biol. Chem. 2004, 279, 53306–53316. [Google Scholar] [CrossRef] [PubMed]
- DeArmond, S.J.; Sanchez, H.; Yehiely, F.; Qiu, Y.; Ninchak-Casey, A.; Daggett, V.; Camerino, A.P.; Cayetano, J.; Rogers, M.; Groth, D.; et al. Selective neuronal targeting in prion disease. Neuron 1997, 19, 1337–1348. [Google Scholar] [CrossRef]
- Budka, H. Neuropathology of prion diseases. Br. Med. Bull. 2003, 66, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Ironside, J.W.; Head, M.W. Biology and neuropathology of prion diseases. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2008; Volume 89, pp. 779–797. [Google Scholar]
- Guenther, K.; Deacon, R.M.; Perry, V.H.; Rawlins, J.N. Early behavioural changes in scrapie-affected mice and the influence of dapsone. Eur. J. Neurosci. 2001, 14, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.; Deacon, R.; Wells, H.; Boche, D.; Waters, S.; Diniz, C.P.; Scott, H.; Rawlins, J.N.; Perry, V.H. Synaptic changes characterize early behavioural signs in the ME7 model of murine prion disease. Eur. J. Neurosci. 2003, 17, 2147–2155. [Google Scholar] [CrossRef] [PubMed]
- Gray, B.C.; Siskova, Z.; Perry, V.H.; O’Connor, V. Selective presynaptic degeneration in the synaptopathy associated with ME7-induced hippocampal pathology. Neurobiol. Dis. 2009, 35, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Siskova, Z.; Page, A.; O’Connor, V.; Perry, V.H. Degenerating synaptic boutons in prion disease: Microglia activation without synaptic stripping. Am. J. Pathol. 2009, 175, 1610–1621. [Google Scholar] [CrossRef] [PubMed]
- Hilton, K.J.; Cunningham, C.; Reynolds, R.A.; Perry, V.H. Early hippocampal synaptic loss precedes neuronal loss and associates with early behavioural deficits in three distinct strains of prion disease. PLoS ONE 2013, 8, e68062. [Google Scholar] [CrossRef] [PubMed]
- Deacon, R.M.; Croucher, A.; Rawlins, J.N. Hippocampal cytotoxic lesion effects on species-typical behaviours in mice. Behav. Brain Res. 2002, 132, 203–213. [Google Scholar] [CrossRef]
- Elmquist, J.K.; Elias, C.F.; Saper, C.B. From lesions to leptin: Hypothalamic control of food intake and body weight. Neuron 1999, 22, 221–232. [Google Scholar] [CrossRef]
- Jeffrey, M.; Halliday, W.G.; Bell, J.; Johnston, A.R.; MacLeod, N.K.; Ingham, C.; Sayers, A.R.; Brown, D.A.; Fraser, J.R. Synapse loss associated with abnormal PrP precedes neuronal degeneration in the scrapie-infected murine hippocampus. Neuropathol. Appl. Neurobiol. 2000, 26, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.C.; Jucker, M. Neurodegenerative diseases: Expanding the prion concept. Annu. Rev. Neurosci. 2015, 38, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. 2003, 4, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Stefani, M.; Dobson, C.M. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 2003, 81, 678–699. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Folding proteins in fatal ways. Nature 2003, 426, 900–904. [Google Scholar] [CrossRef] [PubMed]
- Piccardo, P.; King, D.; Telling, G.; Manson, J.C.; Barron, R.M. Dissociation of prion protein amyloid seeding from transmission of a spongiform encephalopathy. J. Virol. 2013, 87, 12349–12356. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, M.; McGovern, G.; Chambers, E.V.; King, D.; González, L.; Manson, J.C.; Ghetti, B.; Piccardo, P.; Barron, R.M. Mechanism of PrP-amyloid formation in mice without transmissible spongiform encephalopathy. Brain Pathol. 2012, 22, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.; Jucker, M. The amyloid state of proteins in human diseases. Cell 2012, 148, 1188–1203. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.D.; Lansbury, P.T., Jr. Models of amyloid seeding in alzheimer’s disease and scrapie: Mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu. Rev. Biochem. 1997, 66, 385–407. [Google Scholar] [CrossRef] [PubMed]
- Alibhai, J.; Perry, V.H.; Manson, J. Prion diseases in animals: The role of the misfolded prion protein in neurodegeneration. Prion 2014, 8, 59–109. [Google Scholar]
- Watts, J.C.; Giles, K.; Oehler, A.; Middleton, L.; Dexter, D.T.; Gentleman, S.M.; DeArmond, S.J.; Prusiner, S.B. Transmission of multiple system atrophy prions to transgenic mice. Proc. Natl. Acad. Sci. USA 2013, 110, 19555–19560. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B.; Woerman, A.L.; Mordes, D.A.; Watts, J.C.; Rampersaud, R.; Berry, D.B.; Patel, S.; Oehler, A.; Lowe, J.K.; Kravitz, S.N.; et al. Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. USA 2015, 112, E5308–E5317. [Google Scholar] [CrossRef] [PubMed]
- Ridley, R.M.; Baker, H.F.; Windle, C.P.; Cummings, R.M. Very long term studies of the seeding of beta-amyloidosis in primates. J. Neural Transm. 2006, 113, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Iba, M.; Guo, J.L.; McBride, J.D.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of alzheimer’s-like tauopathy. J. Neurosci. 2013, 33, 1024–1037. [Google Scholar] [CrossRef] [PubMed]
- Dujardin, S.; Lecolle, K.; Caillierez, R.; Begard, S.; Zommer, N.; Lachaud, C.; Carrier, S.; Dufour, N.; Auregan, G.; Winderickx, J.; et al. Neuron-to-neuron wild-type Tau protein transfer through a trans-synaptic mechanism: Relevance to sporadic tauopathies. Acta Neuropathol. Commun. 2014, 2, 14. [Google Scholar] [CrossRef] [PubMed]
- Eisele, Y.S.; Obermuller, U.; Heilbronner, G.; Baumann, F.; Kaeser, S.A.; Wolburg, H.; Walker, L.C.; Staufenbiel, M.; Heikenwalder, M.; Jucker, M. Peripherally applied Aβ-containing inoculates induce cerebral β-amyloidosis. Science 2010, 330, 980–982. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Hench, J.; Lavenir, I.; Schweighauser, G.; Frank, S.; Goedert, M.; Tolnay, M. Peripheral administration of tau aggregates triggers intracerebral tauopathy in transgenic mice. Acta Neuropathol. 2014, 127, 299–301. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Lee, V.M. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat. Med. 2014, 20, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, M.; Zurzolo, C. The cell biology of prion-like spread of protein aggregates: Mechanisms and implication in neurodegeneration. Biochem. J. 2013, 452, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Morales, R.; Callegari, K.; Soto, C. Prion-like features of misfolded abeta and tau aggregates. Virus Res. 2015, 207, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, H.A.; Prusiner, S.B.; Stowring, L.E.; DeArmond, S.J. Scrapie prion proteins are synthesized in neurons. Am. J. Pathol. 1986, 122, 1–5. [Google Scholar] [PubMed]
- Manson, J.; West, J.D.; Thomson, V.; McBride, P.; Kaufman, M.H.; Hope, J. The prion protein gene: A role in mouse embryogenesis? Development 1992, 115, 117–122. [Google Scholar] [PubMed]
- Race, R.E.; Priola, S.A.; Bessen, R.A.; Ernst, D.; Dockter, J.; Rall, G.F.; Mucke, L.; Chesebro, B.; Oldstone, M.B. Neuron-specific expression of a hamster prion protein minigene in transgenic mice induces susceptibility to hamster scrapie agent. Neuron 1995, 15, 1183–1191. [Google Scholar] [CrossRef]
- Mallucci, G.; Dickinson, A.; Linehan, J.; Klohn, P.C.; Brandner, S.; Collinge, J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 2003, 302, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Mallucci, G.R.; White, M.D.; Farmer, M.; Dickinson, A.; Khatun, H.; Powell, A.D.; Brandner, S.; Jefferys, J.G.; Collinge, J. Targeting cellular prion protein reverses early cognitive deficits and neurophysiological dysfunction in prion-infected mice. Neuron 2007, 53, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Mirabile, I.; Jat, P.S.; Brandner, S.; Collinge, J. Identification of clinical target areas in the brainstem of prion-infected mice. Neuropathol. Appl. Neurobiol. 2015, 41, 613–630. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.; Bradford, B.; Baybutt, H.; Marshall, A.; Brown, D.; Kisielewski, D.; Alibhai, J.; Barron, R.; Piccardo, P.; Whitehouse, I.; et al. O32 pathways to neurodegeneration associated with protein misfolding. In Presented at the PRION 2011, Montreal, QC, Canada, May 2011.
- Baker, C.A.; Martin, D.; Manuelidis, L. Microglia from creutzfeldt-jakob disease-infected brains are infectious and show specific mrna activation profiles. J. Virol. 2002, 76, 10905–10913. [Google Scholar] [CrossRef] [PubMed]
- Moser, M.; Colello, R.J.; Pott, U.; Oesch, B. Developmental expression of the prion protein gene in glial cells. Neuron 1995, 14, 509–517. [Google Scholar] [CrossRef]
- Van Keulen, L.J.M.; Schreuder, B.E.C.; Meloen, R.H.; Poelen-van den Berg, M.; Mooij-Harkes, G.; Vromans, M.E.W.; Langeveld, J.P.M. Immumohistochemical detection and localization of prion protein in brain tissue of sheep with natural scrapie. Vet. Pathol. 1995, 32, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Raeber, A.J.; Race, R.E.; Brandner, S.; Priola, S.A.; Sailer, A.; Bessen, R.A.; Mucke, L.; Manson, J.; Aguzzi, A.; Oldstone, M.B.; et al. Astrocyte-specific expression of hamster prion protein (PrP) renders PrP knockout mice susceptible to hamster scrapie. EMBO J. 1997, 16, 6057–6065. [Google Scholar] [CrossRef] [PubMed]
- Bruce, M.E.; McBride, P.A.; Jeffrey, M.; Scott, J.R. PrP in pathology and pathogenesis in scrapie-infected mice. Mol. Neurobiol. 1994, 8, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.; Lee, I.Y.; Yoo, H.; Gehlenborg, N.; Cho, J.H.; Petritis, B.; Baxter, D.; Pitstick, R.; Young, R.; Spicer, D.; et al. A systems approach to prion disease. Mol. Syst. Biol. 2009, 5, 252. [Google Scholar] [CrossRef] [PubMed]
- Schenk, D.; Barbour, R.; Dunn, W.; Gordon, G.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; Khan, K.; et al. Immunization with amyloid-β attenuates alzheimer-disease-like pathology in the PDAPP mouse. Nature 1999, 400, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Reactive astrocytes in neural repair and protection. Neuroscience 2005, 11, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Diedrich, J.F.; Bendheim, P.E.; Kim, Y.S.; Carp, R.I.; Haase, A.T. Scrapie-associated prion protein accumulates in astrocytes during scrapie infection. Proc. Natl. Acad. Sci. USA 1991, 88, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Pekny, M.; Nilsson, M. Astrocyte activation and reactive gliosis. Glia 2005, 50, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Perry, V.H.; Cunningham, C.; Boche, D. Atypical inflammation in the central nervous system in prion disease. Curr. Opin. Neurol. 2002, 15, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Boche, D.; Cunningham, C.; Docagne, F.; Scott, H.; Perry, V.H. Tgfbeta1 regulates the inflammatory response during chronic neurodegeneration. Neurobiol. Dis. 2006, 22, 638–650. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Nicola, D.; Fransen, N.L.; Suzzi, S.; Perry, V.H. Regulation of microglial proliferation during chronic neurodegeneration. J. Neurosci. 2013, 33, 2481–2493. [Google Scholar] [CrossRef] [PubMed]
- Bradford, B.M.; Mabbott, N.A. Prion disease and the innate immune system. Viruses 2012, 4, 3389–3419. [Google Scholar] [CrossRef] [PubMed]
- Tamguney, G.; Giles, K.; Glidden, D.V.; Lessard, P.; Wille, H.; Tremblay, P.; Groth, D.F.; Yehiely, F.; Korth, C.; Moore, R.C.; et al. Genes contributing to prion pathogenesis. J. Gen. Virol. 2008, 89, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Felton, L.M.; Cunningham, C.; Rankine, E.L.; Waters, S.; Boche, D.; Perry, V.H. Mcp-1 and murine prion disease: Separation of early behavioural dysfunction from overt clinical disease. Neurobiol. Dis. 2005, 20, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Riemer, C.; Schultz, J.; Burwinkel, M.; Schwarz, A.; Mok, S.W.; Gultner, S.; Bamme, T.; Norley, S.; van Landeghem, F.; Lu, B.; et al. Accelerated prion replication in, but prolonged survival times of, prion-infected cxcr3-/- mice. J. Virol. 2008, 82, 12464–12471. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Hasebe, R.; Takahashi, Y.; Song, C.H.; Suzuki, A.; Yamasaki, T.; Horiuchi, M. Absence of cd14 delays progression of prion diseases accompanied by increased microglial activation. J. Virol. 2013, 87, 13433–13445. [Google Scholar] [CrossRef] [PubMed]
- Thackray, A.M.; McKenzie, A.N.; Klein, M.A.; Lauder, A.; Bujdoso, R. Accelerated prion disease in the absence of interleukin-10. J. Virol. 2004, 78, 13697–13707. [Google Scholar] [CrossRef] [PubMed]
- LaCasse, R.A.; Striebel, J.F.; Favara, C.; Kercher, L.; Chesebro, B. Role of erk1/2 activation in prion disease pathogenesis: Absence of ccr1 leads to increased erk1/2 activation and accelerated disease progression. J. Neuroimmunol. 2008, 196, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Silveira, J.R.; Raymond, G.J.; Hughson, A.G.; Race, R.E.; Sim, V.L.; Hayes, S.F.; Caughey, B. The most infectious prion protein particles. Nature 2005, 437, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Molecular biology of prion diseases. Science 1991, 252, 1515–1522. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.; Liberski, P.P.; Wolff, A.; Gajdusek, D.C. Conservation of infectivity in purified fibrillary extracts of scrapie-infected hamster brain after sequential enzymatic digestion or polyacrylamide gel electrophoresis. Proc. Natl. Acad. Sci. USA 1990, 87, 7240–7244. [Google Scholar] [CrossRef] [PubMed]
- Safar, J.; Wang, W.; Padgett, M.P.; Ceroni, M.; Piccardo, P.; Zopf, D.; Gajdusek, D.C.; Gibbs, C.J., Jr. Molecular mass, biochemical composition, and physicochemical behavior of the infectious form of the scrapie precursor protein monomer. Proc. Natl. Acad. Sci. USA 1990, 87, 6373–6377. [Google Scholar] [CrossRef] [PubMed]
- Hope, J. The nature of the scrapie agent: The evolution of the virino. Ann. N. Y. Acad. Sci. 1994, 724, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Morillas, M.; Vanik, D.L.; Surewicz, W.K. On the mechanism of α-helix to β-sheet transition in the recombinant prion protein. Biochemistry 2001, 40, 6982–6987. [Google Scholar] [CrossRef] [PubMed]
- Alper, T.; Haig, D.A.; Clarke, M.C. The exceptionally small size of the scrapie agent. Biochem. Biophys. Res. Commun. 1966, 22, 278–284. [Google Scholar] [CrossRef]
- Gabizon, R.; McKinley, M.P.; Prusiner, S.B. Purified prion proteins and scrapie infectivity copartition into liposomes. Proc. Natl. Acad. Sci. USA 1987, 84, 4017–4021. [Google Scholar] [CrossRef] [PubMed]
- Legname, G.; Baskakov, I.V.; Nguyen, H.O.B.; Riesner, D.; Cohen, F.E.; DeArmond, S.J.; Prusiner, S.B. Synthetic mammalian prions. Science 2004, 305, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Makarava, N.; Kovacs, G.G.; Bocharova, O.; Savtchenko, R.; Alexeeva, I.; Budka, H.; Rohwer, R.G.; Baskakov, I.V. Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol. 2010, 119, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.I.; Cali, I.; Surewicz, K.; Kong, Q.; Raymond, G.J.; Atarashi, R.; Race, B.; Qing, L.; Gambetti, P.; Caughey, B.; et al. Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J. Biol. Chem. 2010, 285, 14083–14087. [Google Scholar] [CrossRef] [PubMed]
- Makarava, N.; Kovacs, G.G.; Savtchenko, R.; Alexeeva, I.; Ostapchenko, V.G.; Budka, H.; Rohwer, R.G.; Baskakov, I.V. A new mechanism for transmissible prion diseases. J. Neurosci. 2012, 32, 7345–7355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raymond, G.J.; Race, B.; Hollister, J.R.; Offerdahl, D.K.; Moore, R.A.; Kodali, R.; Raymond, L.D.; Hughson, A.G.; Rosenke, R.; Long, D.; et al. Isolation of novel synthetic prion strains by amplification in transgenic mice coexpressing wild-type and anchorless prion proteins. J. Virol. 2012, 86, 11763–11778. [Google Scholar] [CrossRef] [PubMed]
- Colby, D.W.; Giles, K.; Legname, G.; Wille, H.; Baskakov, I.V.; DeArmond, S.J.; Prusiner, S.B. Design and construction of diverse mammalian prion strains. Proc. Natl. Acad. Sci. USA 2009, 106, 20417–20422. [Google Scholar] [CrossRef] [PubMed]
- Deleault, N.R.; Lucassen, R.W.; Supattapone, S. RNA molecules stimulate prion protein conversion. Nature 2003, 425, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Deleault, N.R.; Piro, J.R.; Walsh, D.J.; Wang, F.; Ma, J.; Geoghegan, J.C.; Supattapone, S. Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc. Natl. Acad. Sci. USA 2012, 109, 8546–8551. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, Z.; Wang, X.; Li, J.; Zha, L.; Yuan, C.G.; Weissmann, C.; Ma, J. Genetic informational rna is not required for recombinant prion infectivity. J. Virol. 2012, 86, 1874–1876. [Google Scholar] [CrossRef] [PubMed]
- Saunders, S.E.; Bartelt-Hunt, S.L.; Bartz, J.C. Occurrence, transmission, and zoonotic potential of chronic wasting disease. Emerg. Infect. Dis. 2012, 18, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Detwiler, L.A.; Baylis, M. The epidemiology of scrapie. Rev. Sci. Tech. 2003, 22, 121–143. [Google Scholar] [PubMed]
- Bruce, M.E.; Will, R.G.; Ironside, J.W.; McConnell, I.; Drummond, D.; Suttie, A.; McCardle, L.; Chree, A.; Hope, J.; Birkett, C.; et al. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature 1997, 389, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.F.; Desbruslais, M.; Joiner, S.; Sidle, K.C.; Gowland, I.; Collinge, J.; Doey, L.J.; Lantos, P. The same prion strain causes vCJD and BSE. Nature 1997, 389, 448–450. [Google Scholar] [CrossRef] [PubMed]
- Doherr, M.G. Bovine spongiform encephalopathy (BSE)-infectious, contagious, zoonotic or production disease? Acta Vet. Scand. Suppl. 2003, 98 (Suppl. 1), S33–S42. [Google Scholar] [CrossRef]
- Brown, P.; Gibbs, C.J., Jr.; Rodgers-Johnson, P.; Asher, D.M.; Sulima, M.P.; Bacote, A.; Goldfarb, L.G.; Gajdusek, D.C. Human spongiform encephalopathy: The national institutes of health series of 300 cases of experimentally transmitted disease. Ann. Neurol. 1994, 35, 513–529. [Google Scholar] [CrossRef] [PubMed]
- Diack, A.B.; Ritchie, D.L.; Peden, A.H.; Brown, D.; Boyle, A.; Morabito, L.; Maclennan, D.; Burgoyne, P.; Jansen, C.; Knight, R.S.; et al. Variably protease-sensitive prionopathy, a unique prion variant with inefficient transmission properties. Emerg. Infect. Dis. 2014, 20, 1969–1979. [Google Scholar] [CrossRef] [PubMed]
- Notari, S.; Xiao, X.; Espinosa, J.C.; Cohen, Y.; Qing, L.; Aguilar-Calvo, P.; Kofskey, D.; Cali, I.; Cracco, L.; Kong, Q.; et al. Transmission characteristics of variably protease-sensitive prionopathy. Emerg. Infect. Dis. 2014, 20, 2006–2014. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.; Preece, M.; Brandel, J.P.; Sato, T.; McShane, L.; Zerr, I.; Fletcher, A.; Will, R.G.; Pocchiari, M.; Cashman, N.R.; et al. Iatrogenic creutzfeldt-jakob disease at the millennium. Neurology 2000, 55, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Bruce, M.E. TSE strain variation. Br. Med. Bull. 2003, 66, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, R.G. The scrapie agent: “A virus by any other name”. Curr. Top. Microbiol. Immunol. 1991, 172, 195–232. [Google Scholar] [PubMed]
- Somerville, R.A. Tse agent strains and PrP: Reconciling structure and function. Trends Biochem. Sci. 2002, 27, 606–612. [Google Scholar] [CrossRef]
- Bessen, R.A.; Marsh, R.F. Distinct PrP properties suggest the molecular-basis of strain variation in transmissible mink encephalopathy. J. Virol. 1994, 68, 7859–7868. [Google Scholar] [PubMed]
- Peretz, D.; Williamson, R.A.; Legname, G.; Matsunaga, Y.; Vergara, J.; Burton, D.R.; DeArmond, S.J.; Prusiner, S.B.; Scott, M.R. A change in the conformation of prions accompanies the emergence of a new prion strain. Neuron 2002, 34, 921–932. [Google Scholar] [CrossRef]
- Aguzzi, A.; Heikenwalder, M.; Polymenidou, M. Insights into prion strains and neurotoxicity. Nat. Rev. Mol. Cell Biol. 2007, 8, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Angers, R.C.; Kang, H.E.; Napier, D.; Browning, S.; Seward, T.; Mathiason, C.; Balachandran, A.; McKenzie, D.; Castilla, J.; Soto, C.; et al. Prion strain mutation determined by prion protein conformational compatibility and primary structure. Science 2010, 328, 1154–1158. [Google Scholar] [CrossRef] [PubMed]
- Crowell, J.; Hughson, A.; Caughey, B.; Bessen, R.A. Host determinants of prion strain diversity independent of prion protein genotype. J. Virol. 2015, 89, 10427–10441. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.L.; Kim, C.; Haldiman, T.; ElHag, M.; Mehndiratta, P.; Pichet, T.; Lissemore, F.; Shea, M.; Cohen, Y.; Chen, W.; et al. Rapidly progressive Alzheimer’s disease features distinct structures of amyloid-β. Brain 2015, 138, 1009–1022. [Google Scholar] [CrossRef] [PubMed]
- Sanders, D.W.; Kaufman, S.K.; DeVos, S.L.; Sharma, A.M.; Mirbaha, H.; Li, A.; Barker, S.J.; Foley, A.C.; Thorpe, J.R.; Serpell, L.C.; et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 2014, 82, 1271–1288. [Google Scholar] [CrossRef] [PubMed]
- Peelaerts, W.; Bousset, L.; Van der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; Van den Haute, C.; Melki, R.; Baekelandt, V. α-synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015, 522, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Bousset, L.; Pieri, L.; Ruiz-Arlandis, G.; Gath, J.; Jensen, P.H.; Habenstein, B.; Madiona, K.; Olieric, V.; Böckmann, A.; Meier, B.H.; et al. Structural and functional characterization of two alpha-synuclein strains. Nat. Commun. 2013, 4, 2575. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B.; Scott, M.; Foster, D.; Pan, K.M.; Groth, D.; Mirenda, C.; Torchia, M.; Yang, S.L.; Serban, D.; Carlson, G.A.; et al. Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell 1990, 63, 673–686. [Google Scholar] [CrossRef]
- Collinge, J.; Clarke, A.R. A general model of prion strains and their pathogenicity. Science 2007, 318, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Cancellotti, E.; Barron, R.M.; Bishop, M.T.; Hart, P.; Wiseman, F.; Manson, J.C. The role of host PrP in transmissible spongiform encephalopathies. Biochim. Biophys. Acta 2007, 1772, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Nonno, R.; Di Bari, M.A.; Cardone, F.; Vaccari, G.; Fazzi, P.; Dell’Omo, G.; Cartoni, C.; Ingrosso, L.; Boyle, A.; Galeno, R.; et al. Efficient transmission and characterization of Creutzfeldt-Jakob disease strains in bank voles. PLoS Pathog. 2006, 2, e12. [Google Scholar] [CrossRef] [PubMed]
- Cancellotti, E.; Bradford, B.M.; Tuzi, N.L.; Hickey, R.D.; Brown, D.; Brown, K.L.; Barron, R.M.; Kisielewski, D.; Piccardo, P.; Manson, J.C. Glycosylation of PrPC determines timing of neuroinvasion and targeting in the brain following transmissible spongiform encephalopathy infection by a peripheral route. J. Virol. 2010, 84, 3464–3475. [Google Scholar] [CrossRef] [PubMed]
- Cancellotti, E.; Mahal, S.P.; Somerville, R.; Diack, A.; Brown, D.; Piccardo, P.; Weissmann, C.; Manson, J.C. Post-translational changes to PrP alter transmissible spongiform encephalopathy strain properties. EMBO J. 2013, 32, 756–769. [Google Scholar] [CrossRef] [PubMed]
- Barron, R.M.; Campbell, S.L.; King, D.; Bellon, A.; Chapman, K.E.; Williamson, R.A.; Manson, J.C. High titres of tse infectivity associated with extremely low levels of PrPSc in vivo. J. Biol. Chem. 2007, 282, 35878–35886. [Google Scholar] [CrossRef] [PubMed]
- Lasmezas, C.I.; Deslys, J.P.; Robain, O.; Jaegly, A.; Beringue, V.; Peyrin, J.M.; Fournier, J.G.; Hauw, J.J.; Rossier, J.; Dormont, D. Transmission of the BSE agent to mice in the absence of detectable abnormal prion protein. Science 1997, 275, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Andreoletti, O.; Orge, L.; Benestad, S.L.; Beringue, V.; Litaise, C.; Simon, S.; Le Dur, A.; Laude, H.; Simmons, H.; Lugan, S.; et al. Atypical/nor98 scrapie infectivity in sheep peripheral tissues. PLoS Pathog. 2011, 7, e1001285. [Google Scholar] [CrossRef] [PubMed]
- Balkema-Buschmann, A.; Eiden, M.; Hoffmann, C.; Kaatz, M.; Ziegler, U.; Keller, M.; Groschup, M.H. BSE infectivity in the absence of detectable PrPSc accumulation in the tongue and nasal mucosa of terminally diseased cattle. J. Gen. Virol. 2011, 92, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Tzaban, S.; Friedlander, G.; Schonberger, O.; Horonchik, L.; Yedidia, Y.; Shaked, G.; Gabizon, R.; Taraboulos, A. Protease-sensitive scrapie prion protein in aggregates of heterogeneous sizes. Biochemistry 2002, 41, 12868–12875. [Google Scholar] [PubMed]
- Cronier, S.; Gros, N.; Tattum, M.H.; Jackson, G.S.; Clarke, A.R.; Collinge, J.; Wadsworth, J.D. Detection and characterization of proteinase K-sensitive disease-related prion protein with thermolysin. Biochem. J. 2008, 416, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Cali, I.; Dong, Z.; Puoti, G.; Yuan, J.; Qing, L.; Wang, H.; Kong, Q.; Gambetti, P.; Zou, W.Q. Protease-sensitive prions with 144-bp insertion mutations. Aging 2013, 5, 155–173. [Google Scholar] [PubMed]
- Chiesa, R.; Piccardo, P.; Quaglio, E.; Drisaldi, B.; Si-Hoe, S.L.; Takao, M.; Ghetti, B.; Harris, D.A. Molecular distinction between pathogenic and infectious properties of the prion protein. J. Virol. 2003, 77, 7611–7622. [Google Scholar] [CrossRef] [PubMed]
- Korth, C.; Stierli, B.; Streit, P.; Moser, M.; Schaller, O.; Fischer, R.; Schulz-Schaeffer, W.; Kretzschmar, H.; Raeber, A.; Braun, U.; et al. Prion (PrPSc)-specific epitope defined by a monoclonal antibody. Nature 1997, 390, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Bolmont, T.; Clavaguera, F.; Meyer-Luehmann, M.; Herzig, M.C.; Radde, R.; Staufenbiel, M.; Lewis, J.; Hutton, M.; Tolnay, M.; Jucker, M. Induction of tau pathology by intracerebral infusion of amyloid-β-containing brain extract and by amyloid-β deposition in App × Tau transgenic mice. Am. J. Pathol. 2007, 171, 2012–2020. [Google Scholar] [CrossRef] [PubMed]
- Desplats, P.; Lee, H.J.; Bae, E.J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Lee, V.M. Seeding of normal tau by pathological tau conformers drives pathogenesis of Alzheimer-like tangles. J. Biol. Chem. 2011, 286, 15317–15331. [Google Scholar] [CrossRef] [PubMed]
- Krammer, C.; Schatzl, H.M.; Vorberg, I. Prion-like propagation of cytosolic protein aggregates: Insights from cell culture models. Prion 2009, 3, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Bjorklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Lundmark, K.; Westermark, G.T.; Nystrom, S.; Murphy, C.L.; Solomon, A.; Westermark, P. Transmissibility of systemic amyloidosis by a prion-like mechanism. Proc. Natl. Acad. Sci. USA 2002, 99, 6979–6984. [Google Scholar] [CrossRef] [PubMed]
- Munch, C.; O’Brien, J.; Bertolotti, A. Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3548–3553. [Google Scholar] [CrossRef] [PubMed]
- Sydow, A.; Mandelkow, E.M. “Prion-like” propagation of mouse and human tau aggregates in an inducible mouse model of tauopathy. Neurodegener. Dis. 2010, 7, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Grad, L.I.; Guest, W.C.; Yanai, A.; Pokrishevsky, E.; O’Neill, M.A.; Gibbs, E.; Semenchenko, V.; Yousefi, M.; Wishart, D.S.; Plotkin, S.S.; et al. Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16398–16403. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diack, A.B.; Alibhai, J.D.; Barron, R.; Bradford, B.; Piccardo, P.; Manson, J.C. Insights into Mechanisms of Chronic Neurodegeneration. Int. J. Mol. Sci. 2016, 17, 82. https://doi.org/10.3390/ijms17010082
Diack AB, Alibhai JD, Barron R, Bradford B, Piccardo P, Manson JC. Insights into Mechanisms of Chronic Neurodegeneration. International Journal of Molecular Sciences. 2016; 17(1):82. https://doi.org/10.3390/ijms17010082
Chicago/Turabian StyleDiack, Abigail B., James D. Alibhai, Rona Barron, Barry Bradford, Pedro Piccardo, and Jean C. Manson. 2016. "Insights into Mechanisms of Chronic Neurodegeneration" International Journal of Molecular Sciences 17, no. 1: 82. https://doi.org/10.3390/ijms17010082
APA StyleDiack, A. B., Alibhai, J. D., Barron, R., Bradford, B., Piccardo, P., & Manson, J. C. (2016). Insights into Mechanisms of Chronic Neurodegeneration. International Journal of Molecular Sciences, 17(1), 82. https://doi.org/10.3390/ijms17010082