
Isobaric Tags for Relative and Absolute Quantitation (iTRAQ)-Based Comparative Proteome Analysis of the Response of Ramie under Drought Stress

Abstract
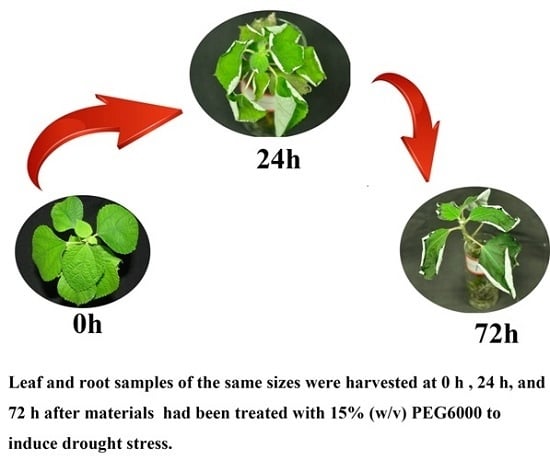
:
1. Introduction
2. Results and Discussion
2.1. Analytical Strategy for Proteome Identification under Drought Stress
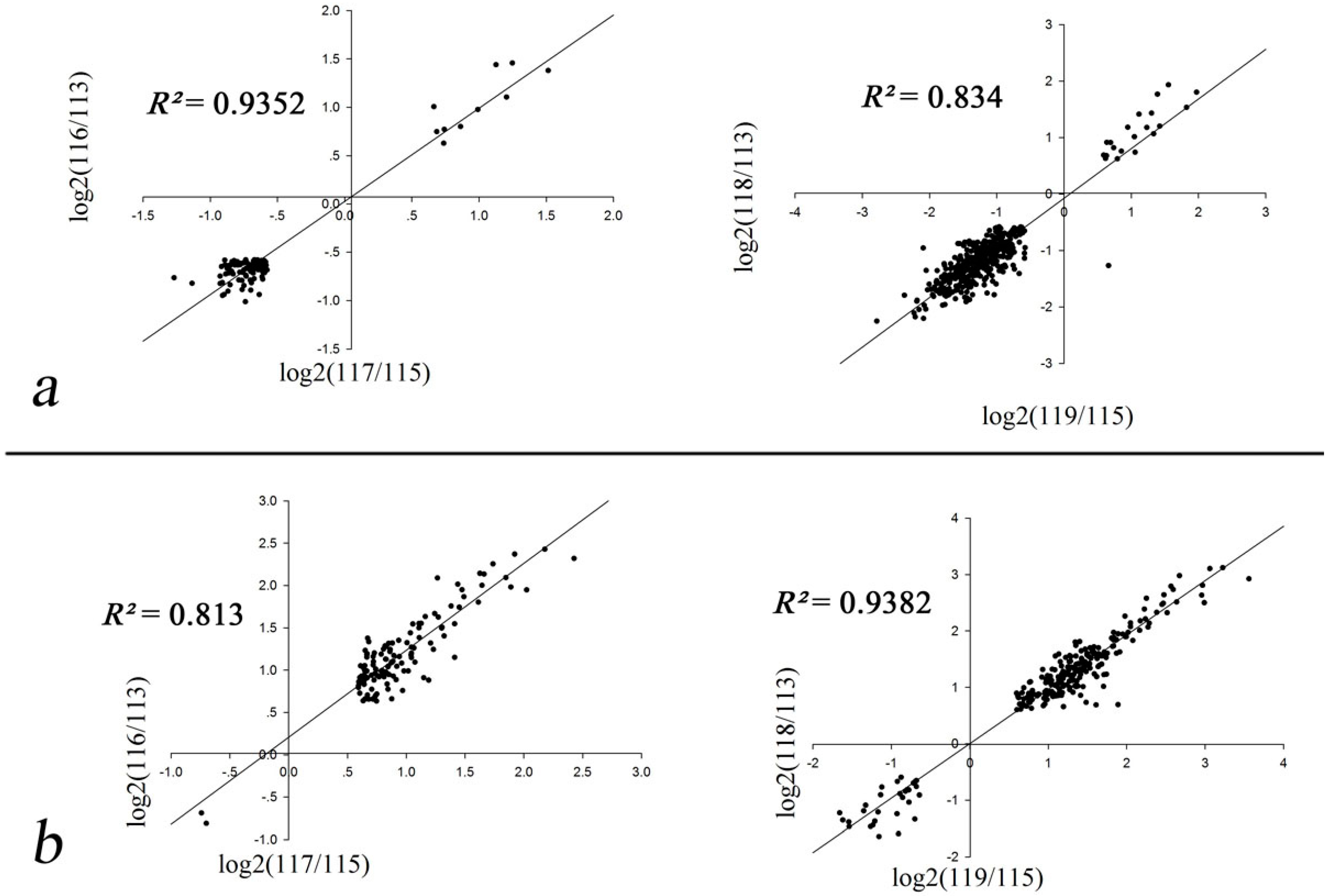
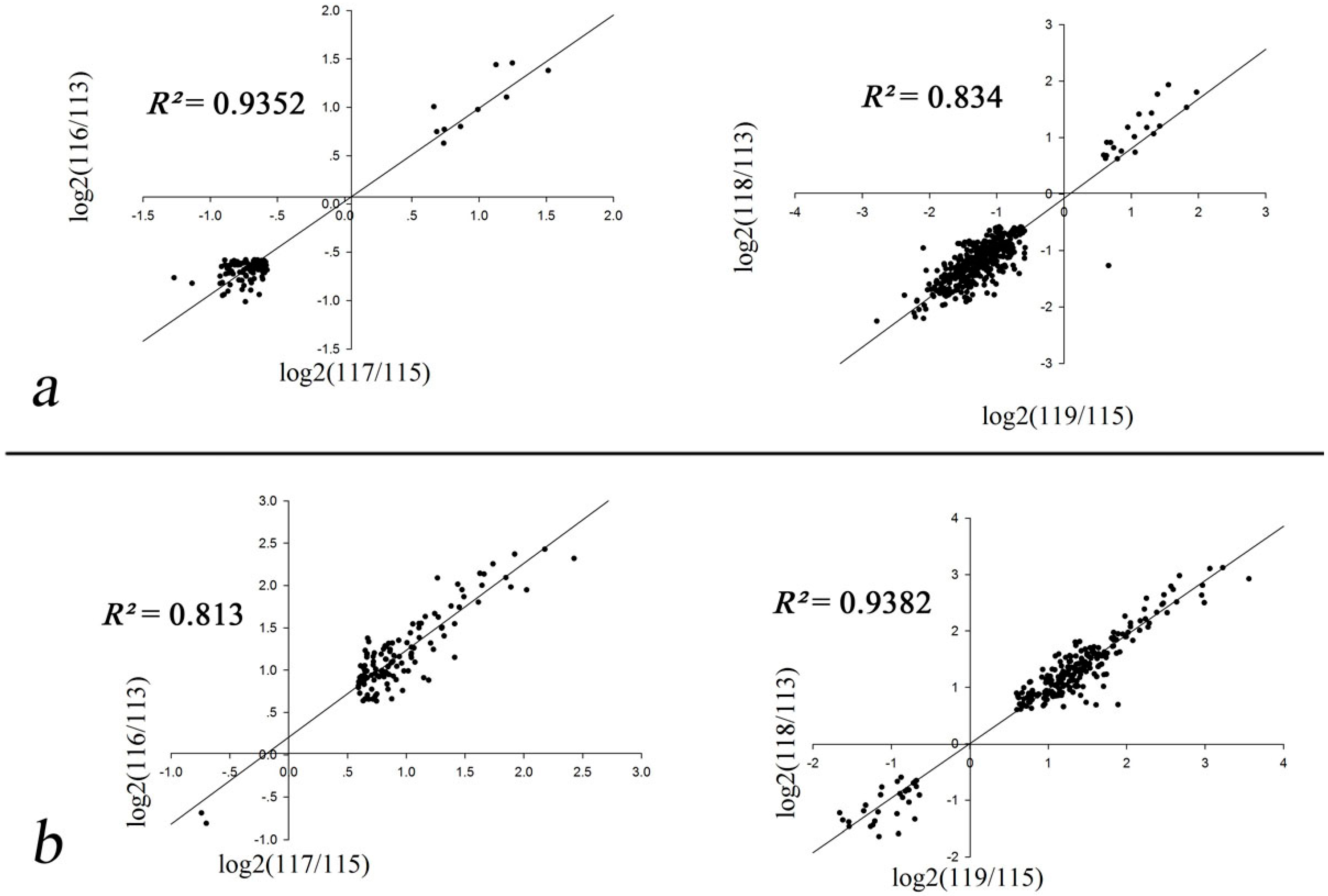
2.2. Correlation Coefficients of Biological Replicates
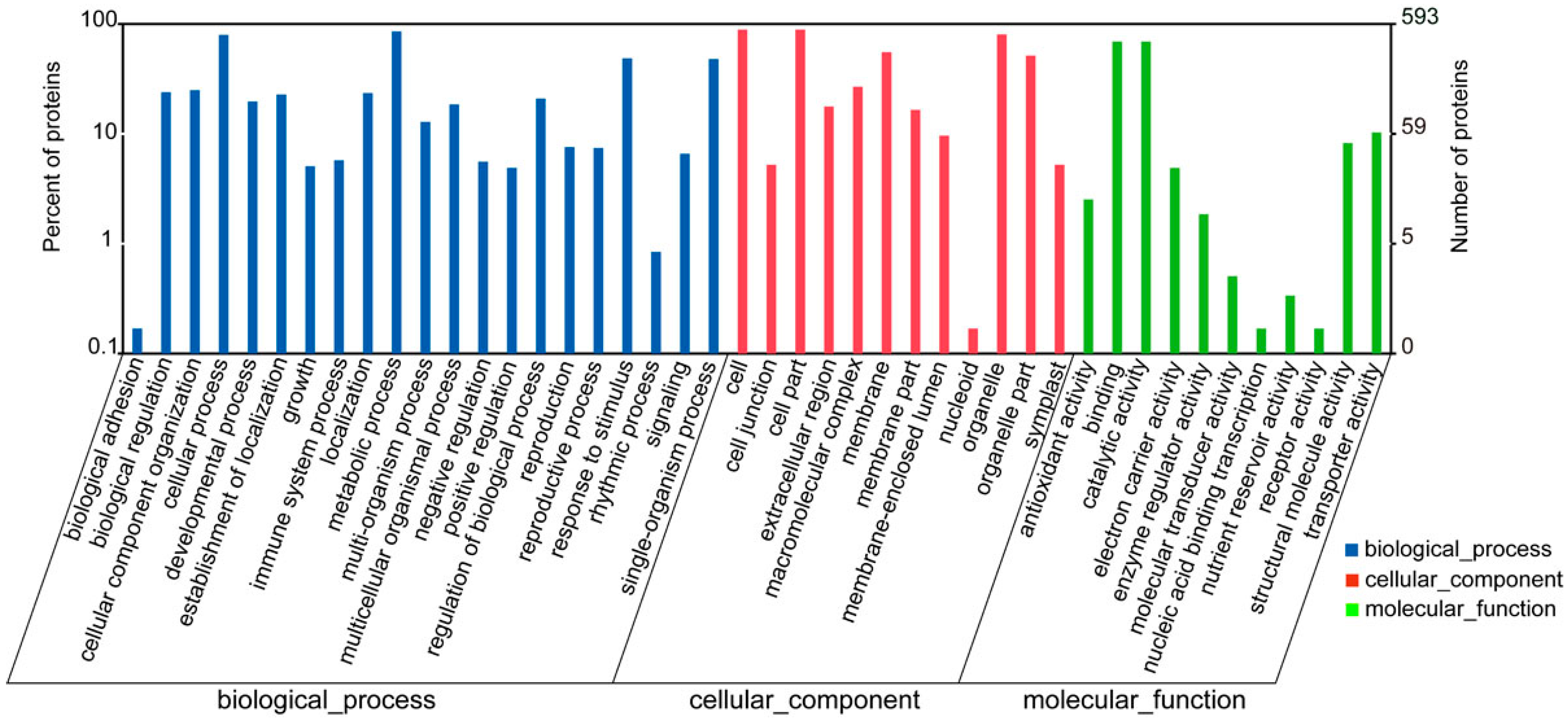
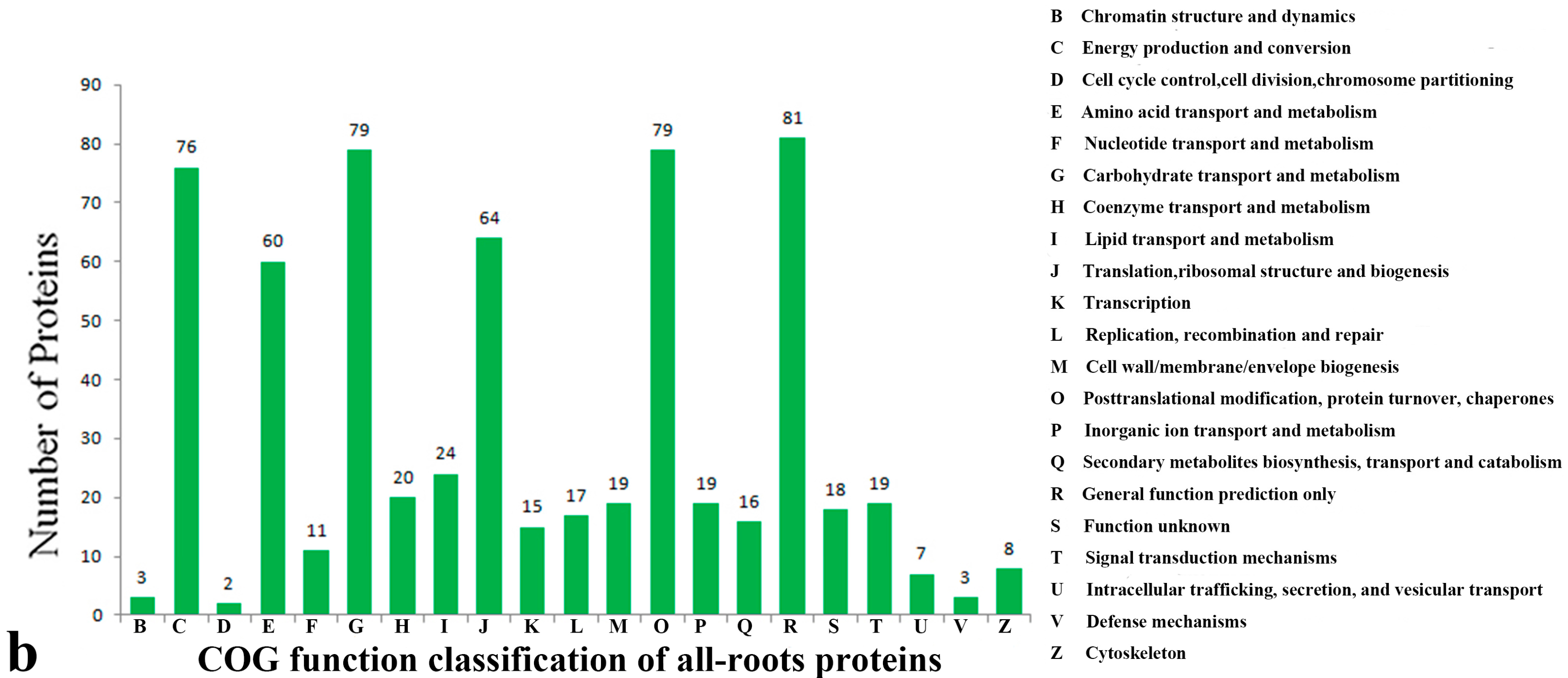
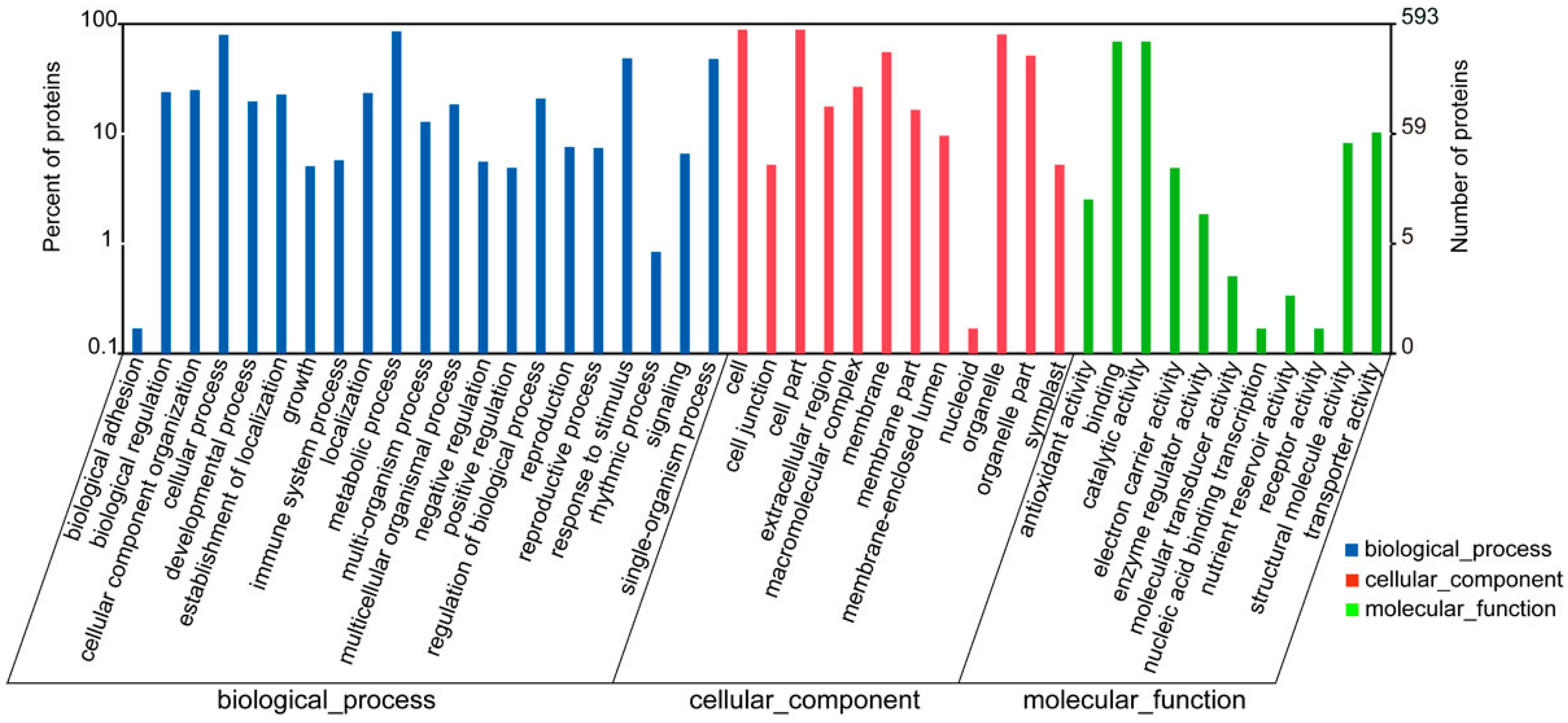
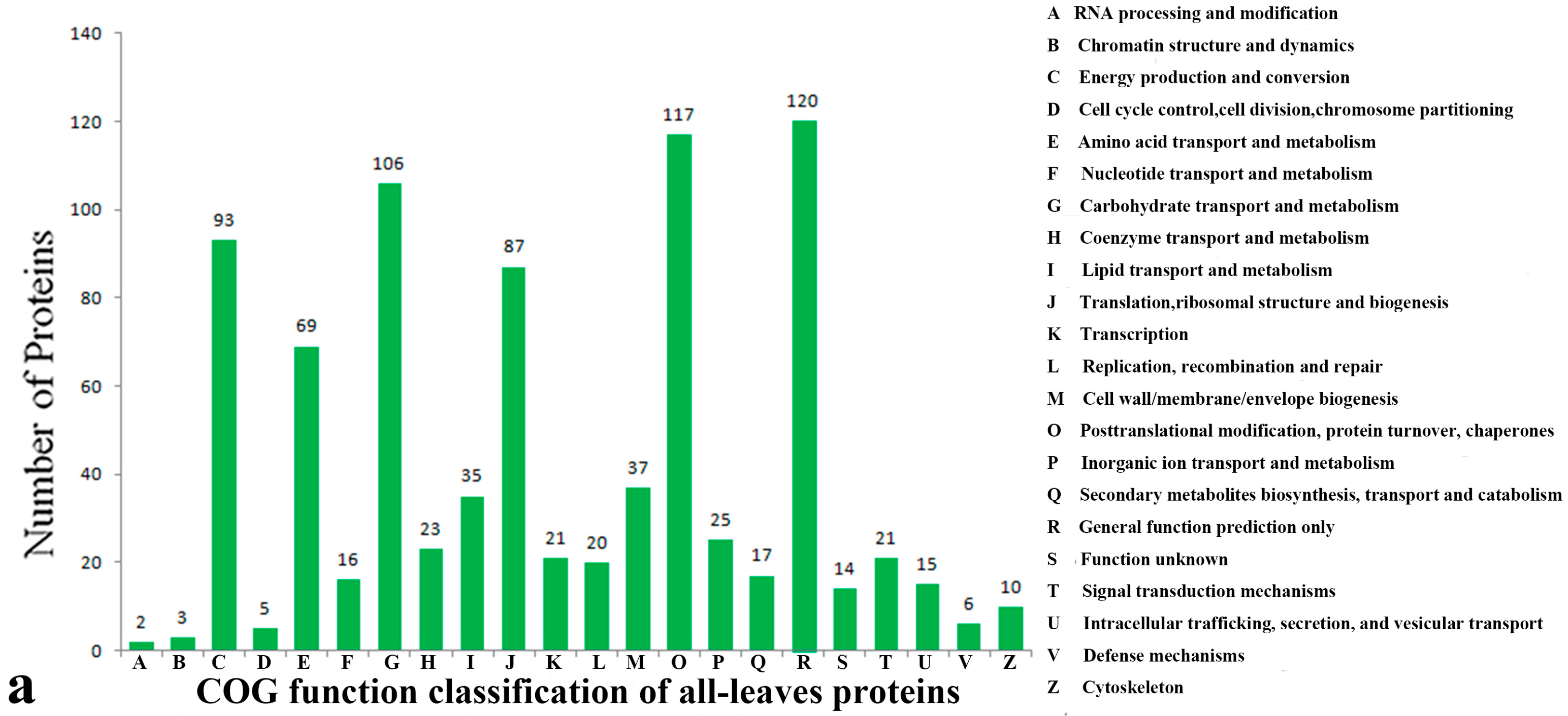
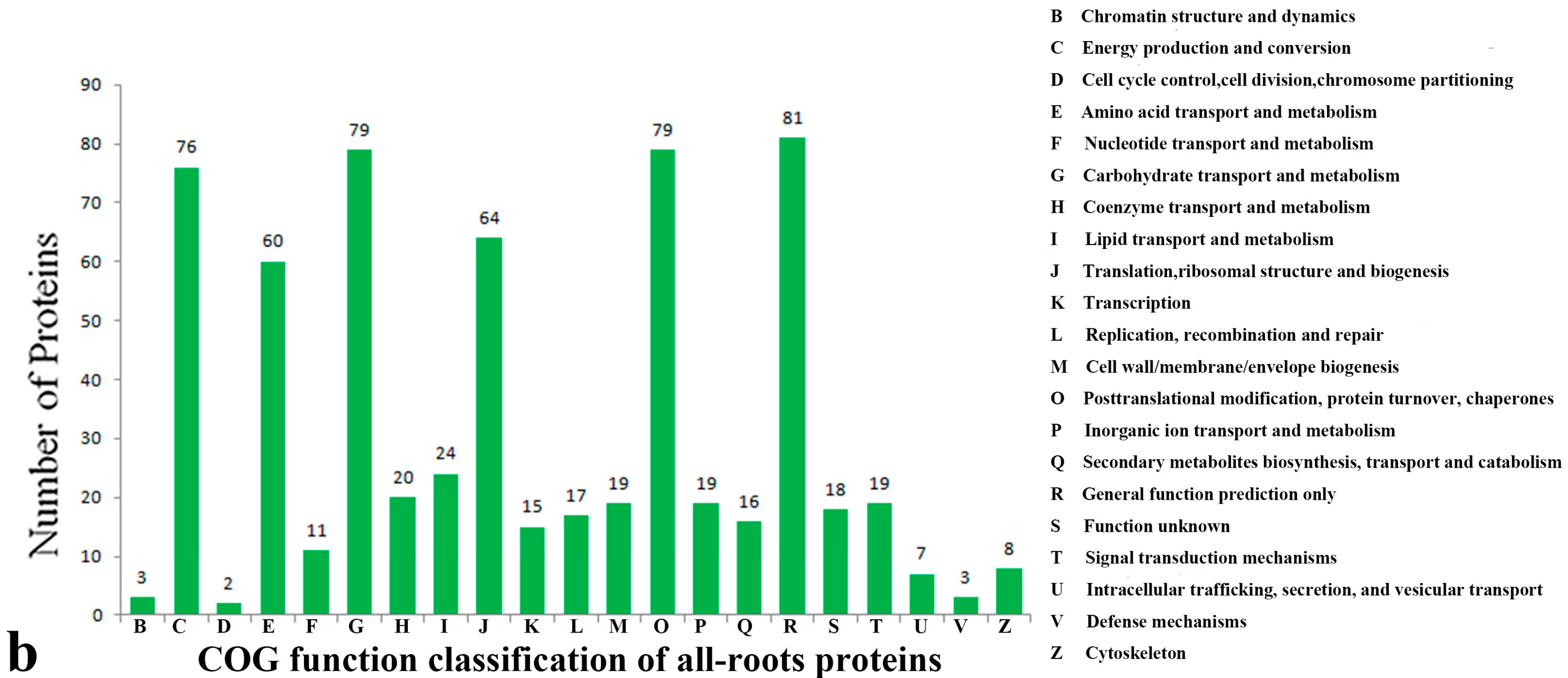
2.3. Functional Classification and Annotation
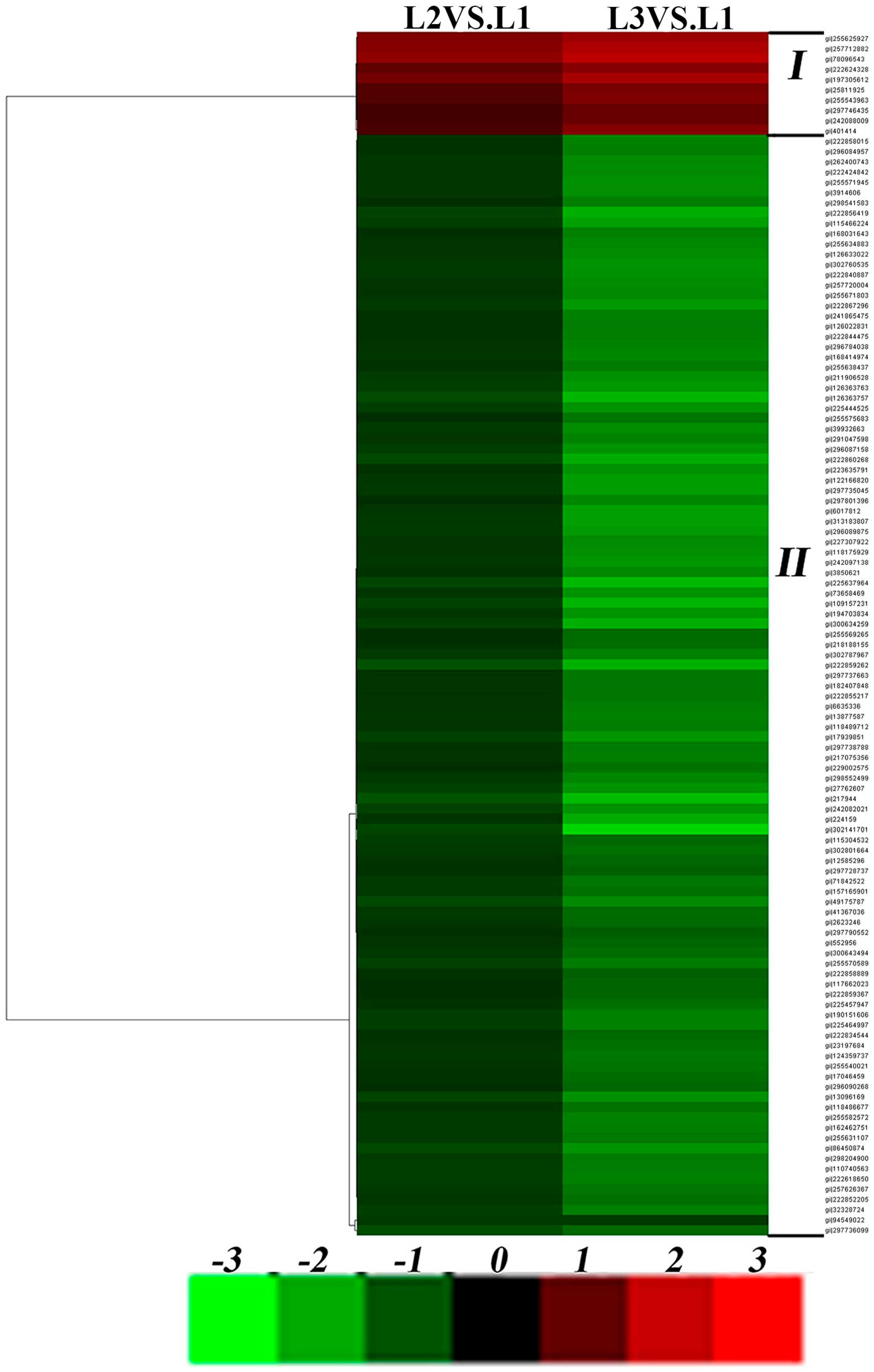
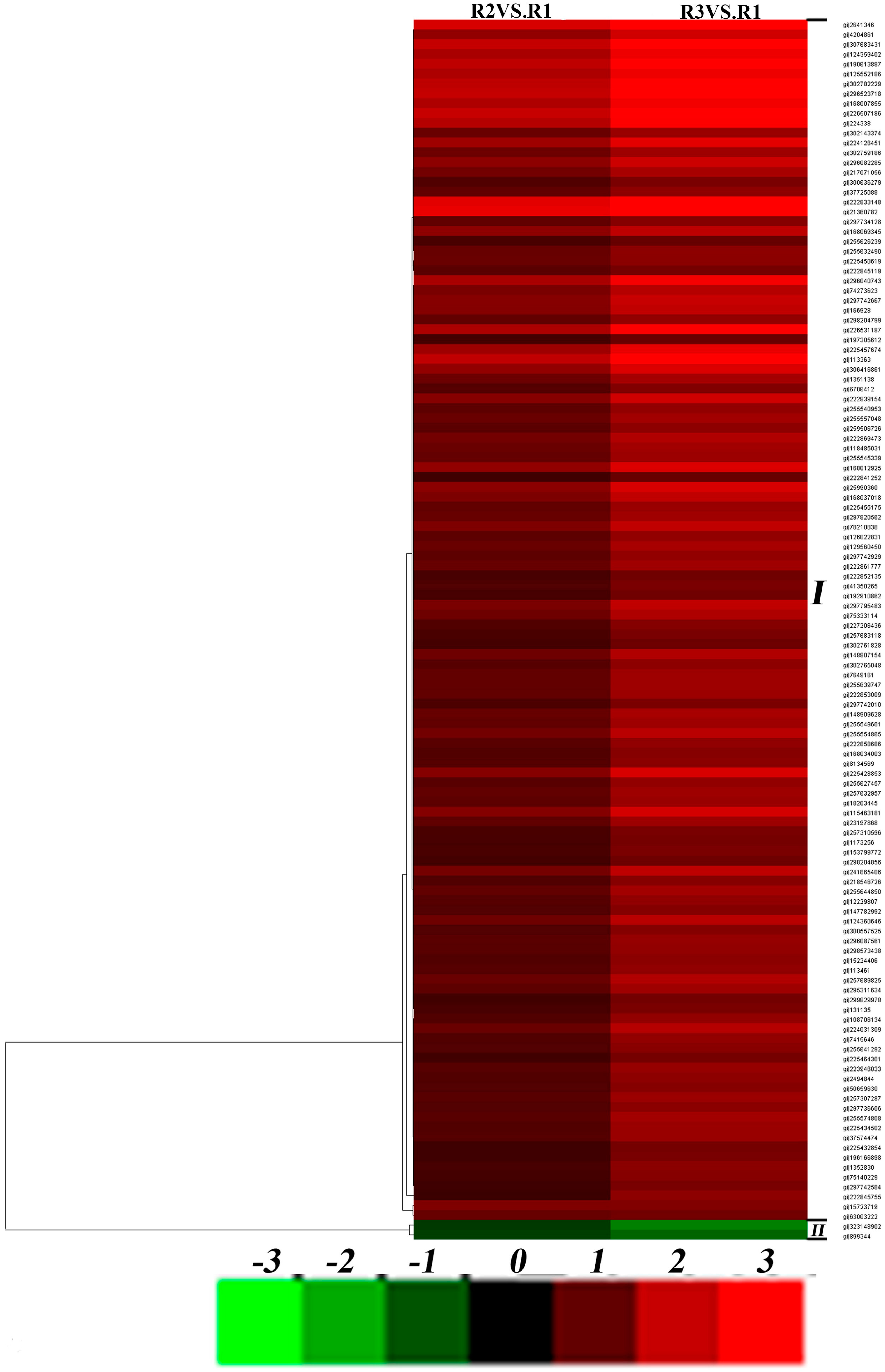
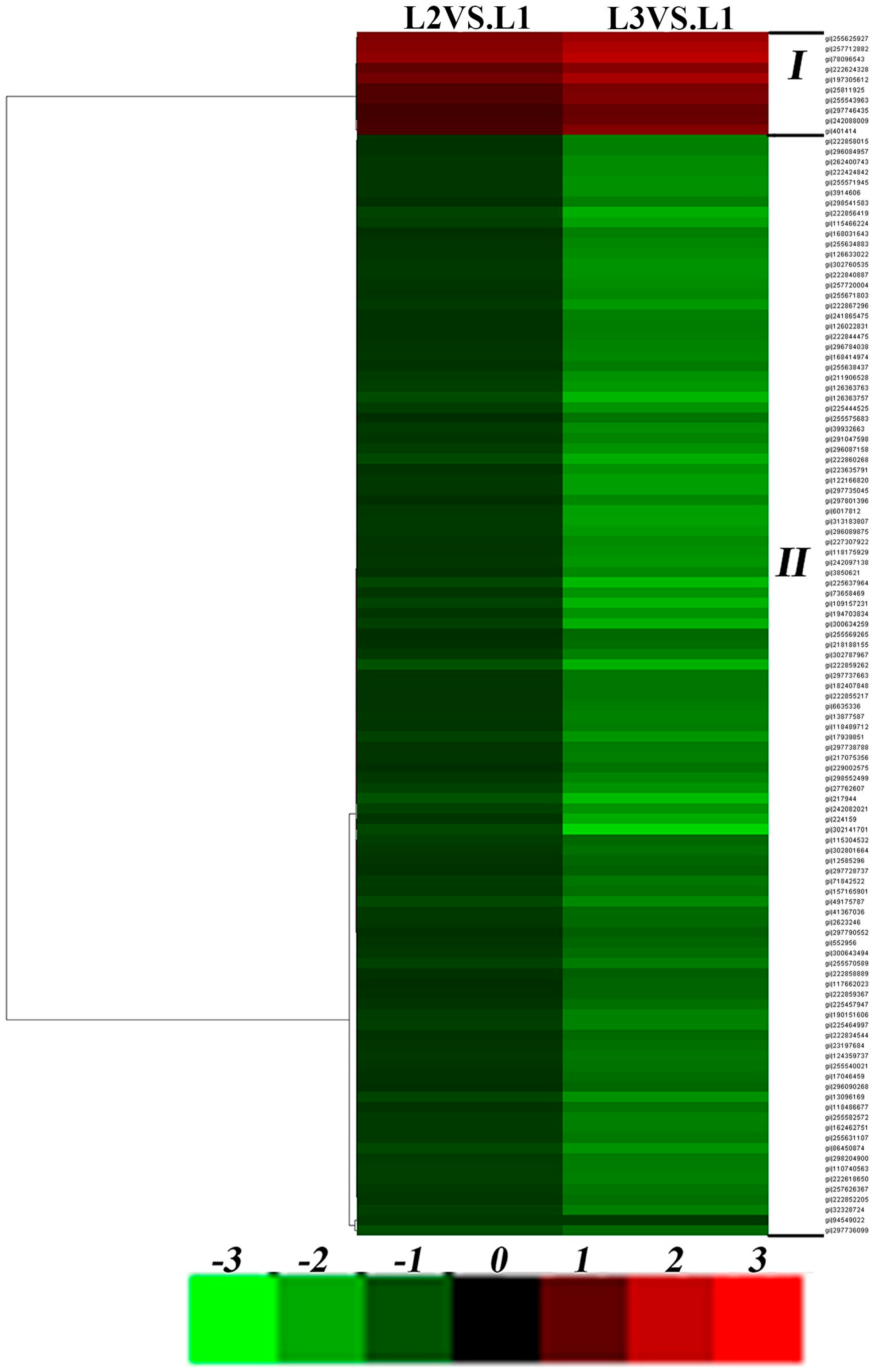
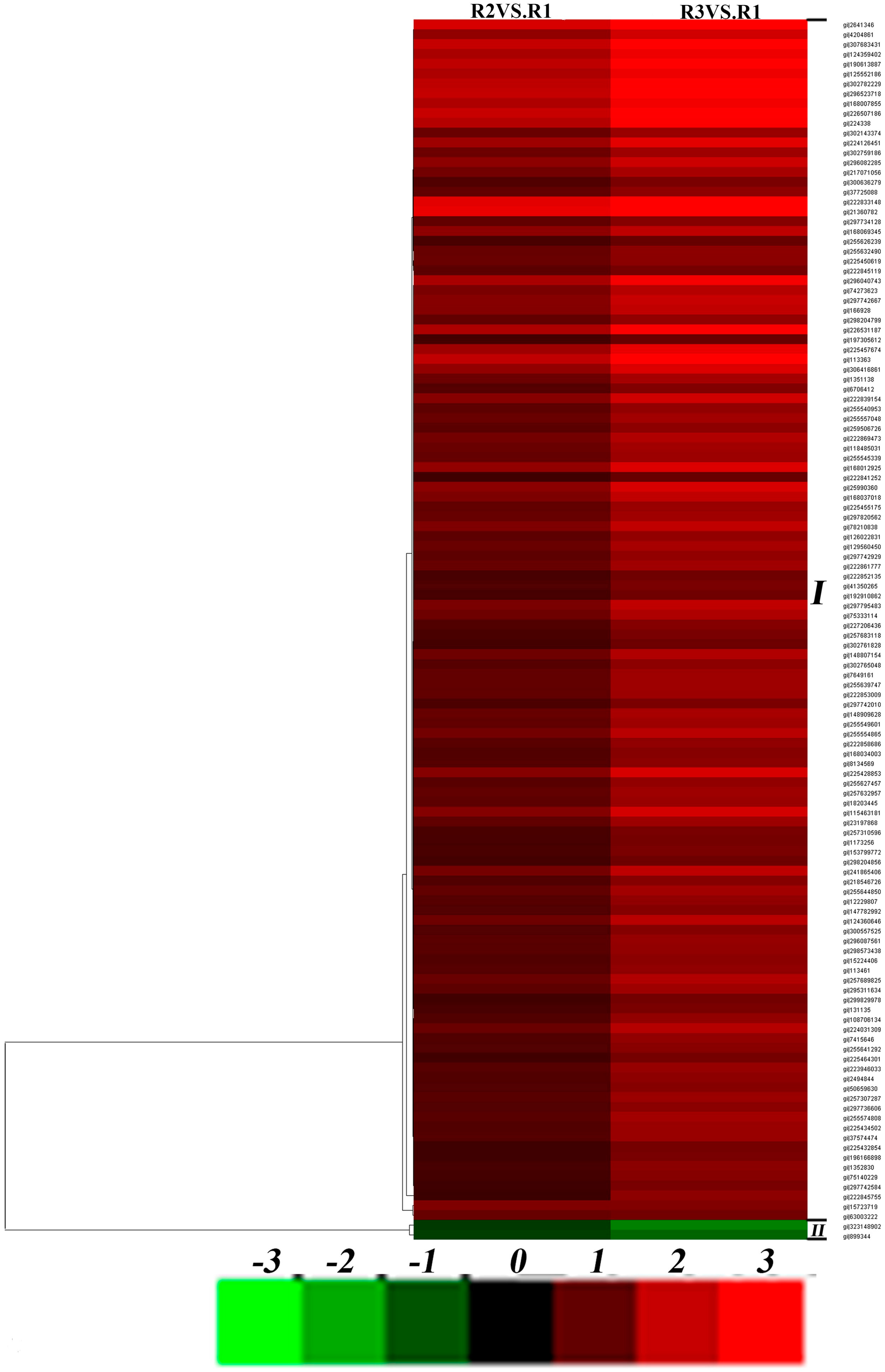
2.4. Effects of Drought Stress on Expression Changes of the Ramie Leaf and Root Proteomes
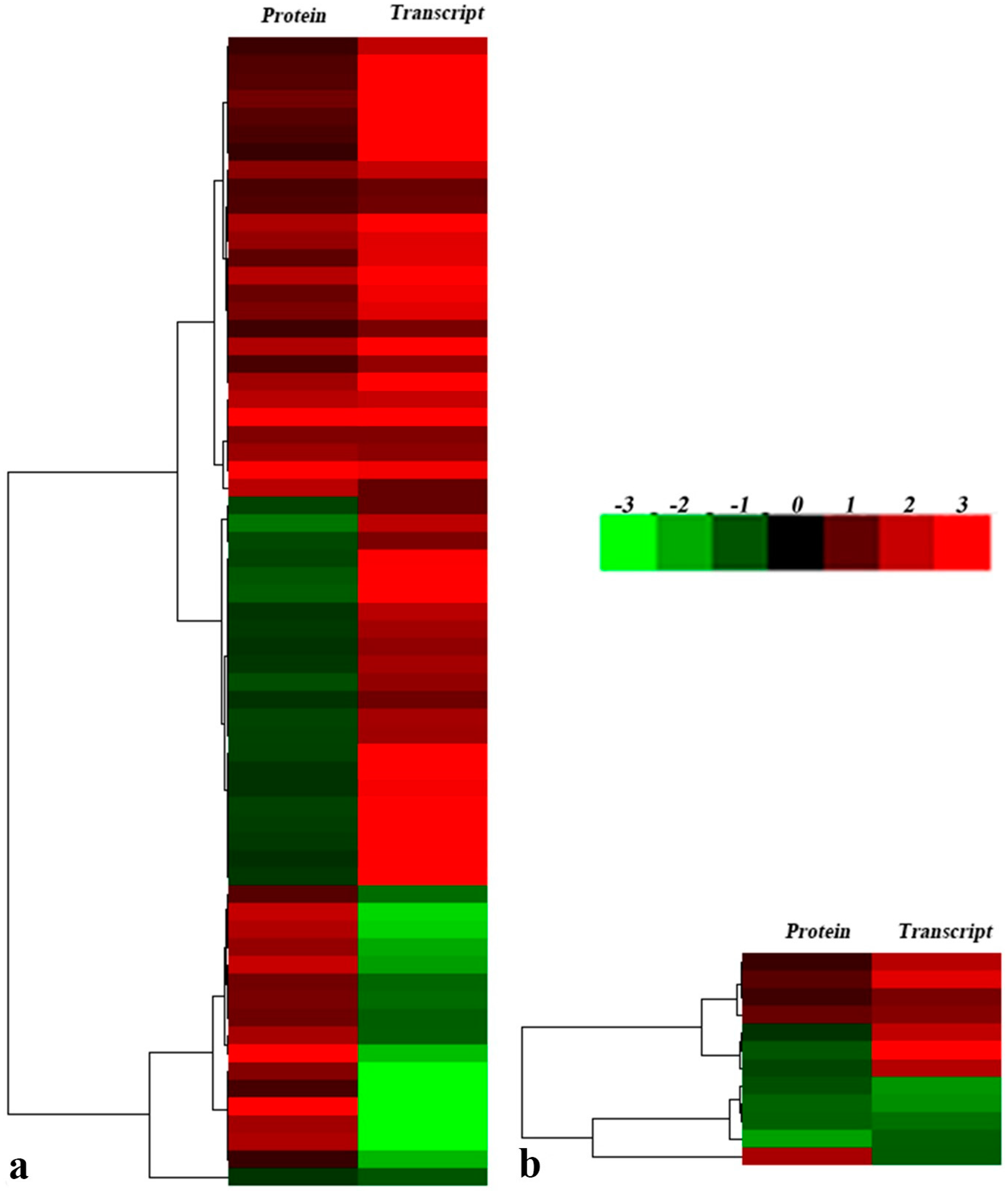
2.5. Association and Differential Expression Analysis of Proteome and Transcriptome Data
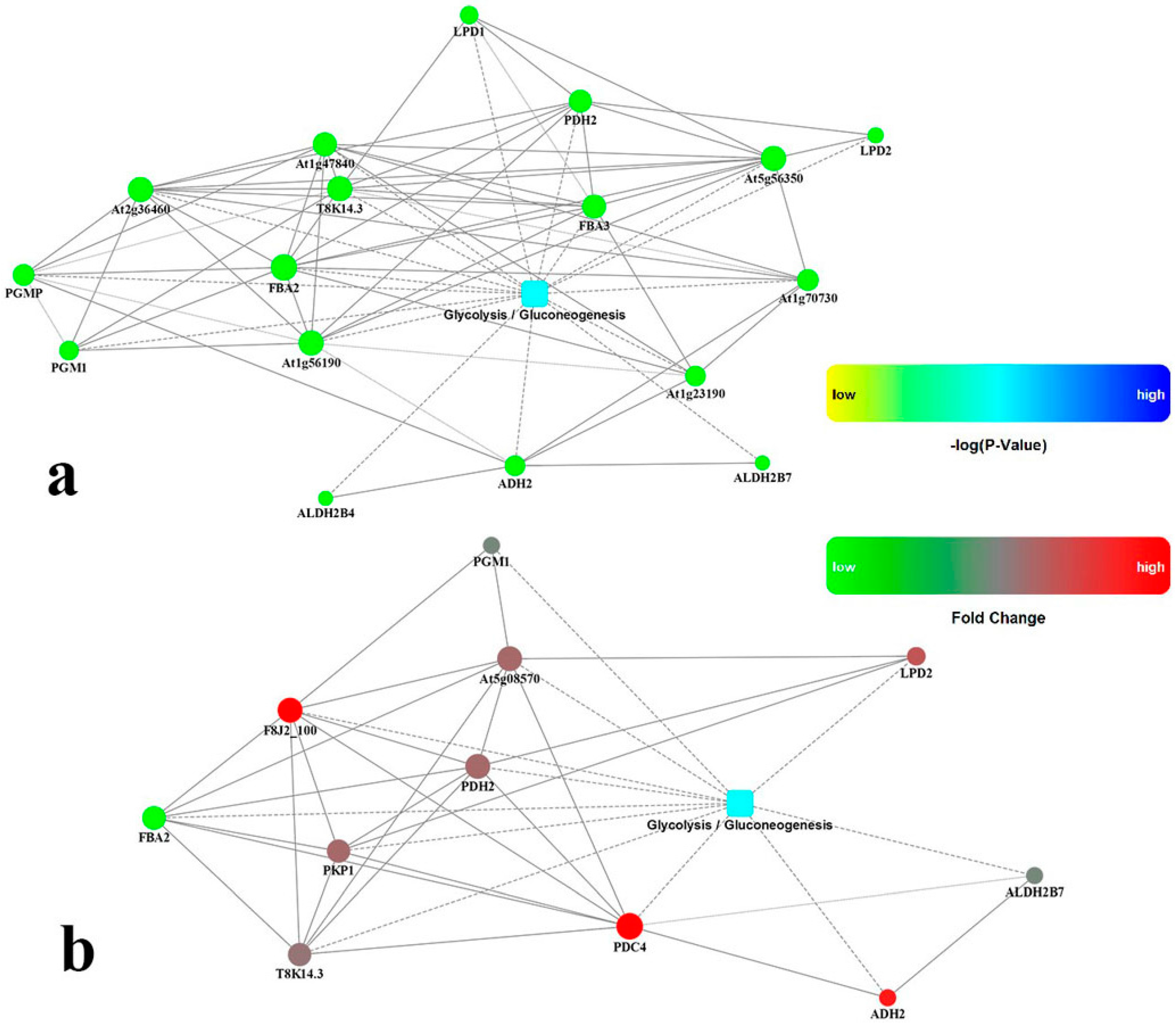
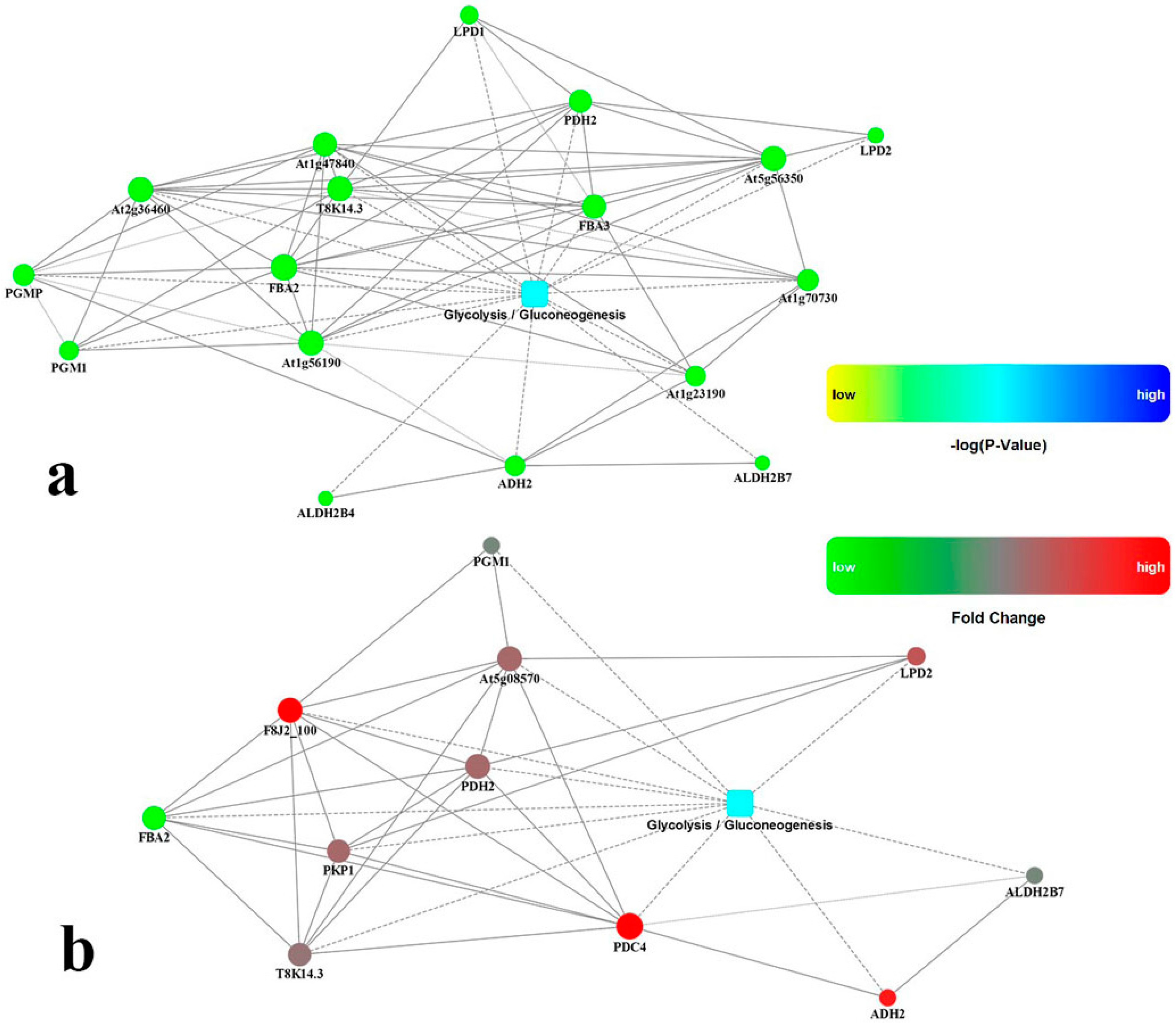
2.6. Identification Biochemical Reactions Significantly Upregulated in Roots by Drought Stress
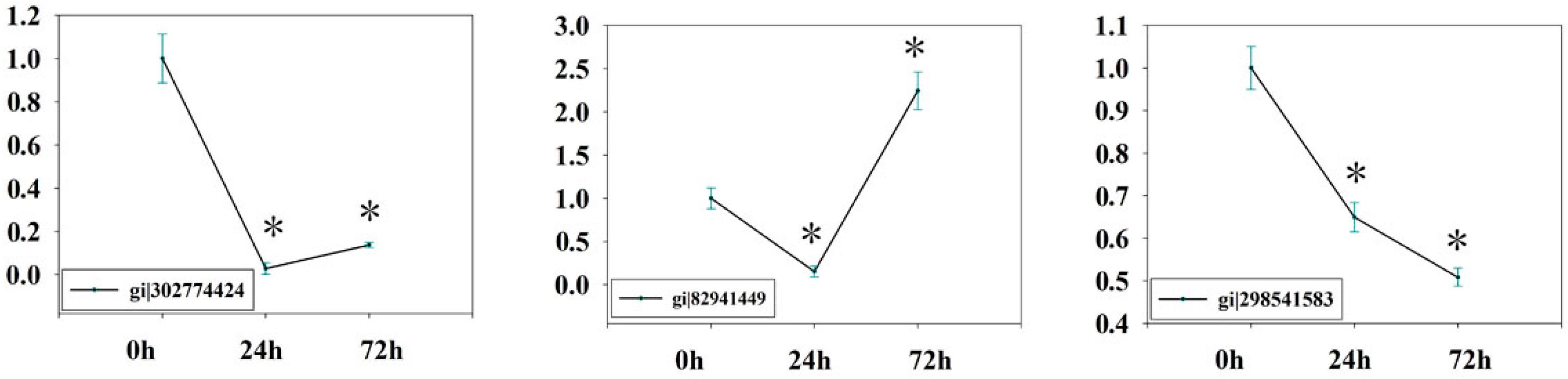
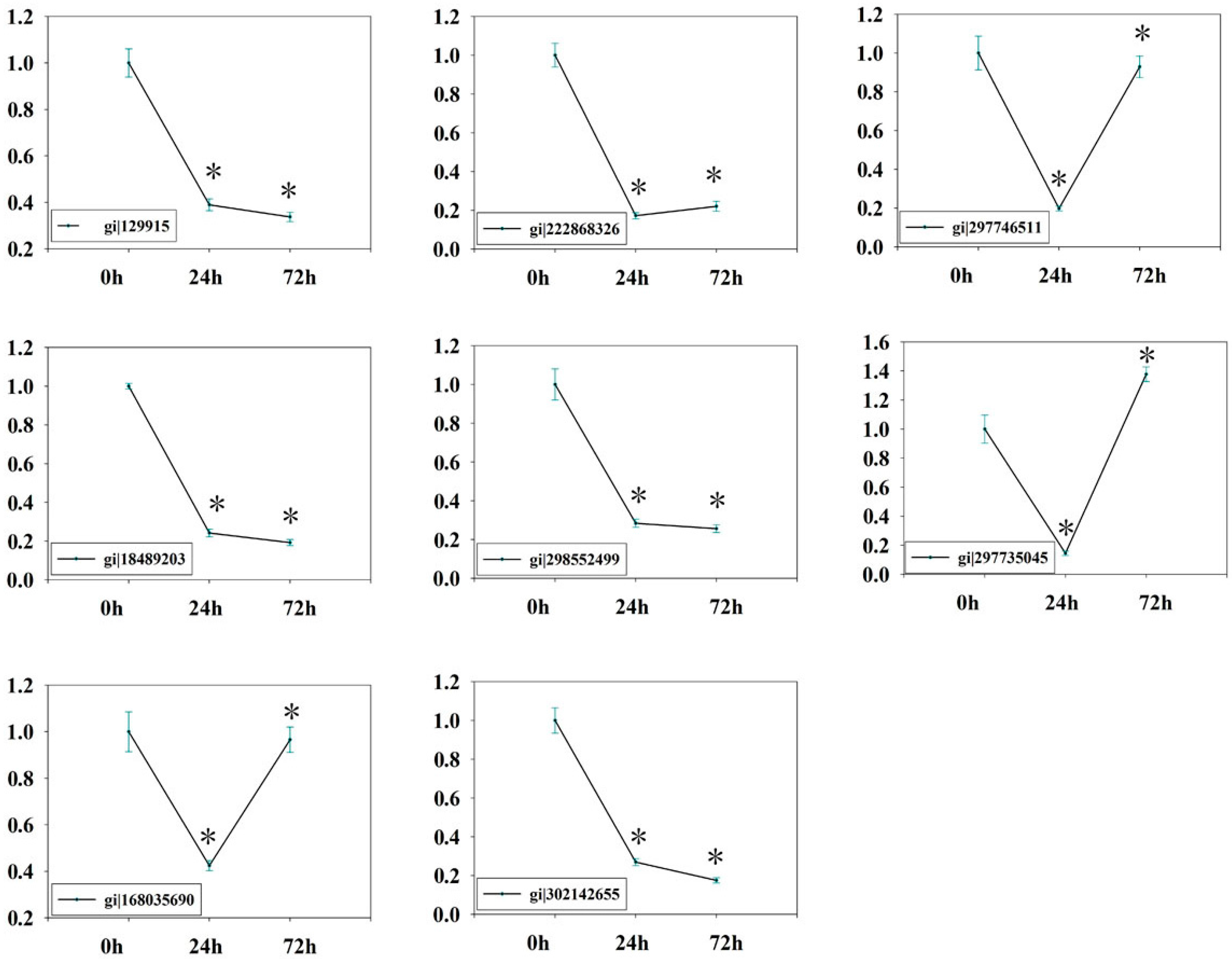
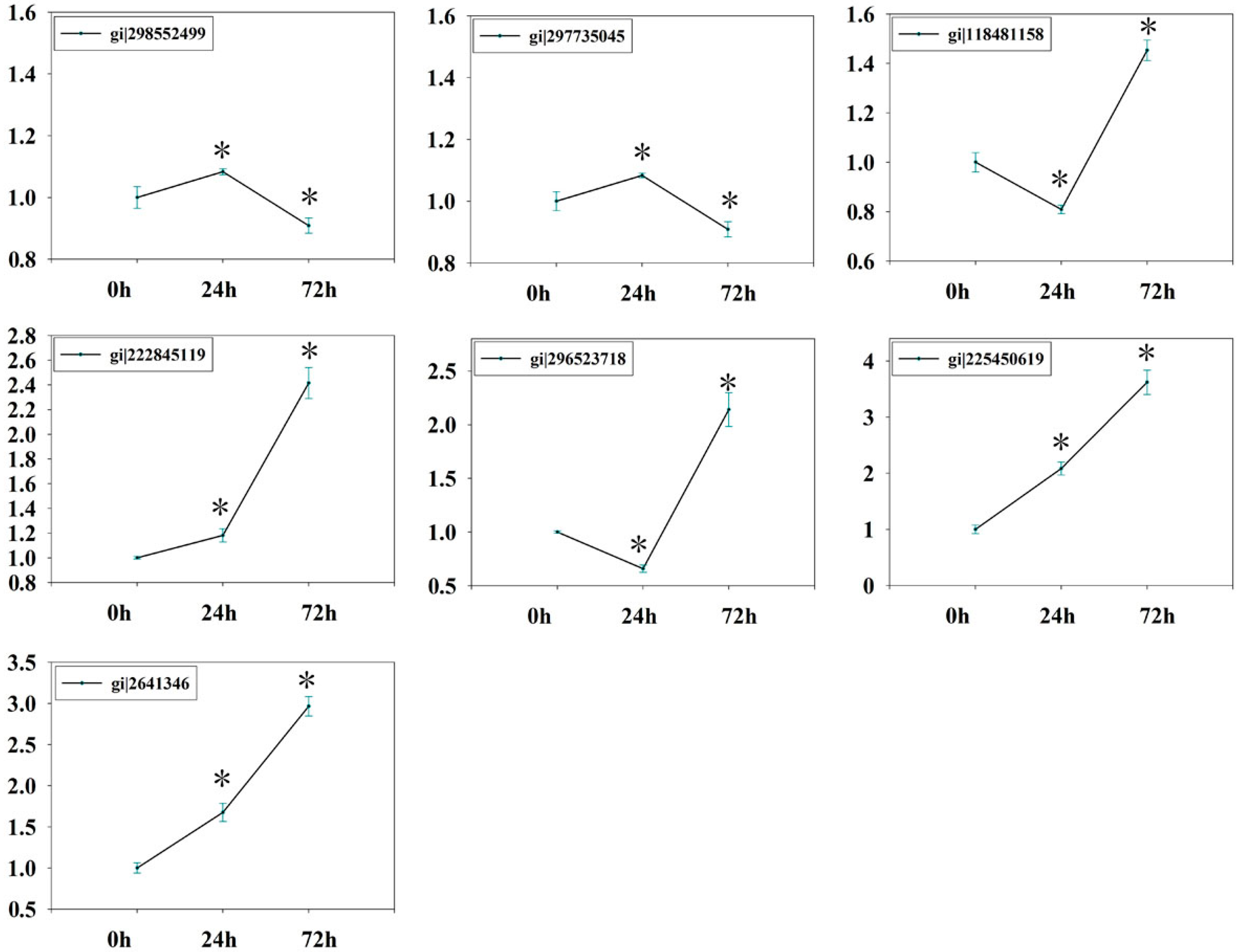
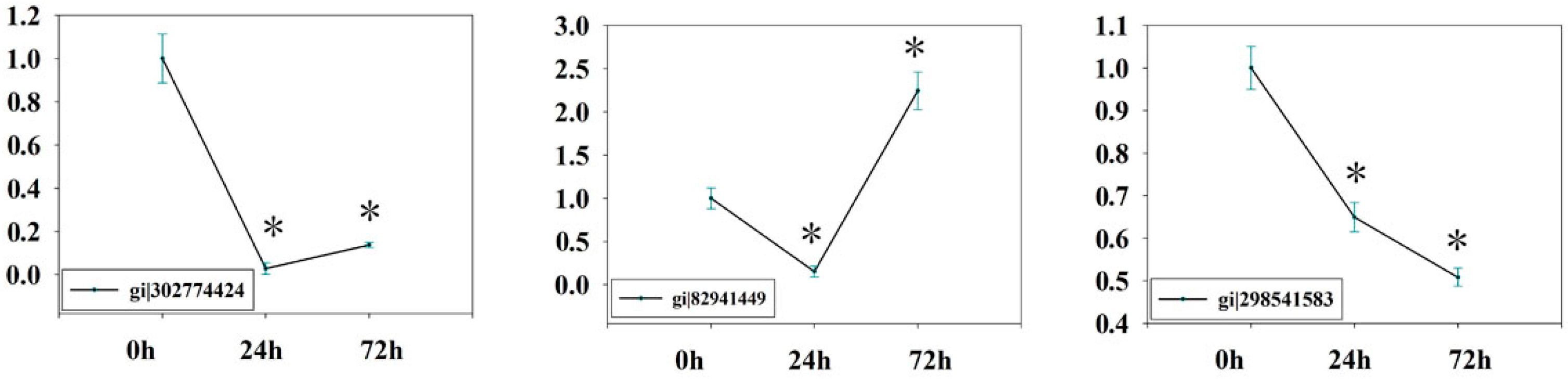
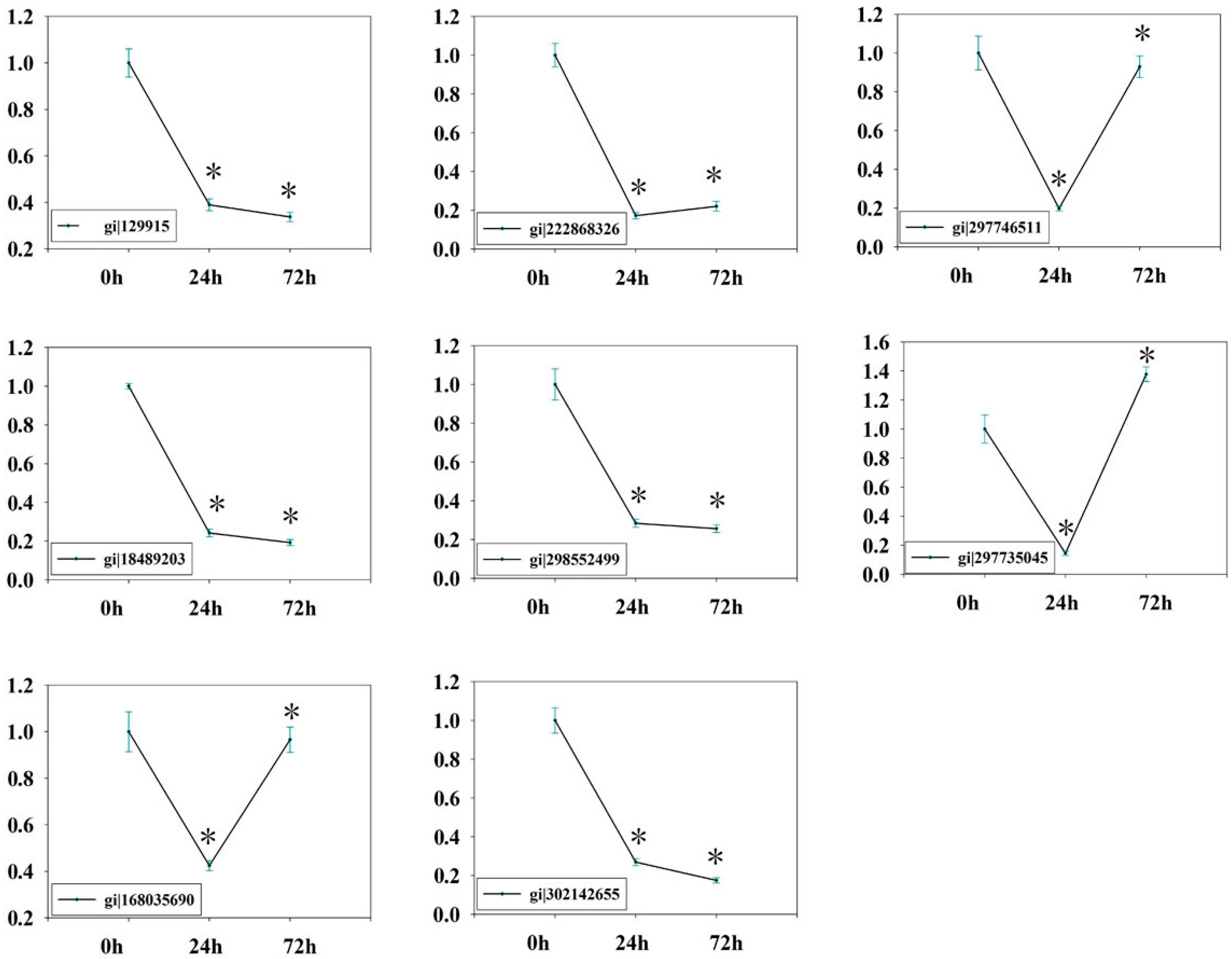
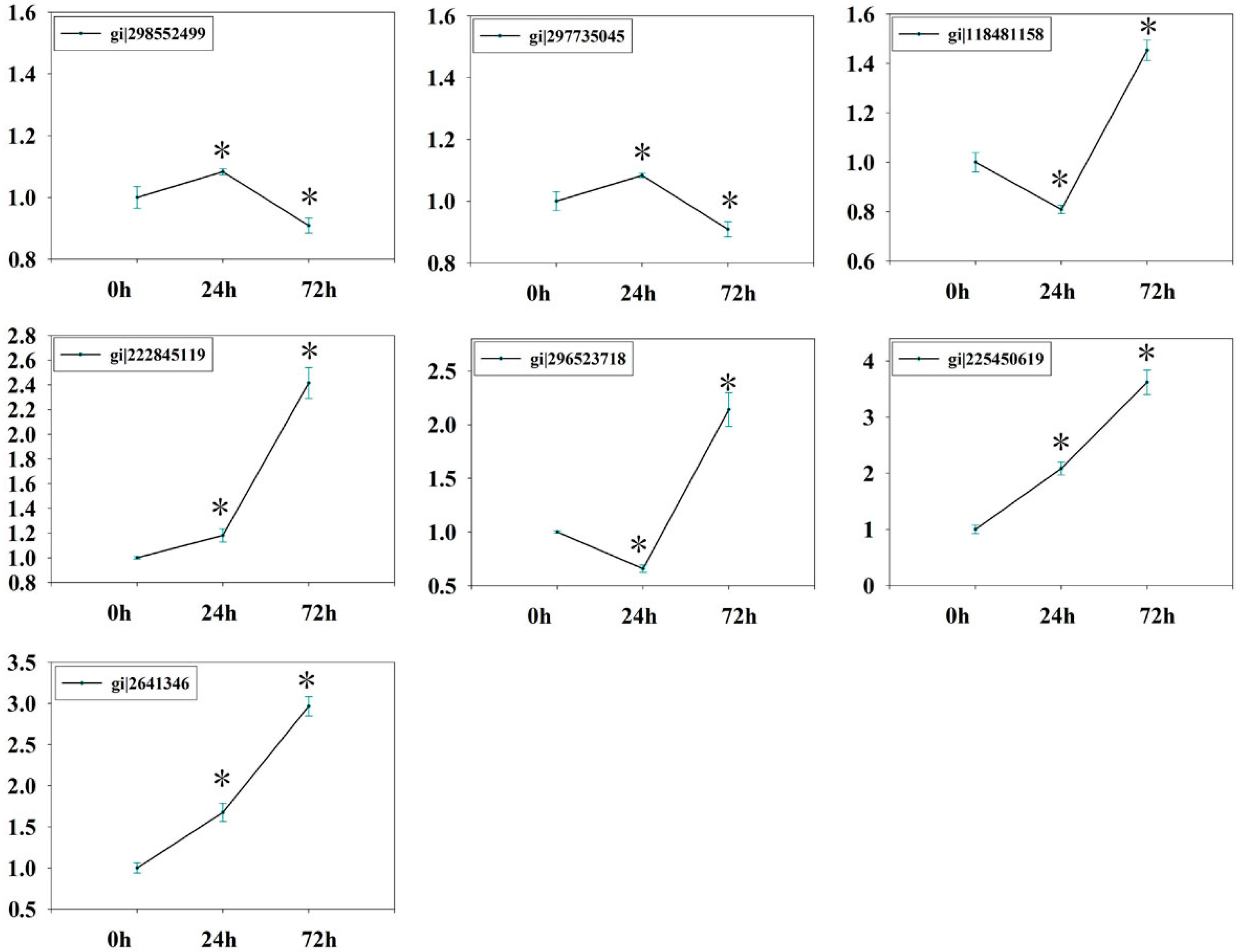
2.7. Verification of Isobaric Tags for Relative and Absolute Quantitation (iTRAQ) Data on Selected Candidates by qPCR
3. Materials and Methods
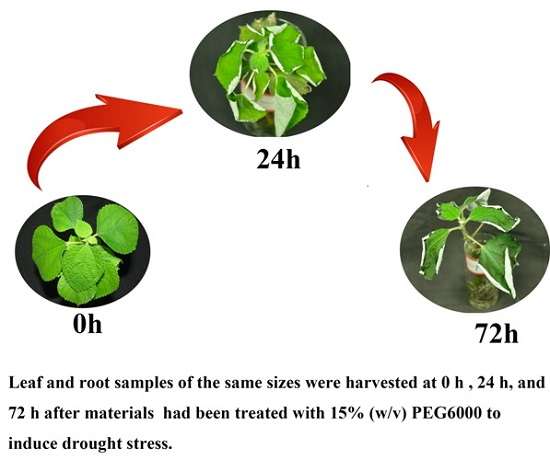
3.1. Plant Materials and Stress Treatments
3.2. Protein Extraction
3.3. Digestion and iTRAQ Labeling
3.4. Fractionation by Strong Cationic Exchange (SCX) Chromatography
3.5. Liquid Chromatography-Electrospray Ionization-Tandem Mass Spectrometry (LC-ESI-MS/MS) Analysis
3.6. Association Analysis of Proteomics and Transcriptomics
3.7. Quantitative Realtime PCR (qRT-PCR)
3.8. Data Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| iTRAQ | isobaric tag for relative and absolute quantification |
| SCX | strong cation exchange |
| HPLC | high pressure liquid chromatography |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KOBAS | KEGG Orthology-Based Annotation System |
| PEG | polyethylene glycol |
| RWC | relative water content |
| POD | peroxidaseactivity |
| qRT-PCR | quantitative realtime PCR |
References
- Mohammadi, P.P.; Moieni, A.; Komatsu, S. Comparative proteome analysis of drought-sensitive and drought-tolerant rapeseed roots and their hybrid F1 line under drought stress. Amino Acids 2012, 43, 2137–2152. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Dai, M.; Yao, J.; Xiao, B.; Li, X.; Zhang, Q.; Xiong, L. Overexpressing a NAM, ATAF, and CUC (NAC) transcription factor enhances drought resistance and salt tolerance in rice. Proc. Natl. Acad. Sci. USA 2006, 103, 12987–12992. [Google Scholar] [CrossRef] [PubMed]
- Alam, I.; Lee, D.G.; Kim, K.H.; Park, C.H.; Sharmin, S.A.; Lee, H.; Oh, K.W.; Yun, B.W.; Lee, B.H. Proteome analysis of soybean roots under waterlogging stress at an early vegetative stage. J. Biosci. 2010, 35, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q. Strategies for developing green super rice. Proc. Natl. Acad. Sci. USA 2007, 104, 16402–16409. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.K. Salt and drought stress signal transduction in plants. Annu. Rev. Plant Biol. 2002, 53, 247–273. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, K.; Yamaguchi-Shinozaki, K.; Seki, M. Regulatory network of gene expression in the drought and cold stress responses. Curr. Opin. Plant Biol. 2003, 6, 410–417. [Google Scholar] [CrossRef]
- Seki, M.; Kamei, A.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Molecular responses to drought, salinity and frost: Common and different paths for plant protection. Curr. Opin. Biotechnol. 2003, 14, 194–199. [Google Scholar] [CrossRef]
- Plomion, C.; Lalanne, C.; Claverol, S.; Meddour, H.; Kohler, A.; Bogeat-Triboulot, M.B.; Barre, A.; Le Provost, G.; Dumazet, H.; Jacob, D.; et al. Mapping the proteome of poplar and application to the discovery of drought-stress responsive proteins. Proteomics 2006, 6, 6509–6527. [Google Scholar] [CrossRef] [PubMed]
- Aranjuelo, I.; Molero, G.; Erice, G.; Avice, J.C.; Nogues, S. Plant physiology and proteomics reveals the leaf response to drought in alfalfa (Medicago sativa L.). J. Exp. Bot. 2011, 62, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Ashoub, A.; Beckhaus, T.; Berberich, T.; Karas, M.; Bruggemann, W. Comparative analysis of barley leaf proteome as affected by drought stress. Planta 2013, 237, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Matsui, A.; Ishida, J.; Morosawa, T.; Mochizuki, Y.; Kaminuma, E.; Endo, T.A.; Okamoto, M.; Nambara, E.; Nakajima, M.; Kawashima, M.; et al. Arabidopsis transcriptome analysis under drought, cold, high-salinity and aba treatment conditions using a tiling array. Plant Cell Physiol. 2008, 49, 1135–1149. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Fu, J.; Gou, M.; Huai, J.; Liu, Y.; Jian, M.; Huang, Q.; Guo, X.; Dong, Z.; Wang, H.; et al. Genome-wide transcriptome analysis of two maize inbred lines under drought stress. Plant Mol. Biol. 2010, 72, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Cal, A.J.; Liu, D.; Mauleon, R.; Hsing, Y.I.; Serraj, R. Transcriptome profiling of leaf elongation zone under drought in contrasting rice cultivars. PLoS ONE 2013, 8, e54537. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Nishiyama, R.; Watanabe, Y.; Tanaka, M.; Seki, M.; Ham le, H.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Tran, L.S. Differential gene expression in soybean leaf tissues at late developmental stages under drought stress revealed by genome-wide transcriptome analysis. PLoS ONE 2012, 7, e49522. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhu, S.; Tang, Q.; Yu, Y.; Tang, S. Identification of drought stress-responsive transcription factors in ramie (Boehmeria nivea L. Gaud.). BMC Plant Biol. 2013, 13, 130. [Google Scholar] [CrossRef] [PubMed]
- Budak, H.; Akpinar, B.A.; Unver, T.; Turktas, M. Proteome changes in wild and modern wheat leaves upon drought stress by two-dimensional electrophoresis and nanolc-esi-ms/ms. Plant Mol. Biol. 2013, 83, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Liu, Q.; Liang, X.; Huang, H.; Zhang, S. Morphological, anatomical, and physiological assessment of ramie (Boehmeria nivea (L.) Gaud.) tolerance to soil drought. Genet. Resour. Crop Evol. 2005, 52, 497–506. [Google Scholar] [CrossRef]
- Bonhomme, L.; Monclus, R.; Vincent, D.; Carpin, S.; Lomenech, A.M.; Plomion, C.; Brignolas, F.; Morabito, D. Leaf proteome analysis of eight populus xeuramericana genotypes: Genetic variation in drought response and in water-use efficiency involves photosynthesis-related proteins. Proteomics 2009, 9, 4121–4142. [Google Scholar] [CrossRef] [PubMed]
- Ali, G.M.; Komatsu, S. Proteomic analysis of rice leaf sheath during drought stress. J. Proteome Res. 2006, 5, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Zang, X.; Komatsu, S. A proteomics approach for identifying osmotic-stress-related proteins in rice. Phytochemistry 2007, 68, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Nouri, M.Z.; Komatsu, S. Comparative analysis of soybean plasma membrane proteins under osmotic stress using gel-based and LC MS/MS-based proteomics approaches. Proteomics 2010, 10, 1930–1945. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.T.; Qi, Y.P.; Lu, Y.B.; Guo, P.; Sang, W.; Feng, H.; Zhang, H.X.; Chen, L.S. iTRAQ protein profile analysis of citrus sinensis roots in response to long-term boron-deficiency. J. Proteom. 2013, 93, 179–206. [Google Scholar] [CrossRef] [PubMed]
- Zieske, L.R. A perspective on the use of itraq reagent technology for protein complex and profiling studies. J. Exp. Bot. 2006, 57, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Verslues, P.E.; Agarwal, M.; Katiyar-Agarwal, S.; Zhu, J.; Zhu, J.K. Methods and concepts in quantifying resistance to drought, salt and freezing, abiotic stresses that affect plant water status. Plant J. 2006, 45, 523–539. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Chen, J.; Zhang, J.; Liao, Y.; Dai, L.; Wang, B.; Liu, L.; Peng, D. Transcriptome profiling and identification of transcription factors in ramie (Boehmeria nivea L. Gaud) in response to peg treatment, using illumina paired-end sequencing technology. Int. J. Mol. Sci. 2015, 16, 3493–3511. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, M. Biotechnological approach of improving plant salt tolerance using antioxidants as markers. Biotechnol. Adv. 2009, 27, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Cui, X.; Chen, Y.; Gu, C.; Miao, H.; Gao, H.; Chen, F.; Liu, Z.; Guan, Z.; Fang, W. Cgdreba transgenic chrysanthemum confers drought and salinity tolerance. Environ. Exp. Bot. 2011, 74, 255–260. [Google Scholar] [CrossRef]
- Zi, J.; Zhang, J.; Wang, Q.; Zhou, B.; Zhong, J.; Zhang, C.; Qiu, X.; Wen, B.; Zhang, S.; Fu, X.; et al. Stress responsive proteins are actively regulated during rice (Oryza sativa) embryogenesis as indicated by quantitative proteomics analysis. PLoS ONE 2013, 8, e74229. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Liu, L.J.; Zhong, X.Y.; Lao, C.Y.; Wang, H.Y.; Wang, B.; Zhu, C.; Shah, F.; Peng, D.X. Comparative proteome analysis of the response of ramie under N, P and K deficiency. Planta 2014, 239, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Yang, B.; Harris, N.S.; Deyholos, M.K. Comparative proteomic analysis of nacl stress-responsive proteins in Arabidopsis roots. J. Exp. Bot. 2007, 58, 3591–3607. [Google Scholar] [CrossRef] [PubMed]
- Ge, P.; Ma, C.; Wang, S.; Gao, L.; Li, X.; Guo, G.; Ma, W.; Yan, Y. Comparative proteomic analysis of grain development in two spring wheat varieties under drought stress. Anal. Bioanal. Chem. 2012, 402, 1297–1313. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhao, J.; He, X.; Sun, H.; Zhang, G.; Wu, F. Comparative proteomic analysis of drought tolerance in the two contrasting tibetan wild genotypes and cultivated genotype. BMC Genom. 2015, 16, 432. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Wang, M.; Li, F.; Lv, H.; Li, C.; Xia, G. A proteomic study of the response to salinity and drought stress in an introgression strain of bread wheat. Mol. Cell. Proteom. 2009, 8, 2676–2686. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Q.; Xu, X.Y.; Gong, Q.Q.; Xie, C.; Fan, W.; Yang, J.L.; Lin, Q.S.; Zheng, S.J. Root proteome of rice studied by iTRAQ provides integrated insight into aluminum stress tolerance mechanisms in plants. J. Proteom. 2014, 98, 189–205. [Google Scholar] [CrossRef] [PubMed]
- Nogues, S.; Baker, N.R. Effects of drought on photosynthesis in mediterranean plants grown under enhanced uv-b radiation. J. Exp. Bot. 2000, 51, 1309–1317. [Google Scholar] [CrossRef] [PubMed]
- Ramachandra Reddy, A.; Chaitanya, K.V.; Vivekanandan, M. Drought-induced responses of photosynthesis and antioxidant metabolism in higher plants. J. Plant Physiol. 2004, 161, 1189–1202. [Google Scholar] [CrossRef] [PubMed]
- Maksup, S.; Roytrakul, S.; Supaibulwatana, K. Physiological and comparative proteomic analyses of thai jasmine rice and two check cultivars in response to drought stress. J. Plant Int. 2012, 9, 43–55. [Google Scholar] [CrossRef]
- Vassileva, V.; Demirevska, K.; Simova-Stoilova, L.; Petrova, T.; Tsenov, N.; Feller, U. Long-term field drought affects leaf protein pattern and chloroplast ultrastructure of winter wheat in a cultivar-specific manner. J. Agron. Crop Sci. 2012, 198, 104–117. [Google Scholar] [CrossRef]
- Caruso, G.; Cavaliere, C.; Foglia, P.; Gubbiotti, R.; Samperi, R.; Laganà, A. Analysis of drought responsive proteins in wheat (Triticum durum) by 2D-page and maldi-tof mass spectrometry. Plant Sci. 2009, 177, 570–576. [Google Scholar] [CrossRef]
- Guo, X.L.; Bai, L.R.; Su, C.Q.; Shi, L.R.; Wang, D.W. Molecular cloning and expression of drought-induced protein 3 (DIP3) encoding a class III chitinase in upland rice. Genet. Mol. Res. 2013, 12, 6860–6870. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, K.; Yamaguchi-Shinozaki, K. Molecular responses to dehydration and low temperature: Differences and cross-talk between two stress signaling pathways. Curr. Opin. Plant Biol. 2000, 3, 217–223. [Google Scholar] [CrossRef]
- Finkelstein, R.R.; Gampala, S.S.; Rock, C.D. Abscisic acid signaling in seeds and seedlings. Plant Cell 2002, 14, S15–S45. [Google Scholar] [PubMed]
- Xiong, L.; Schumaker, K.S.; Zhu, J.K. Cell signaling during cold, drought, and salt stress. Plant Cell 2002, 14, S165–S183. [Google Scholar] [PubMed]
- Yao, Y.; Ni, Z.; Du, J.; Wang, X.; Wu, H.; Sun, Q. Isolation and characterization of 15 genes encoding ribosomal proteins in wheat (Triticum aestivum L.). Plant Sci. 2006, 170, 579–586. [Google Scholar] [CrossRef]
- Swindell, W.R.; Huebner, M.; Weber, A.P. Transcriptional profiling of Arabidopsis heat shock proteins and transcription factors reveals extensive overlap between heat and non-heat stress response pathways. BMC Genom. 2007, 8, 125. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. Kobas 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Baena-Gonzalez, E.; Rolland, F.; Thevelein, J.M.; Sheen, J. A central integrator of transcription networks in plant stress and energy signalling. Nature 2007, 448, 938–942. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Stitt, M. Coordination of carbon supply and plant growth. Plant Cell Environ. 2007, 30, 1126–1149. [Google Scholar] [CrossRef] [PubMed]
- Baena-Gonzalez, E.; Sheen, J. Convergent energy and stress signaling. Trends Plant Sci. 2008, 13, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, C.A.; Good, A.G.; Taylor, G.J. Induction of vacuolar atpase and mitochondrial ATP synthase by aluminum in an aluminum-resistant cultivar of wheat. Plant Physiol. 2001, 125, 2068–2077. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.Y.; Xie, F.Z.; Zhai, C.; Stern, J.S.; Liu, Y.; Liu, S.L. The role of cellular oxidative stress in regulating glycolysis energy metabolism in hepatoma cells. Mol. Cancer 2009, 8, 32. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, S.Q.; Liu, Y.F.; Liu, P.; Lei, G.; He, S.J.; Ma, B.; Zhang, W.K.; Zhang, J.S.; Chen, S.Y. Receptor-like kinase ossik1 improves drought and salt stress tolerance in rice (Oryza sativa) plants. Plant J. 2010, 62, 316–329. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Wang, B.; Liu, L.; Jiang, H.; Chen, J.; Ye, S.; Chen, L.; Guo, P.; Huang, X.; Peng, D. Agrobacterium-mediated genetic transformation and regeneration of transgenic plants using leaf midribs as explants in ramie (Boehmeria nivea (L.) Gaud). Mol. Biol. Rep. 2014, 41, 3257–3269. [Google Scholar] [CrossRef] [PubMed]
- Villeneuve, L.M.; Stauch, K.L.; Fox, H.S. Proteomic analysis of the mitochondria from embryonic and postnatal rat brains reveals response to developmental changes in energy demands. J. Proteom. 2014, 109, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Vizcaino, J.A.; Deutsch, E.W.; Wang, R.; Csordas, A.; Reisinger, F.; Rios, D.; Dianes, J.A.; Sun, Z.; Farrah, T.; Bandeira, N.; et al. Proteomexchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Fan, N.J.; Gao, C.F.; Wang, C.S.; Zhao, G.; Lv, J.J.; Wang, X.L.; Chu, G.H.; Yin, J.; Li, D.H.; Chen, X.; et al. Identification of the up-regulation of TP-α, collagen α-1(VI) chain, and S100A9 in esophageal squamous cell carcinoma by a proteomic method. J. Proteom. 2012, 75, 3977–3986. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Upregulated | Downregulated |
|---|---|---|
| L1–L2 | 10 | 108 |
| L2–L3 | 0 | 216 |
| L1–L3 | 20 | 413 |
| R1–R2 | 122 | 2 |
| R2–R3 | 20 | 7 |
| R1–R3 | 211 | 29 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
An, X.; Zhang, J.; Dai, L.; Deng, G.; Liao, Y.; Liu, L.; Wang, B.; Peng, D. Isobaric Tags for Relative and Absolute Quantitation (iTRAQ)-Based Comparative Proteome Analysis of the Response of Ramie under Drought Stress. Int. J. Mol. Sci. 2016, 17, 1607. https://doi.org/10.3390/ijms17101607
An X, Zhang J, Dai L, Deng G, Liao Y, Liu L, Wang B, Peng D. Isobaric Tags for Relative and Absolute Quantitation (iTRAQ)-Based Comparative Proteome Analysis of the Response of Ramie under Drought Stress. International Journal of Molecular Sciences. 2016; 17(10):1607. https://doi.org/10.3390/ijms17101607
Chicago/Turabian StyleAn, Xia, Jingyu Zhang, Lunjin Dai, Gang Deng, Yiwen Liao, Lijun Liu, Bo Wang, and Dingxiang Peng. 2016. "Isobaric Tags for Relative and Absolute Quantitation (iTRAQ)-Based Comparative Proteome Analysis of the Response of Ramie under Drought Stress" International Journal of Molecular Sciences 17, no. 10: 1607. https://doi.org/10.3390/ijms17101607
APA StyleAn, X., Zhang, J., Dai, L., Deng, G., Liao, Y., Liu, L., Wang, B., & Peng, D. (2016). Isobaric Tags for Relative and Absolute Quantitation (iTRAQ)-Based Comparative Proteome Analysis of the Response of Ramie under Drought Stress. International Journal of Molecular Sciences, 17(10), 1607. https://doi.org/10.3390/ijms17101607