
Identification and Functional Analysis of microRNAs Involved in the Anther Development in Cotton Genic Male Sterile Line Yu98-8A

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Morphological and Histological Identification of Male Genic Sterile Buds
2.2. Analysis of the Small RNA Library and Expression Profile of Small RNAs during Anther Development
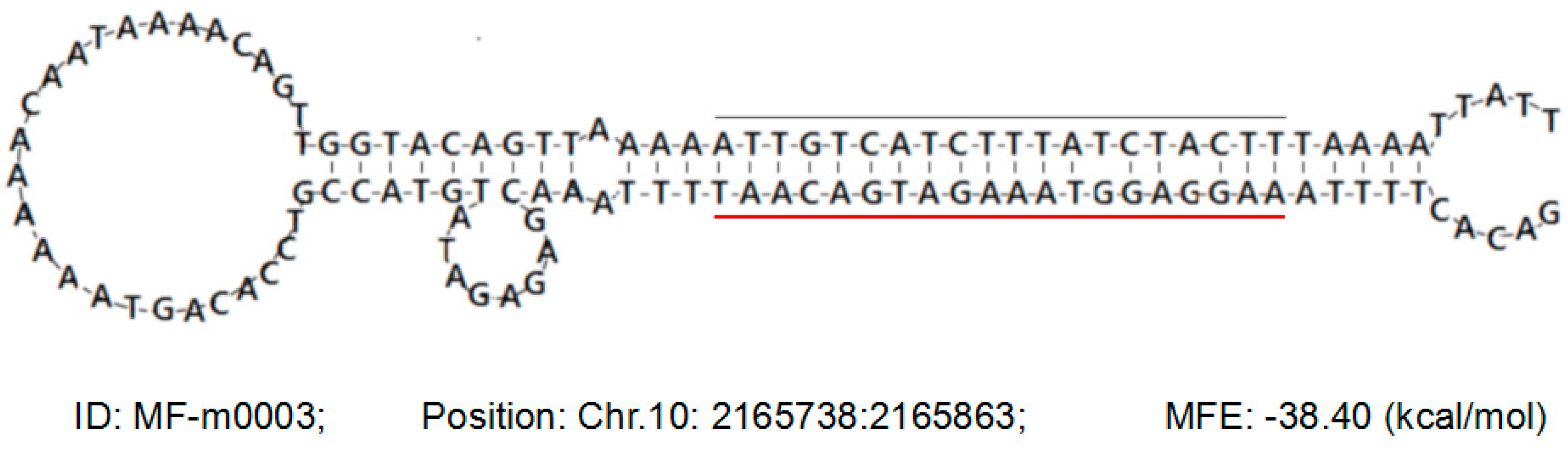
2.3. Identification and Prediction of Known miRNAs and Novel miRNAs
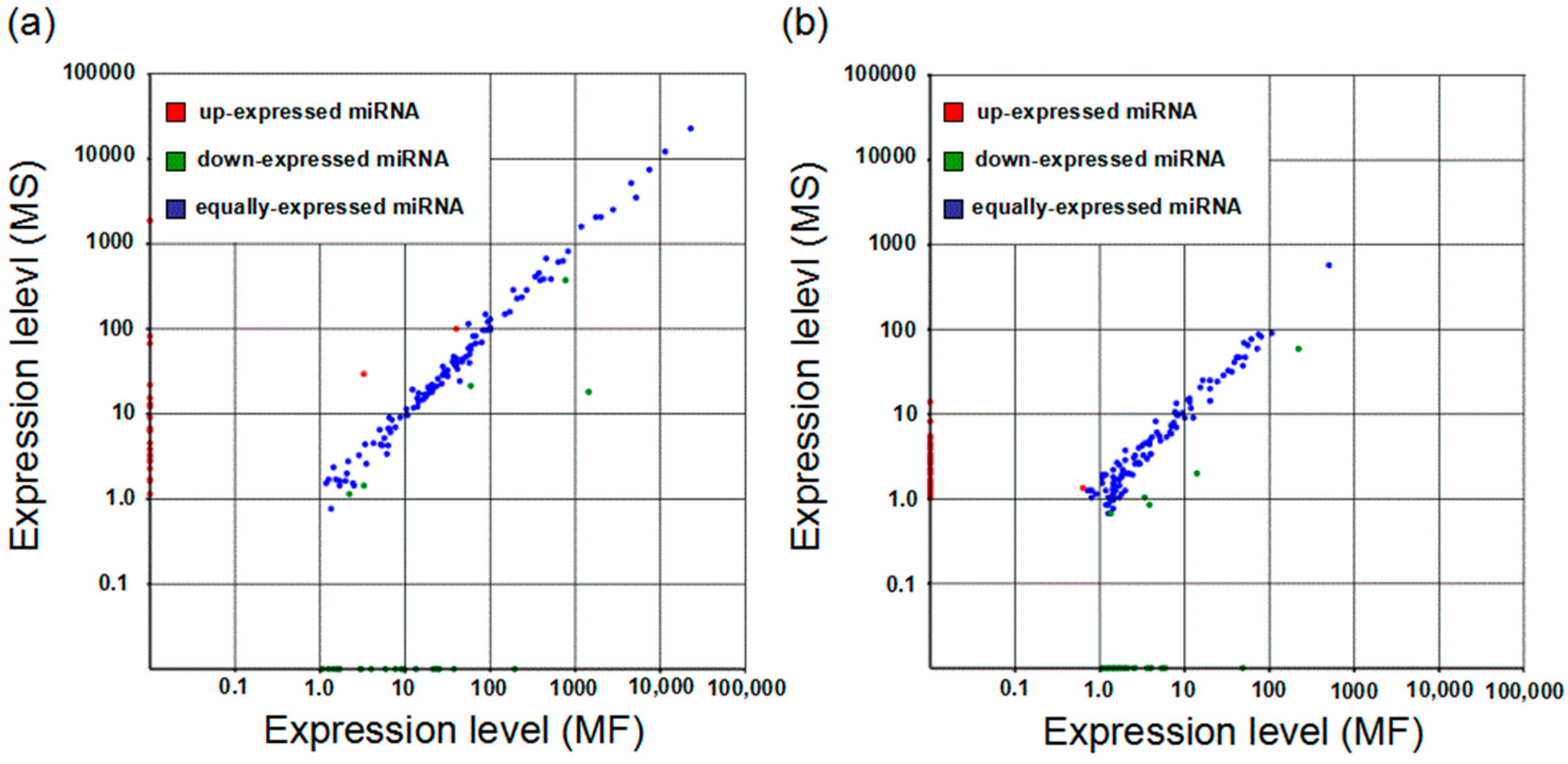
2.4. Differentially-Expressed Analysis and Target Prediction of Known and Novel miRNAs
2.5. Experimental Identification of miRNA Target Genes by Degradome Analysis
2.6. Expression Pattern Analysis of miRNAs by RT-qPCR
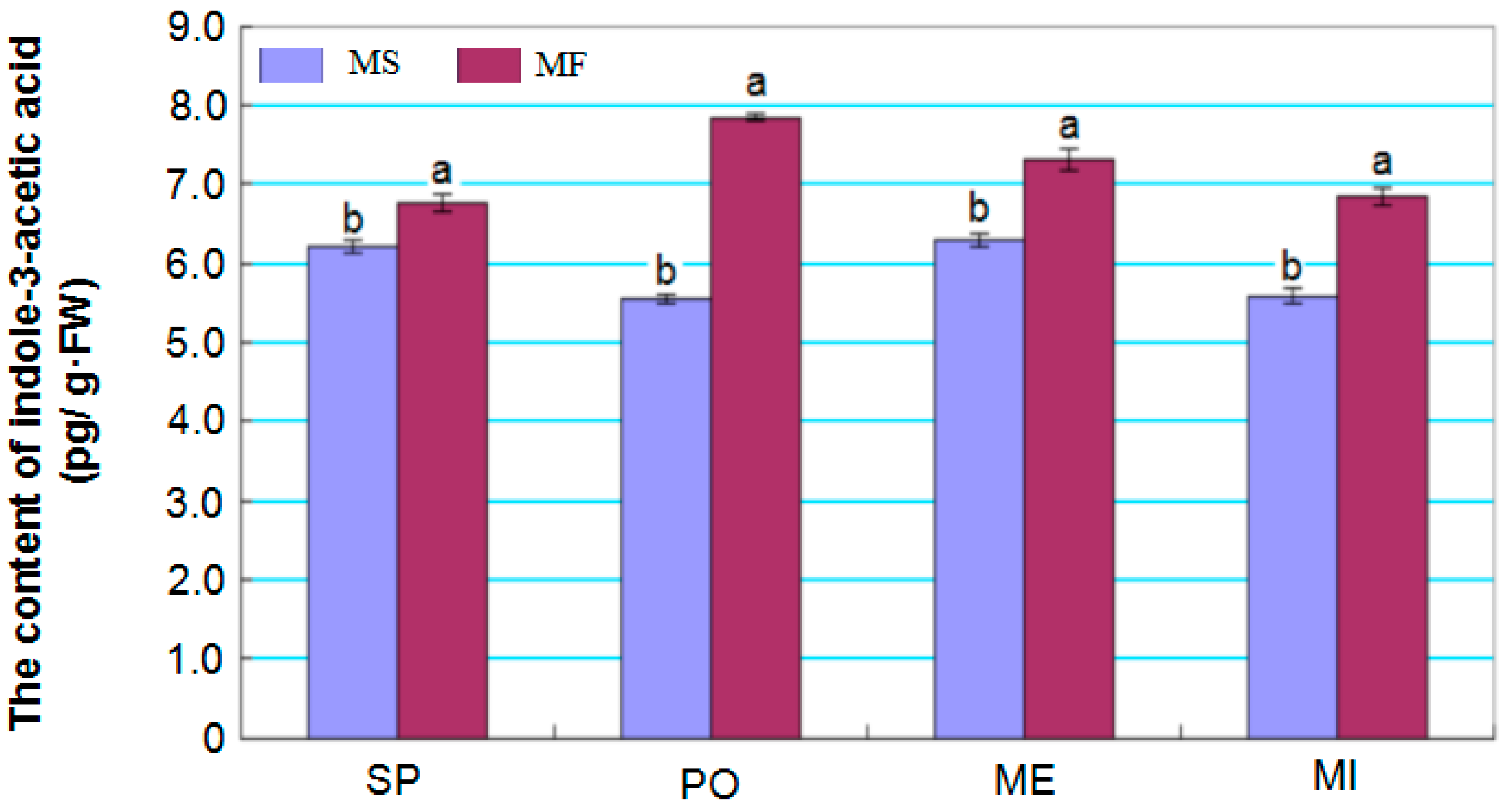
2.7. Dynamics of Indole-3-Acetic Acid Levels in Young Buds during Different Developmental Stages
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Histological Analysis
4.3. Extraction, Library Construction and Sequencing of Small RNAs
4.4. Computational Analysis of Sequencing Data and Identification of Known Conserved and Novel microRNAs
4.5. Target Prediction and Identification of miRNA by Informatics and Degradome Sequencing
4.6. Expression Analysis of miRNAs by RT-qPCR
4.7. Measurement of the Indole-3 Acetic Acid Content of Young Buds
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sunilkumar, G.; Campbell, L.M.; Puckhaber, L.; Stipanovic, R.D.; Rathore, K.S. Engineering cottonseed for use in human nutrition by tissue-specific reduction of toxic gossypol. Proc. Natl. Acad. Sci. USA 2006, 103, 18054–18059. [Google Scholar] [CrossRef] [PubMed]
- Jones-Rhoades, M.W.; Bartel, D.P.; Bartel, B. MicroRNAs and their regulatory roles in plants. Annu. Rev. Plant Biol. 2006, 57, 19–53. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhou, H.; Zhang, Q.; Zhang, J.; Ni, F.; Liu, C.; Qi, Y. DNA methylation mediated by a microRNA pathway. Mol. Cell 2010, 38, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Reyes, J.L.; Chua, N.H. ABA induction of miR159 controls transcript levels of two MYB factors during Arabidopsis seed germination. Plant J. 2007, 49, 592–606. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, L.; Bussell, J.D.; Pacurar, D.I.; Schwambach, J.; Pacurar, M.; Bellini, C. Phenotypic plasticity of adventitious rooting in Arabidopsis is controlled by complex regulation of AUXIN RESPONSE FACTOR transcripts and microRNA abundance. Plant Cell 2009, 21, 3119–3132. [Google Scholar] [CrossRef] [PubMed]
- Floyd, S.K.; Bowman, J.L. Gene regulation: Ancient microRNA target sequences in plants. Nature 2004, 428, 485–486. [Google Scholar] [CrossRef] [PubMed]
- Chen, X. A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science 2004, 303, 2022–2025. [Google Scholar] [CrossRef] [PubMed]
- Millar, A.A.; Gubler, F. The Arabidopsis GAMYB-like genes, MYB33 and MYB65, are microRNA-regulated genes that redundantly facilitate anther development. Plant Cell 2005, 17, 705–721. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.Z.; Chen, L.; Zhao, M.G.; Tian, Q.Y.; Zhang, W.H. Identification of drought-responsive microRNAs and their targets in Medicago truncatula by genome-wide high-throughput sequencing and degradome analysis. BMC Genom. 2011, 12, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Zhang, L.; Wang, H.; Liu, Z.; Zhang, Z.; Zheng, Y. Differential expression of miRNAs in response to salt stress in maize roots. Ann. Bot. 2009, 103, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.J.; Li, Y.; Han, X.L.; Shen, F.F. Genome-wide profiling of miRNAs and other small non-coding RNAs in the Verticillium dahliae-inoculated Cotton Roots. PLoS ONE 2012, 7, e35765. [Google Scholar] [CrossRef] [PubMed]
- Aukerman, M.J.; Sakai, H. Regulation of flowering time and floral organ identity by a microRNA and its APETALA2 -like target genes. Plant Cell 2003, 15, 2730–2741. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.P.; Salinas, M.; Höhmann, S.; Berndtgen, R.; Huijser, P. MiR156-targeted and nontargeted SBP-box transcription factors act in concert to secure male fertility in Arabidopsis. Plant Cell 2010, 22, 3935–3950. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.H.; Guo, S.Y.; Xu, Y.Y.; Li, H.; Zhang, Z.Y.; Zhang, J.; Xu, S.J.; Zhang, C.; Chong, K. OsmiR396d-regulated OsGRFs function in floral organogenesis in rice through binding to their targets OsJMJ706 and OsCR4. Plant Physiol. 2014, 165, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, H.; Aya, K.; Ueguchi-Tanaka, M.; Shimada, Y.; Nakazono, M.; Watanabe, R.; Nishizawa, N.K.; Gomi, K.; Shimada, A.; Kitano, H.; et al. GAMYB controls different sets of genes and is differentially regulated by microRNA in aleurone cells and anthers. Plant J. 2006, 47, 427–444. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.L.; Chen, X.M.; McCormick, S. The anaphase-promoting complex is a dual integrator that regulates both microRNA-mediated transcriptional regulation of cyclin B1 and degradation of cyclin B1 during Arabidopsis male gametophyte development. Plant Cell 2011, 23, 1033–1046. [Google Scholar] [CrossRef] [PubMed]
- Mallory, A.C.; Bartel, D.P.; Bartel, B. MicroRNA-directed regulation of Arabidopsis AUXIN RESPONSE FACTOR17 is essential for proper development and modulates expression of early auxin response genes. Plant Cell 2005, 17, 1360–1375. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, P.; Ellis, C.M.; Weber, H.; Ploense, S.E.; Barkawi, L.S.; Guilfoyle, T.J.; Hagen, G.; Alonso, J.M.; Cohen, J.D.; Farmer, E.E.; et al. Auxin response factors ARF6 and ARF8 promote jasmonic acid production and flower maturation. Development 2005, 132, 4107–4118. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.F.; Tian, Q.; Reed, J.W. Arabidopsis microRNA167 controls patterns of ARF6 and ARF8 expression, and regulates both female and male reproduction. Development 2006, 133, 4211–4218. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.O.; Zhang, Z.M.; Lin, H.J.; Liu, H.L.; Chen, J.; Peng, H.; Cao, M.J.; Rong, T.Z.; Pan, G.T. Cytoplasmic male sterility-regulated novel microRNAs from maize. Funct. Integr. Genom. 2011, 11, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.J.; Zhang, H.Y.; Zheng, Y.Z.; Ding, Y. Comparative expression profiling of miRNAs between the cytoplasmic male sterile line MeixiangA and its maintainer line MeixiangB during rice anther development. Planta 2015, 241, 109–123. [Google Scholar]
- Yang, J.H.; Liu, X.Y.; Xu, B.C.; Zhao, N.; Yang, X.D.; Zhang, M.F. Identification of miRNAs and their targets using high-throughput sequencing and degradome analysis in cytoplasmic male-sterile and its maintainer fertile lines of Brassica juncea. BMC Genom. 2013, 14, 9–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, M.M.; Wei, H.L.; Wu, M.; Song, M.Z.; Zhang, J.F.; Yu, J.W.; Fan, S.L.; Yu, S.X. Comparative expression profiling of miRNA during anther development in genetic male sterile and wild type cotton. BMC Plant Biol. 2013, 13, 66–80. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.X.; Jiang, J.J.; Yang, Y.F.; Cao, J.S. Identification of microRNAs potentially involved in male sterility of Brassica campestris ssp. chinensis using microRNA array and quantitative RT-PCR assays. Cell Mol. Biol. Lett. 2013, 18, 416–432. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.X.; Lv, M.L.; Liang, Y.; Ma, Z.M.; Cao, J.S. Identification of novel and conserved miRNAs involved in pollen development in Brassica campestris ssp. chinensis by high-throughput sequencing and degradome analysis. BMC Genom. 2014, 15, 146–159. [Google Scholar]
- Zhang, W.; Xie, Y.; Xu, L.; Wang, Y.; Zhu, X.W.; Wang, R.H.; Zhang, Y.; Muleke, E.M.; Liu, L.W. Identification of microRNAs and their target genes explores miRNA-mediated regulatory network of cytoplasmic male sterility occurrence during anther development in Radish (Raphanus sativus L.). Front. Plant Sci. 2016, 22, 1054–1070. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.L.; Li, J.J.; Zhang, H.; He, T.T.; Han, S.H.; Li, Y.W.; Yang, S.P.; Gai, J.Y. Identification of miRNAs and their targets by high-throughput sequencing and degradome analysis in cytoplasmic male-sterile line NJCMS1A and its maintainer NJCMS1B of soybean. BMC Genom. 2016, 17, 24–40. [Google Scholar] [CrossRef] [PubMed]
- Markakis, M.N.; Boron, A.K.; Van Loock, B.; Saini, K.; Cirera, S.; Verbelen, J.P.; Vissenberg, K. Characterization of a small auxin-up RNA (SAUR)-like gene involved in Arabidopsis thaliana development. PLoS ONE 2013, 8, e82596. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Gray, W.M. SAUR Proteins as effectors of hormonal and environmental signals in plant growth. Mol. Plant 2015, 8, 1153–1164. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Xu, S.B.; Li, J. BAK1 directly regulates brassinosteroid perception and BRI1 activation. J. Integr. Plant Biol. 2013, 55, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Li, J. Multi-tasking of somatic embryogenesis receptor-like protein kinases. Curr. Opin. Plant Biol. 2010, 13, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.Y.; Sae-Seaw, J.; Wang, Z.Y. Brassinosteroid signalling. Development 2013, 140, 1615–1620. [Google Scholar] [CrossRef] [PubMed]
- Mutasa-Göttgens, E.; Hedden, P. Gibberellin as a factor in floral regulatory networks. J. Exp. Bot. 2009, 60, 1979–1989. [Google Scholar] [CrossRef] [PubMed]
- Nag, A.; King, S.; Jack, T. miR319a targeting of TCP4 is critical for petal growth and development in Arabidopsis. Proc. Natl. Acad. Sci. USA 2009, 106, 22534–22539. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Somoza, I.; Weigel, D. Coordination of flower maturation by a regulatory circuit of three microRNAs. PLoS Genet. 2013, 9, e1003374. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Mao, Y.F.; Yang, J.; He, Y.K. TCP 24 modulates secondary cell wall thickening and anther endothecium and anther development. Front. Plant Sci. 2015, 6, 436–446. [Google Scholar]
- Liu, X.; Huang, J.; Wang, Y.; Khanna, K.; Xie, Z.; Owen, H.A.; Zhao, D. The role of floral organs in carpels, an Arabdopsis loss-of-function mutation in microRNA160a, in organogenesis and the mechanism regulating its expression. Plant J. 2010, 62, 416–428. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.B.; Zhang, D.; Zhao, C.P.; Li, Y.M.; Ma, J.J.; An, N.; Han, M.Y. Shoot bending promotes flower bud formation by miRNA-mediated regulation in apple (Malus domestica Borkh.). Plant Biotechnol. J. 2016, 14, 749–770. [Google Scholar] [CrossRef] [PubMed]
- Achard, P.; Herr, A.; Baulcombe, D.C.; Harberd, N.P. Modulation of floral development by a gibberellin-regulated microRNA. Development 2004, 131, 3357–3365. [Google Scholar] [CrossRef] [PubMed]
- Ariizumi, T.; Lawrence, P.K.; Steber, C.M. The role of two f-box proteins, SLEEPY1 and SNEEZY, in Arabidopsis gibberellin signaling. Plant Physiol. 2011, 155, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.P.; Zhao, F.A.; Sun, Y.; Xie, D.Y.; Sun, L.; Xu, Z.Z.; Zhu, W.; Yang, L.R.; Zhao, Y.M.; Lv, S.P.; et al. Transcriptomic profiling reveals complex molecular regulation in cotton genic male sterile mutant Yu98-8A. PLoS ONE 2015, 10, e0133425. [Google Scholar] [CrossRef] [PubMed]
- Chaabane, S.B.; Liu, R.Y.; Chinnusamy, V.; Kwon, Y.; Park, J.H.; Kim, S.Y.; Zhu, J.K.; Yang, S.W.; Lee, B.H. STA1, an Arabidopsis pre-mRNA processing factor 6 homolog, is a new player involved in miRNA biogenesis. Nucleic Acids Res. 2013, 41, 1984–1997. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. microRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef] [PubMed]
- Meyers, B.C.; Axtell, M.J.; Bartel, B.; Bartel, D.P.; Baulcombe, D.; Bowman, J.L.; Cao, X.; Carrington, J.C.; Chen, X.; Green, P.J.; et al. Criteria for annotation of plant MicroRNAs. Plant Cell 2008, 20, 3186–3190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.H.; Pan, X.P.; Cox, S.B.; Cobb, G.P.; Anderson, T.A. Evidence that miRNAs are different from other RNAs. Cell Mol. Life Sci. 2006, 63, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Audic, S.; Claverie, J.M. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [PubMed]
- Addo-Quaye, C.; Eshoo, T.W.; Bartel, D.P.; Axtell, M.J. Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr. Biol. 2008, 18, 758–762. [Google Scholar] [CrossRef] [PubMed]
- German, M.A.; Pillay, M.; Jeong, D.H.; Hetawal, A.; Luo, S.; Janardhanan, P.; Kannan, V.; Rymarquis, L.A.; Nobuta, K.; German, R.; et al. Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends. Nat. Biotechnol. 2008, 26, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Addo-Quaye, C.; Miller, W.; Axtell, M.J. CleaveLand: A pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 2009, 25, 130–131. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Zhou, X.; Ling, Y.; Zhang, Z.H.; Su, Z. AgriGO: A GO analysis tool kit for the agricultural community. Nucleic Acids Res. 2010, 38, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Lu, Y.; Bai, S.L.; Zhang, W.N.; Duan, X.W.; Meng, D.; Wang, Z.G.; Wang, A.D.; Zhou, Z.S.; Li, T.Z. Cloning and characterization of miRNAs and their targets, including a novel miRNA-targeted NBS-LRR protein class gene in apple (Golden Delicious). Mol. Plant 2014, 7, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Zhang, J.H.; Wang, Z.Q.; Zhu, Q.S.; Wang, W. Hormonal changes in the grains of rice subjected to water stress during grain filling. Plant Physiol. 2001, 127, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Neill, S.J.; Tang, Z.; Cai, W. Gibberellin-regulated XET is differentially induced by auxin in rice leaf sheath bases during gravitropic bending. J. Exp. Bot. 2005, 56, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.P.; Xie, D.Y.; Zhu, H.Q.; Li, W.; Xu, Z.Z.; Yang, L.R.; Li, Z.F.; Sun, L.; Wang, J.X.; Nie, L.H.; et al. Comparative proteomic analysis of Gossypium thurberi in response to Verticillium dahliae inoculation. Int. J. Mol. Sci. 2015, 16, 25121–25140. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.Y.; Zhang, C.X. Data Processing System (DPS) software with experimental design, statistical analysis and data mining developed for use in entomological research. Insect Sci. 2013, 20, 254–260. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Library of MS Buds | Library of MF Buds | ||
|---|---|---|---|---|
| Unique sRNAs (%) | Total sRNAs (%) | Unique sRNAs (%) | Total sRNAs (%) | |
| Total | 5,410,937 (100) | 11,002,282 (100) | 5,151,550 (100) | 10,663,659 (100) |
| Exon_antisense | 49,962 (0.92) | 99,307 (0.9) | 44,463 (0.86) | 91,843 (0.86) |
| Exon_sense | 70,831 (1.31) | 120,224 (1.09) | 6,5017 (1.26) | 110,560 (1.04) |
| Intron_antisense | 44,021 (0.81) | 116,204 (1.06) | 42,055 (0.82) | 114,356 (1.07) |
| Intron_sense | 54,670 (1.01) | 136,727 (1.24) | 52,371 (1.02) | 133,319 (1.25) |
| miRNA | 15,511 (0.29) | 763,877 (6.94) | 15,476 (0.3) | 718,317 (6.74) |
| rRNA | 28,523 (0.53) | 179,086 (1.63) | 28,326 (0.55) | 189,958 (1.78) |
| repeat | 227,017 (4.2) | 635,535 (5.78) | 213,793 (4.15) | 598,611 (5.61) |
| snRNA | 2119 (0.04) | 5134 (0.05) | 1801 (0.03) | 4478 (0.04) |
| snoRNA | 850 (0.02) | 2271 (0.02) | 691 (0.01) | 2441 (0.02) |
| tRNA | 6206 (0.11) | 61,552 (0.56) | 4,459 (0.09) | 46,324 (0.43) |
| Unannotated | 4,911,227 (90.76) | 8,882,365 (80.73) | 4,683,098 (90.91) | 8,653,452 (81.15) |
| miRNA | Target | |
|---|---|---|
| Gene ID | Function Annotation | |
| miR160a | Cotton_D_gene_10025306 | auxin response factor 18-like |
| Cotton_D_gene_10006397 | putative auxin response factor ARF16 | |
| Cotton_D_gene_10005922 | auxin response factor 18-like | |
| Cotton_D_gene_10019554 | auxin response factor | |
| Cotton_D_gene_10010678 | putative auxin response factor | |
| Cotton_D_gene_10008456 | auxin response factor | |
| Cotton_D_gene_10002224 | auxin response factor | |
| miR2118a_3p | Cotton_D_gene_10027482 | DNA-directed RNA polymerases I, II, and III subunit RPABC1 |
| Cotton_D_gene_10027759 | phospholipase D | |
| Cotton_D_gene_10025494 | pre-mRNA-processing factor 40 | |
| Cotton_D_gene_10027502 | DNA-directed RNA polymerases I, II, and III subunit RPABC1 | |
| Cotton_D_gene_10027449 | DNA-directed RNA polymerases I, II, and III subunit RPABC1 | |
| Cotton_D_gene_10027508 | maintenance of ploidy protein MOB1 | |
| Cotton_D_gene_10027458 | NBS-LRR resistance protein-like protein | |
| Cotton_D_gene_10027448 | TIR-NBS-LRR resistance protein | |
| Cotton_D_gene_10027489 | DNA-directed RNA polymerases I, II, and III subunit RPABC1 | |
| Cotton_D_gene_10039426 | nitrate reductase (NADH) | |
| Cotton_D_gene_10017763 | uncharacterized protein | |
| Cotton_D_gene_10027457 | DNA-directed RNA polymerases I, II, and III subunit RPABC1 | |
| Cotton_D_gene_10027494 | DNA-directed RNA polymerases I, II, and III subunit RPABC1 | |
| Cotton_D_gene_10017214 | uncharacterized protein | |
| Cotton_D_gene_10012644 | DNA-directed RNA polymerases I, II, and III subunit RPABC1 | |
| Cotton_D_gene_10031090 | protein phosphatase 1, catalytic subunit | |
| Cotton_D_gene_10027490 | ATP binding protein | |
| Cotton_D_gene_10027493 | DNA-directed RNA polymerases I, II, and III subunit RPABC1 | |
| Cotton_D_gene_10027495 | maintenance of ploidy protein MOB1 | |
| Cotton_D_gene_10027490 | ATP binding protein | |
| Cotton_D_gene_10027485 | DNA-directed RNA polymerases I, II, and III subunit RPABC1 | |
| Cotton_D_gene_10027456 | NBS-LRR resistance protein-like protein | |
| Cotton_D_gene_10027452 | TIR-NBS-LRR resistance protein | |
| Cotton_D_gene_10027491 | hypothetical protein | |
| Cotton_D_gene_10027497 | DNA-directed RNA polymerases I, II, and III subunit RPABC1 | |
| Cotton_D_gene_10027484 | hypothetical protein | |
| miR394a | Cotton_D_gene_10011419 | F-box family protein |
| miR397a | Cotton_D_gene_10033270 | laccase 1b |
| miR5224a | Cotton_D_gene_10020595 | cysteine-rich receptor-like protein kinase 25-like |
| novel_mir_104 | Cotton_D_gene_10039906 | TCP4 |
| Cotton_D_gene_10003308 | TCP4 | |
| novel_mir_168 | Cotton_D_gene_10018721 | predicted protein |
| novel_mir_20 | Cotton_D_gene_10009655 | chromatin licensing and DNA replication factor 1 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, X.; Zhao, Y.; Xie, D.; Sun, Y.; Zhu, X.; Esmaeili, N.; Yang, Z.; Wang, Y.; Yin, G.; Lv, S.; et al. Identification and Functional Analysis of microRNAs Involved in the Anther Development in Cotton Genic Male Sterile Line Yu98-8A. Int. J. Mol. Sci. 2016, 17, 1677. https://doi.org/10.3390/ijms17101677
Yang X, Zhao Y, Xie D, Sun Y, Zhu X, Esmaeili N, Yang Z, Wang Y, Yin G, Lv S, et al. Identification and Functional Analysis of microRNAs Involved in the Anther Development in Cotton Genic Male Sterile Line Yu98-8A. International Journal of Molecular Sciences. 2016; 17(10):1677. https://doi.org/10.3390/ijms17101677
Chicago/Turabian StyleYang, Xiaojie, Yuanming Zhao, Deyi Xie, Yao Sun, Xunlu Zhu, Nardana Esmaeili, Zuoren Yang, Ye Wang, Guo Yin, Shuping Lv, and et al. 2016. "Identification and Functional Analysis of microRNAs Involved in the Anther Development in Cotton Genic Male Sterile Line Yu98-8A" International Journal of Molecular Sciences 17, no. 10: 1677. https://doi.org/10.3390/ijms17101677
APA StyleYang, X., Zhao, Y., Xie, D., Sun, Y., Zhu, X., Esmaeili, N., Yang, Z., Wang, Y., Yin, G., Lv, S., Nie, L., Tang, Z., Zhao, F., Li, W., Mishra, N., Sun, L., Zhu, W., & Fang, W. (2016). Identification and Functional Analysis of microRNAs Involved in the Anther Development in Cotton Genic Male Sterile Line Yu98-8A. International Journal of Molecular Sciences, 17(10), 1677. https://doi.org/10.3390/ijms17101677