
Upregulated MicroRNA-25 Mediates the Migration of Melanoma Cells by Targeting DKK3 through the WNT/β-Catenin Pathway

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
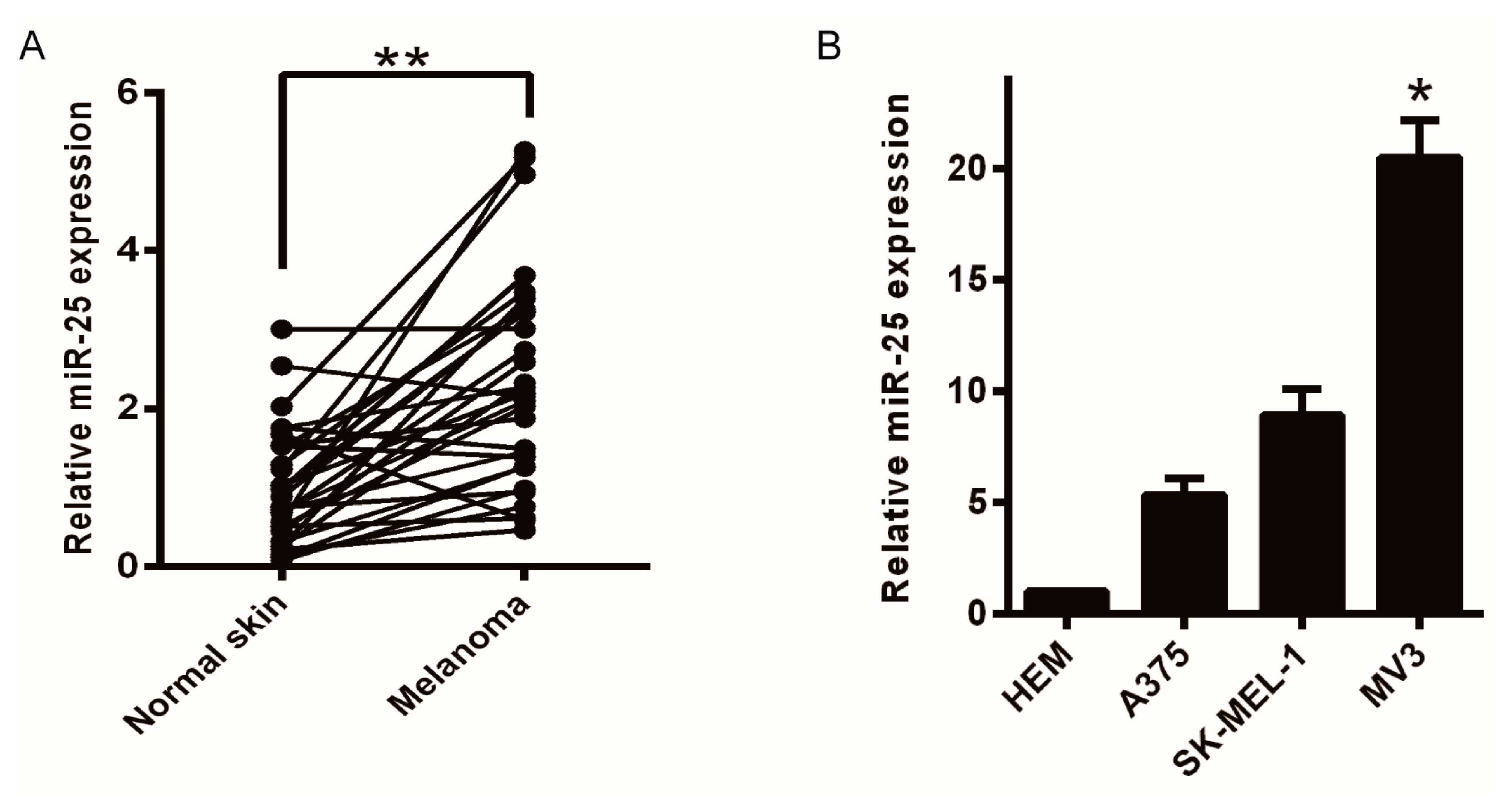
2.1. Upregulation of miR-25 Is Associated with Melanoma
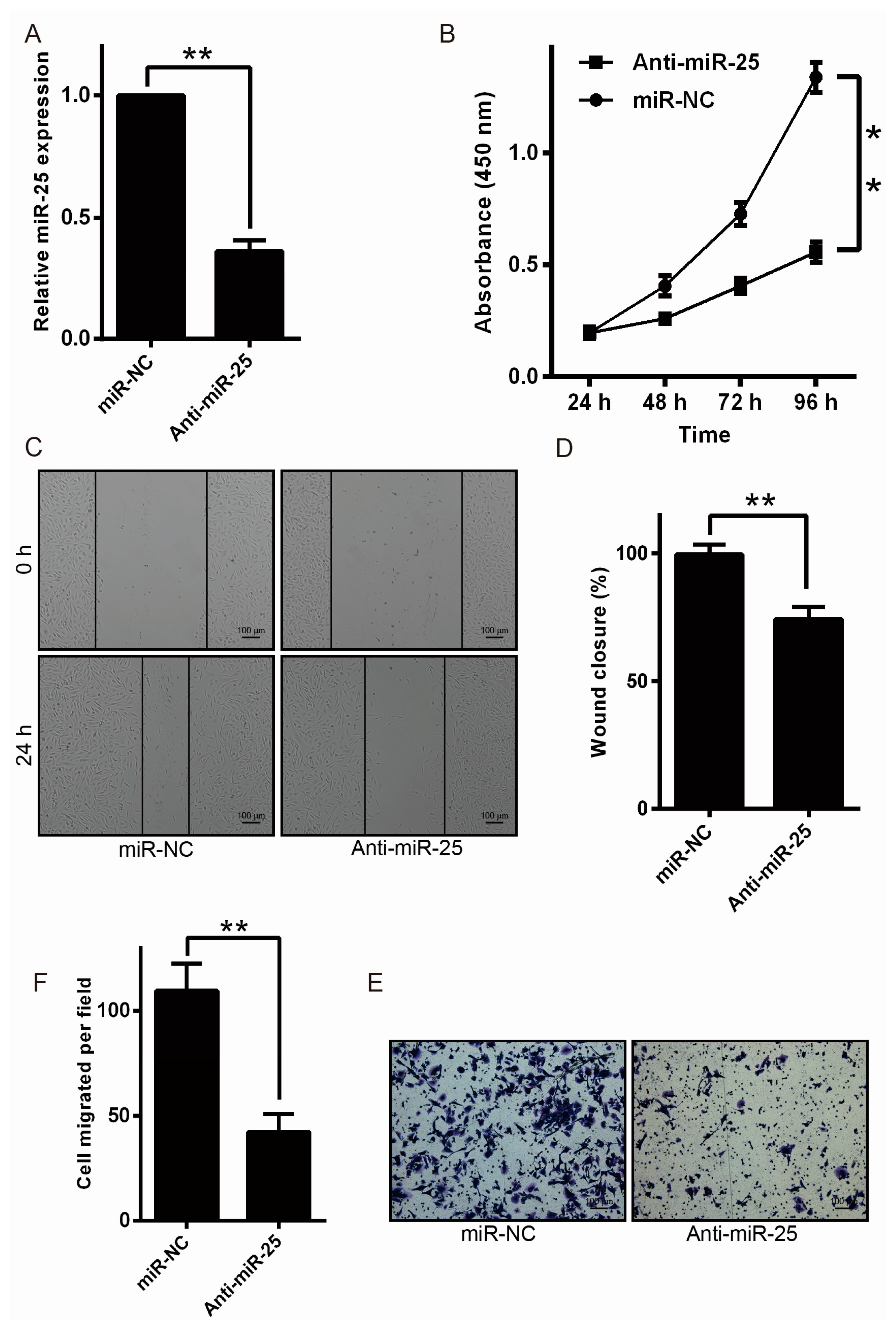
2.2. Downregulation of miR-25 Inhibits Melanoma Cell Proliferation and Motility
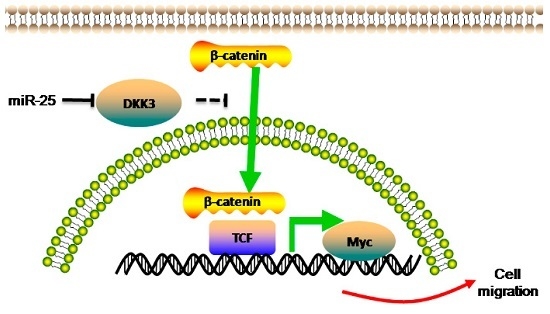
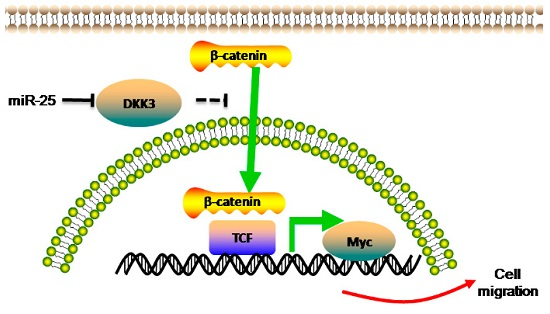
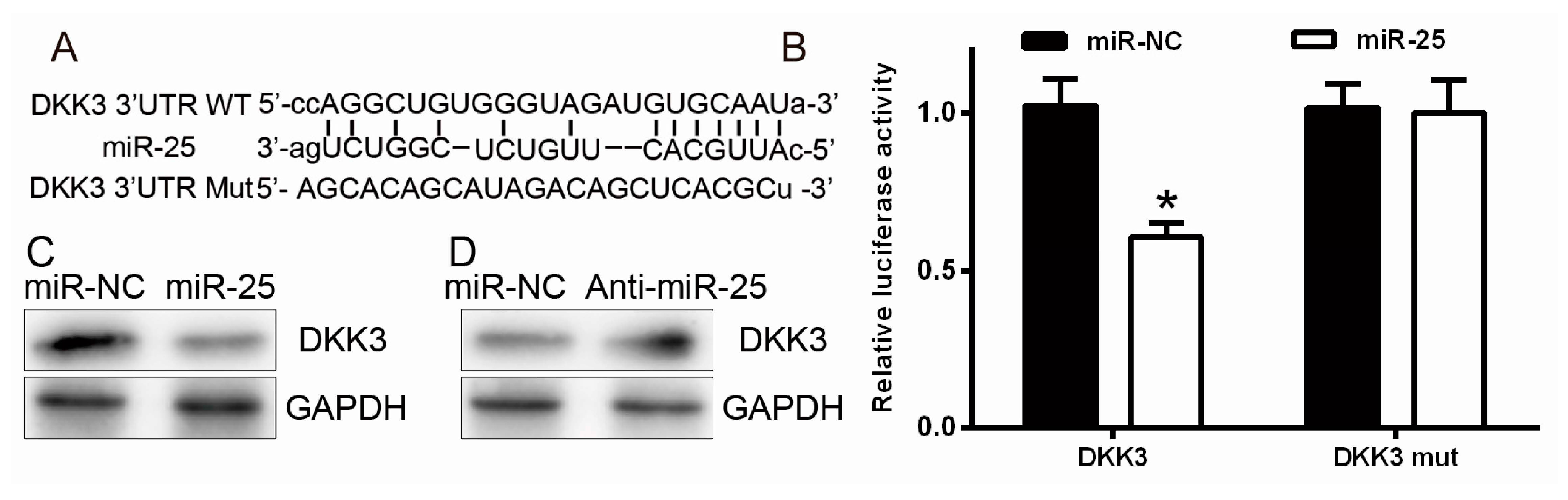
2.3. miR-25 Targets DKK3 (Dickkopf-Related Protein 3)
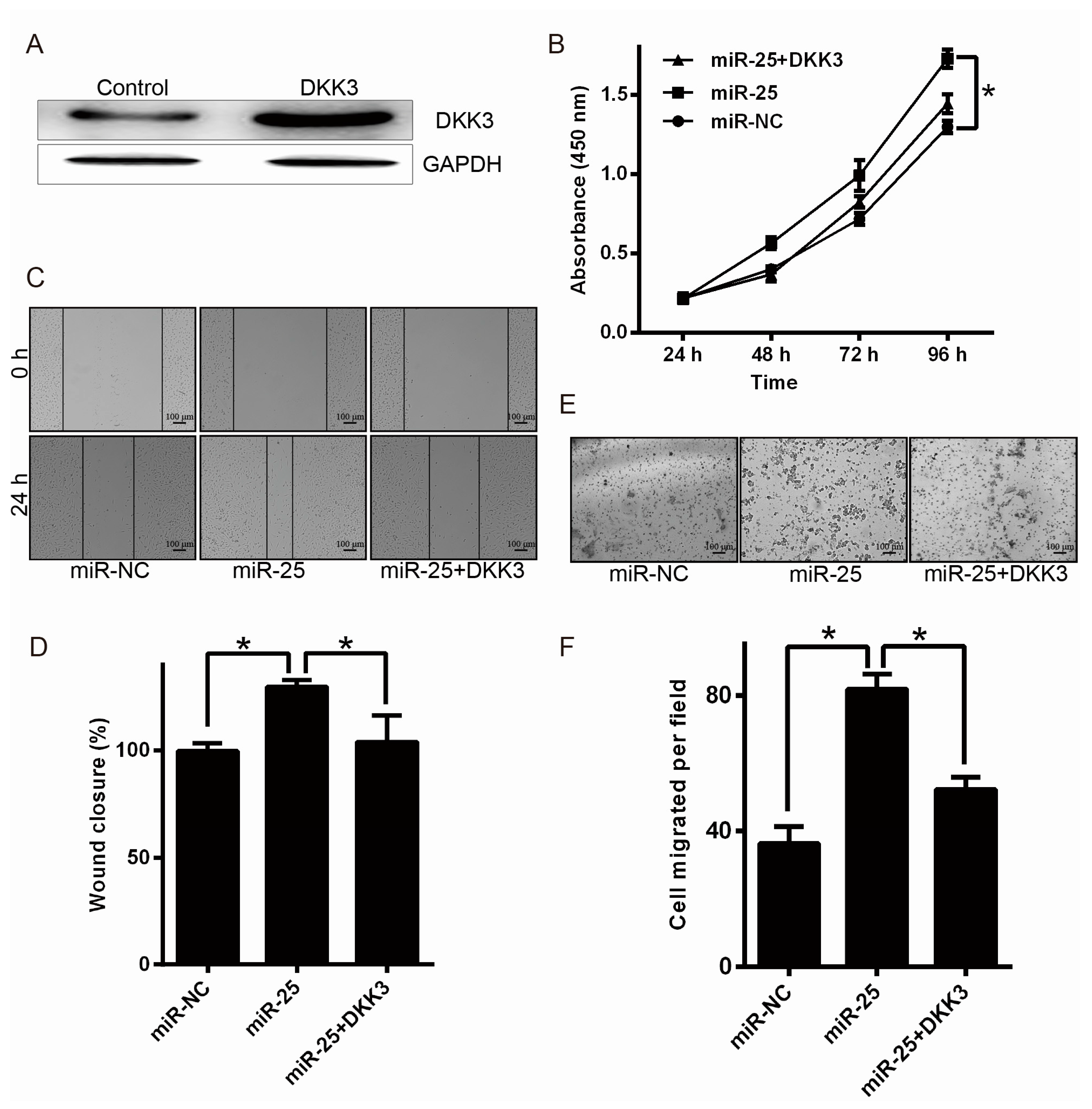
2.4. Upregulation of DKK3 Partially Attenuates the Oncogenic of miR-25
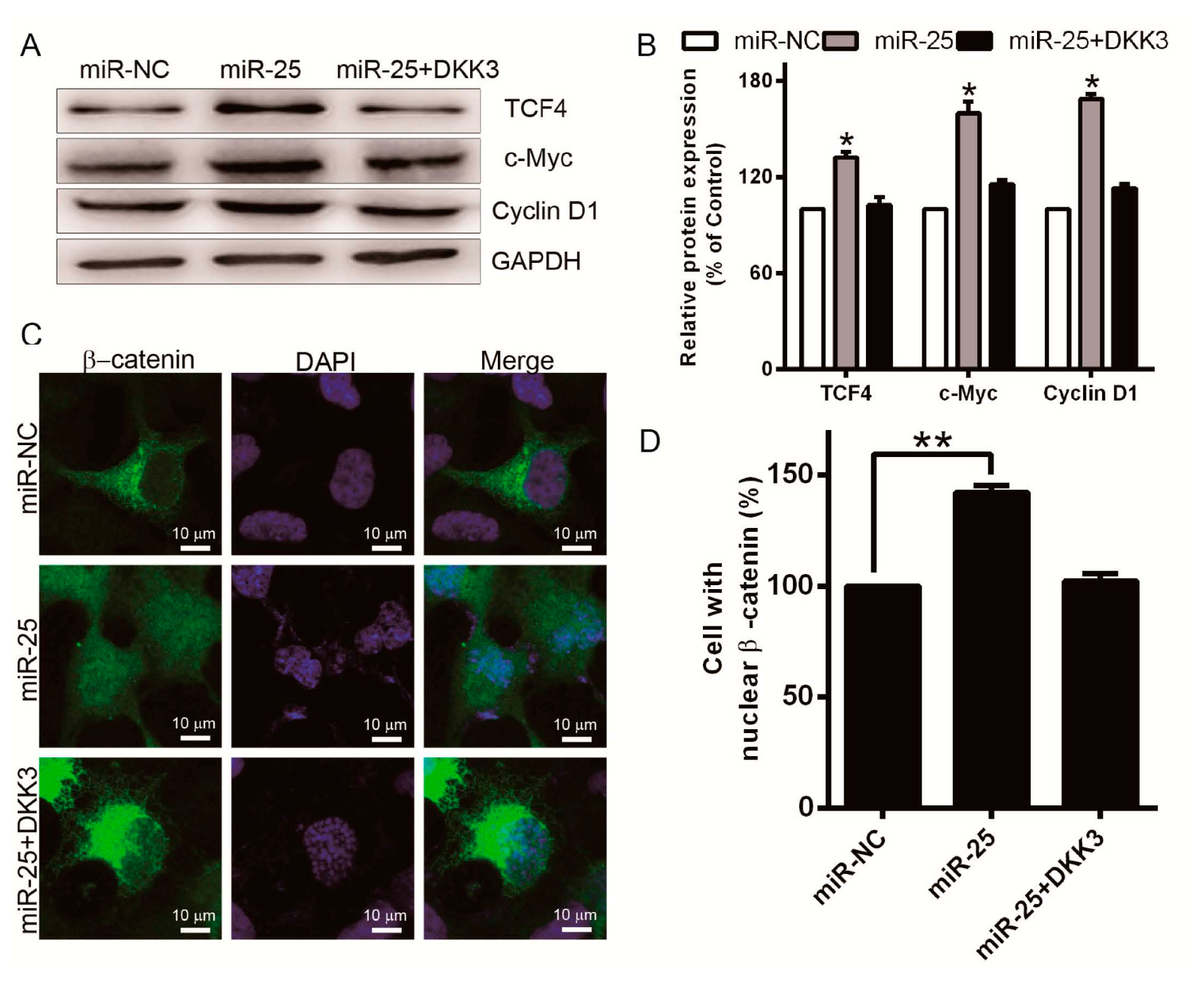
2.5. Overexpreesion of miR-25 Promotes Cell Proliferation and Induces β-Catenin/TCF4 Signaling Activation
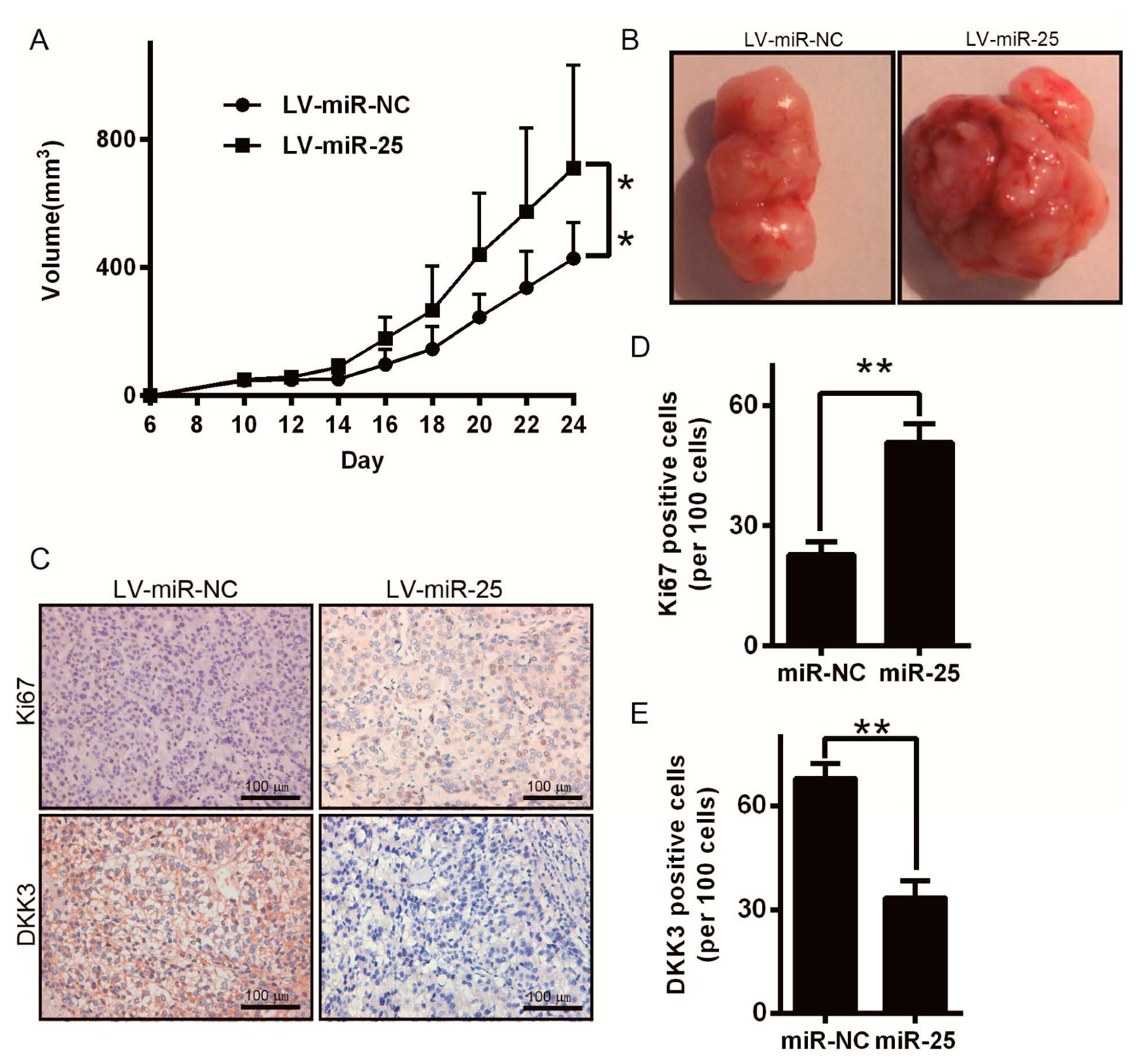
2.6. miR-25 Promotes Melanoma Tumor Growth in Nude Mice
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Tissue Samples
4.3. RNA Isolation, Reverse Transcription and Quantitative Real-Time PCR (qRT-PCR)
4.4. Analysis of 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT)
4.5. Cell Migration Assays
4.6. Wound-Healing Assay
4.7. Transient Transfection with miR-25 or miRNA Inhibitors
4.8. Protein Isolation and Western Blot
4.9. 3′-Untranslated Region (3′-UTR) Luciferase Reporter Assays
4.10. Lentivirus-Based MiR-25 Overexpression
4.11. Tumorigenicity Assays in Nude Mice
4.12. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Global cancer statistics, 2002. CA Cancer J. Clin. 2005, 55, 74–108. [Google Scholar] [CrossRef] [PubMed]
- Brozyna, A.A.; Jochymski, C.; Janjetovic, Z.; Jozwicki, W.; Tuckey, R.C.; Slominski, A.T. CYP24A1 expression inversely correlates with melanoma progression: Clinic-pathological studies. Int. J. Mol. Sci. 2014, 15, 19000–19017. [Google Scholar] [CrossRef] [PubMed]
- Melnikova, V.O.; Bar-Eli, M. Transcriptional control of the melanoma malignant phenotype. Cancer Biol. Ther. 2008, 7, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Chin, L. The genetics of malignant melanoma: Lessons from mouse and man. Nat. Rev. Cancer 2003, 3, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Micrornas: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Shukla, G.C.; Singh, J.; Barik, S. Micrornas: Processing, maturation, target recognition and regulatory functions. Mol. Cell. Pharmacol. 2011, 3, 83–92. [Google Scholar] [PubMed]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by micrornas: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Valastyan, S.; Weinberg, R.A. MicroRNAs: Crucial multi-tasking components in the complex circuitry of tumor metastasis. Cell Cycle 2009, 8, 3506–3512. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.P.; Cost, N.G.; Hegde, S.; Kellner, E.; Mikhaylova, O.; Stratton, Y.; Ehmer, B.; Abplanalp, W.A.; Pandey, R.; Biesiada, J.; et al. TRPM3 and miR-204 establish a regulatory circuit that controls oncogenic autophagy in clear cell renal cell carcinoma. Cancer Cell 2014, 26, 738–753. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.J.; Bahal, R.; Babar, I.A.; Pincus, Z.; Barrera, F.; Liu, C.; Svoronos, A.; Braddock, D.T.; Glazer, P.M.; Engelman, D.M.; et al. MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature 2015, 518, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, X.; Su, Z.; Fei, H.; Liu, X.; Pan, Q. miR-25 promotes hepatocellular carcinoma cell growth, migration and invasion by inhibiting RhoGDI1. Oncotarget 2015, 6, 36231–36244. [Google Scholar] [PubMed]
- Wang, Z.; Wang, N.; Liu, P.; Chen, Q.; Situ, H.; Xie, T.; Zhang, J.; Peng, C.; Lin, Y.; Chen, J. MicroRNA-25 regulates chemoresistance-associated autophagy in breast cancer cells, a process modulated by the natural autophagy inducer isoliquiritigenin. Oncotarget 2014, 5, 7013–7026. [Google Scholar] [CrossRef] [PubMed]
- Razumilava, N.; Bronk, S.F.; Smoot, R.L.; Fingas, C.D.; Werneburg, N.W.; Roberts, L.R.; Mott, J.L. miR-25 targets TNF-related apoptosis inducing ligand (trail) death receptor-4 and promotes apoptosis resistance in cholangiocarcinoma. Hepatology 2012, 55, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Pan, W.; Jin, Y.; Zheng, J. miR-25 promotes ovarian cancer proliferation and motility by targeting LATS2. Tumour Biol. 2014, 35, 12339–12344. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Liu, J.; Wang, C.; Wang, Y.; Jiang, Y.; Guo, M. MicroRNA-25 regulates small cell lung cancer cell development and cell cycle through cyclin E2. Int. J. Clin. Exp. Pathol. 2014, 7, 7726–7734. [Google Scholar] [PubMed]
- Li, B.S.; Zuo, Q.F.; Zhao, Y.L.; Xiao, B.; Zhuang, Y.; Mao, X.H.; Wu, C.; Yang, S.M.; Zeng, H.; Zou, Q.M.; et al. MicroRNA-25 promotes gastric cancer migration, invasion and proliferation by directly targeting transducer of ERBB2, 1 and correlates with poor survival. Oncogene 2015, 34, 2556–2565. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tan, W.; Neo, T.W.; Aung, M.O.; Wasser, S.; Lim, S.G.; Tan, T.M. Role of the miR-106b-25 microRNA cluster in hepatocellular carcinoma. Cancer Sci. 2009, 100, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, J.; Wu, S.; Zhu, S.; Wang, S.; Zhou, H.; Tian, X.; Tang, N.; Nie, S. Angiopoietin-like protein 2 negatively regulated by microRNA-25 contributes to the malignant progression of colorectal cancer. Int. J. Mol. Med. 2014, 34, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Veeck, J.; Dahl, E. Targeting the WNT pathway in cancer: The emerging role of Dickkopf-3. Biochim. Biophys. Acta 2012, 1825, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Kuphal, S.; Lodermeyer, S.; Bataille, F.; Schuierer, M.; Hoang, B.H.; Bosserhoff, A.K. Expression of Dickkopf genes is strongly reduced in malignant melanoma. Oncogene 2006, 25, 5027–5036. [Google Scholar] [CrossRef] [PubMed]
- Hoang, B.H.; Kubo, T.; Healey, J.H.; Yang, R.; Nathan, S.S.; Kolb, E.A.; Mazza, B.; Meyers, P.A.; Gorlick, R. Dickkopf 3 inhibits invasion and motility of Saos-2 osteosarcoma cells by modulating the WNT-β-catenin pathway. Cancer Res. 2004, 64, 2734–2739. [Google Scholar] [CrossRef] [PubMed]
- Yue, W.; Sun, Q.; Dacic, S.; Landreneau, R.J.; Siegfried, J.M.; Yu, J.; Zhang, L. Downregulation of Dkk3 activates β-catenin/TCF-4 signaling in lung cancer. Carcinogenesis 2008, 29, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Yin, Y.; Yuan, H.; Sakamaki, T.; Okano, H.; Glazer, R.I. Musashi1 modulates mammary progenitor cell expansion through proliferin-mediated activation of the WNT and notch pathways. Mol. Cell. Biol. 2008, 28, 3589–3599. [Google Scholar] [CrossRef] [PubMed]
- Damsky, W.E.; Curley, D.P.; Santhanakrishnan, M.; Rosenbaum, L.E.; Platt, J.T.; Gould Rothberg, B.E.; Taketo, M.M.; Dankort, D.; Rimm, D.L.; McMahon, M.; et al. β-Catenin signaling controls metastasis in Braf-activated Pten-deficient melanomas. Cancer Cell 2011, 20, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Scherr, M.; Venturini, L.; Battmer, K.; Schaller-Schoenitz, M.; Schaefer, D.; Dallmann, I.; Ganser, A.; Eder, M. Lentivirus-mediated antagomir expression for specific inhibition of miRNA function. Nucleic Acids Res. 2007, 35, e149. [Google Scholar] [CrossRef] [PubMed]






© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huo, J.; Zhang, Y.; Li, R.; Wang, Y.; Wu, J.; Zhang, D. Upregulated MicroRNA-25 Mediates the Migration of Melanoma Cells by Targeting DKK3 through the WNT/β-Catenin Pathway. Int. J. Mol. Sci. 2016, 17, 1124. https://doi.org/10.3390/ijms17111124
Huo J, Zhang Y, Li R, Wang Y, Wu J, Zhang D. Upregulated MicroRNA-25 Mediates the Migration of Melanoma Cells by Targeting DKK3 through the WNT/β-Catenin Pathway. International Journal of Molecular Sciences. 2016; 17(11):1124. https://doi.org/10.3390/ijms17111124
Chicago/Turabian StyleHuo, Jia, Yanfei Zhang, Ruilian Li, Yuan Wang, Jiawen Wu, and Dingwei Zhang. 2016. "Upregulated MicroRNA-25 Mediates the Migration of Melanoma Cells by Targeting DKK3 through the WNT/β-Catenin Pathway" International Journal of Molecular Sciences 17, no. 11: 1124. https://doi.org/10.3390/ijms17111124
APA StyleHuo, J., Zhang, Y., Li, R., Wang, Y., Wu, J., & Zhang, D. (2016). Upregulated MicroRNA-25 Mediates the Migration of Melanoma Cells by Targeting DKK3 through the WNT/β-Catenin Pathway. International Journal of Molecular Sciences, 17(11), 1124. https://doi.org/10.3390/ijms17111124