Abstract
There are numerous gene rearrangements and transfer RNA gene absences existing in mitochondrial (mt) genomes of Aleyrodidae species. To understand how mt genomes evolved in the family Aleyrodidae, we have sequenced the complete mt genome of Aleurocanthus camelliae and comparatively analyzed all reported whitefly mt genomes. The mt genome of A. camelliae is 15,188 bp long, and consists of 13 protein-coding genes, two rRNA genes, 21 tRNA genes and a putative control region (GenBank: KU761949). The tRNA gene, trnI, has not been observed in this genome. The mt genome has a unique gene order and shares most gene boundaries with Tetraleurodes acaciae. Nineteen of 21 tRNA genes have the conventional cloverleaf shaped secondary structure and two (trnS1 and trnS2) lack the dihydrouridine (DHU) arm. Using ARWEN and homologous sequence alignment, we have identified five tRNA genes and revised the annotation for three whitefly mt genomes. This result suggests that most absent genes exist in the genomes and have not been identified, due to be lack of technology and inference sequence. The phylogenetic relationships among 11 whiteflies and Drosophila melanogaster were inferred by maximum likelihood and Bayesian inference methods. Aleurocanthus camelliae and T. acaciae form a sister group, and all three Bemisia tabaci and two Bemisia afer strains gather together. These results are identical to the relationships inferred from gene order. We inferred that gene rearrangement plays an important role in the mt genome evolved from whiteflies.
1. Introduction
Over the last decade, the mitochondrial (mt) genome sequences have been frequently used to study species discrimination [1,2], molecule evolution [3,4,5,6], phylogenetic inferences [3,4,7,8] and population genetics [9,10], due to their small genome size, rapid evolutionary rate, low level or absence of sequence recombination and evolutionary conserved gene products [11,12]. With a few exceptions, animal mt genome is typically a circular double strand DNA molecule, with a size of 13–20 kb, consisting of a putative control region (CR) and 37 genes: 13 protein-coding genes (PCG), two ribosomal RNA genes (rRNA), and 22 transfer RNA genes (tRNA) [13,14,15,16,17]. The animal ancestral genome organization is retained in most insects, although minor changes were observed in some species [14,17]. In several groups of insects, mt genome organizations are not conservative and contain a lot of gene absences and rearrangements, pseudogenes or have been fragmented [5,8,18,19].
In recent years, whiteflies have become an almost worldwide problem for agriculturalists. Family Aleyrodidae, with around 1450 named species, belongs to the order Hemiptera and comprise a single superfamily, Aleyrodoidea, within the suborder Sternorrhyncha [20]. Recent studies showed that mt genome of whiteflies have numerous gene rearrangements and transfer RNA gene absences [21,22,23,24]. Transposition of gene block cox3-trnG-nad3 and the position of trnY and trnC are typical in whitefly mt genomes. The number of absent genes ranges from one to five, and the mt genome of Neomaskellia andropogonis has lost the most tRNAs, e.g., trnA, trnR, trnN, trnI and trnS2 [21]. The gene, trnI, is missed frequently and not found in the mt genomes of Aleurochiton aceris, Tetraleurodes acaciae and N. andropogonis [21].
The camellia spiny whitefly, Aleurocanthus camelliae Kanmiya and Kasai (Hemiptera: Aleyrodidae), is an important pest in tea plantations and poses a risk to tea quality and production. To understand how mt genomes evolved in the family Aleyrodidae, the complete sequence of the mt genome of A. camelliae was sequenced and comparative analyses of all reported whitefly mt genomes were conducted.
2. Results and Discussion
2.1. Genome Content and Nucleotide Composition
The mt genome of A. camelliae (GenBank accession KU761949) is a typical closed-circular and double stranded DNA molecule. The genome contains 36 of 37 genes usually found in animal mt genomes, including 13 protein-coding genes (PCG), two ribosomal RNAs, 21 transfer RNAs, and a putative control region (CR) (Figure 1 and Table 1). Genes are encoded by both strands of this mt genome: 14 genes on one strand whereas the rest on the other strand. The camellia spiny whitefly mt genome is in size of 15,188 bp, which is comparable to mt genome of other whiteflies ranging from 14,496 bp (N. andropogonis) to 18,414 bp (Trialeurodes vaporariorum) [21]. The major reason for the variation of genome size is the variable number of tandemly repeated elements in the putative control region. The control region of A. camelliae includes a poly-T sequence and two tandem repeat sequences, one repeat region consists of two 128 bp repeat units (13,423–13,678), and the other contains eight 19 bp repeat units (13,737–13,888). Similarly, the mt genome of T. vaporariorum has five 721 bp repeat units [21].
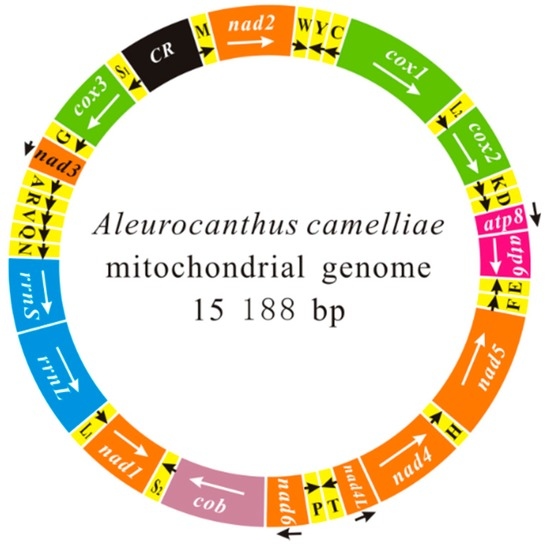
Figure 1.
The mitochondrial genomes of Aleurocanthus camelliae. The transcriptional orientation is indicated with arrows. Protein-coding genes, ribosomal RNA genes and transfer RNA genes are shown in bright colors. All of them in the map follow standard abbreviations. tRNA genes for the two serine and two leucine tRNAs: S1 = AGN, S2 = UCN, L1 = CUN and L2 = UUR. The putative control region is indicated in black.

Table 1.
Annotation and gene organization of the mitochondrial genome of Aleurocanthus camelliae.
A total of 28 bp overlaps have been found at eight gene junctions of this mt genome, and the size of overlaps are ranging from 1 to 7 bp. Except for the putative control region, the genome has a total of 254 bp of intergenic sequence at 16 locations ranging from 1 to 94 bp, and the longest intergenic sequence is located between rrnL and rrnS. Unlike A. camelliae, a 53 bp long intergenic sequence locates between rrnL and trnV in the mt genome of T. acaciae [21].
As for all other insects, the nucleotide composition of the mt genome of A. camelliae is biased toward A and T nucleotides, with an A + T content of 70.90%. It consists of 69.49% in the protein coding genes, 76.61% in the ribosomal RNA genes, 77.19% in the transfer RNA genes and 66.86% in the putative control region, respectively (Table 2). Excluding the incomplete stop codons, a total of 3610 amino acids of protein-coding genes are encoded in the camellia spiny whitefly mt genome. The high A+T content is reflected in the codon usage, especially in the third position: codons for A or T (A = 33.88%, T = 40.46%) are strongly preferred over C or G (C = 14.19%, G = 11.47%) in the third codon position. The most frequent amino acids in the PCGs of the camellia spiny whitefly are Leucine (13.95%), Serine (10.37%), Phenylalanine (9.69%) and Isoleucine (9.36%) (Table 3).
Table 2.
Nucleotide composition of the mitochondrial genome of Aleurocanthus camelliae.
Table 3.
Codon usage for the 13 mitochondrial protein-coding genes of Aleurocanthus camelliae.
The total length of all 13 protein-coding genes is 10,833 bp, which accounts for 71.33% of the whole length of the mt genome of A. camelliae. All PCGs initiate translation using an ATN codon (ATA for atp8, nad2 and nad4–6; ATT for atp6, cox2, nad1 and nad3; ATG for cob, cox1, cox3 and nad4L). Three PCGs (cox2, nad1 and nad5) have incomplete terminal codons consisting of “T-” nucleotide, and other stop with TAA and TAG (Table 1). Incomplete stop codons are common in animal mt genomes and could produce functional stop codons in polycistronic transcription cleavage and polyadenylation mechanisms [25,26].
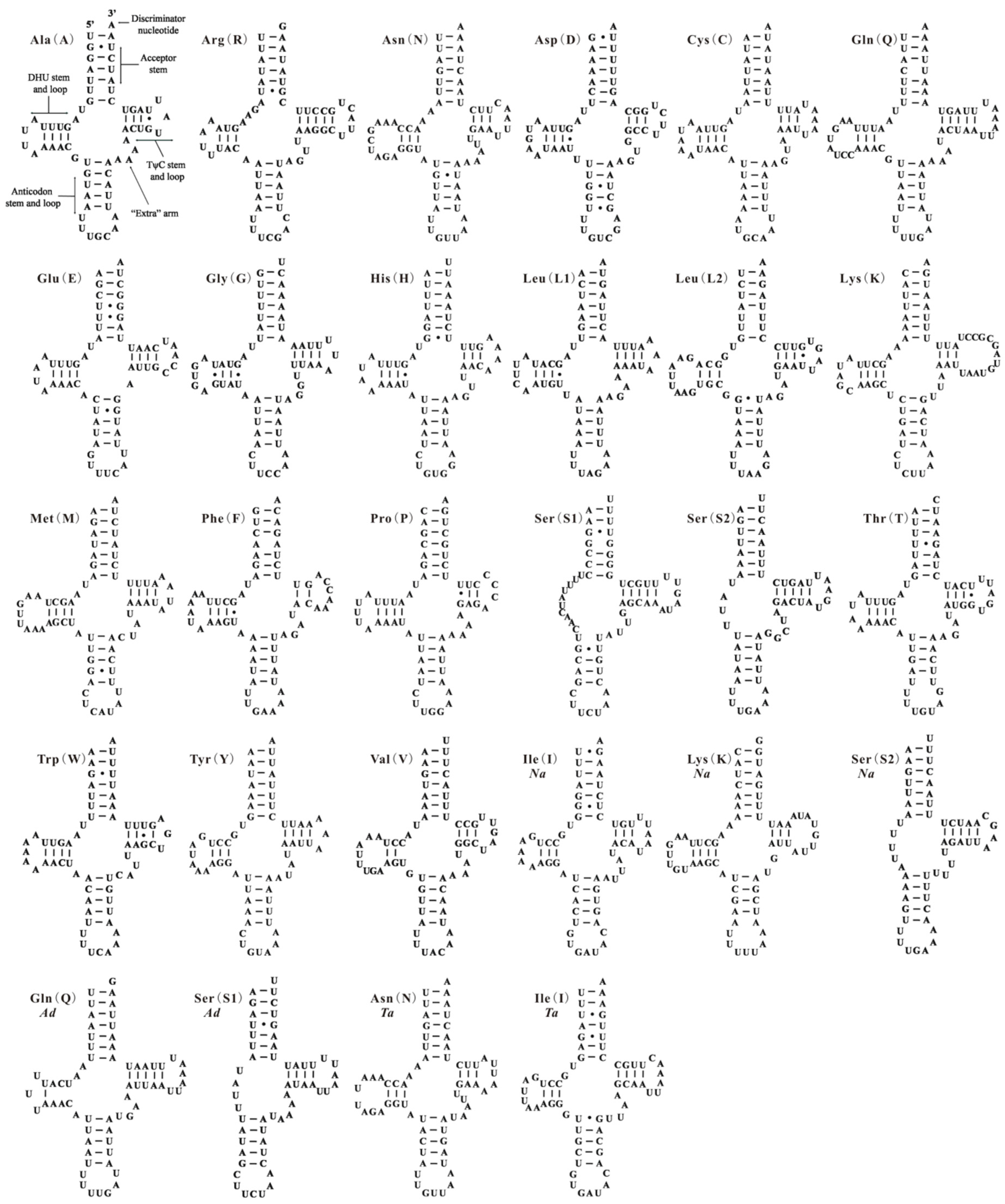
All 22 tRNA genes usually found in the mt genomes of insects are present in A. camelliae, except for trnI. Seven of the 21 tRNA genes are encoded by one strand and the remaining genes are encoded by the other strand. The nucleotide length of tRNA genes are ranging from 56 bp (trnS2) to 71 bp (trnQ), and A + T content are ranging from 61.29% (trnS1) to 92.06% (trnC) (Table 1). In the mt genome of A. camelliae, 19 of 21 tRNA genes have the conventional cloverleaf shaped secondary structure and two (trnS1 and trnS2) lacks the dihydrouridine (DHU) arm (Figure 2). The gene trnS1 almost lacks the DHU arm in all metazoans [3,4,6], and trnS2 without DHU arm also exists in B. tabaci Asia I [27] and N. andropogonis (Figure 2). In the 21 tRNA genes of A. camelliae, 39 unmatched base pairs were found, and they can be corrected through RNA-editing mechanisms that are well known for insect mtDNA [28].
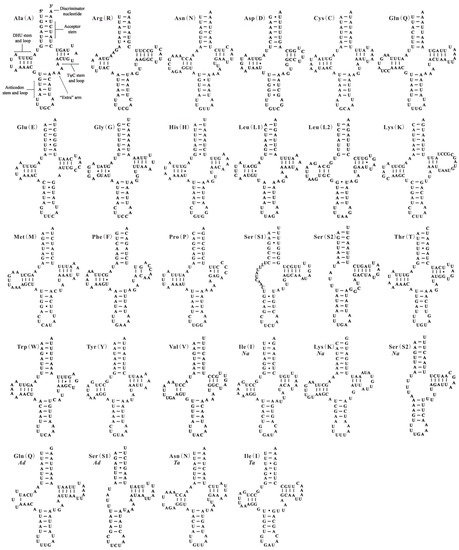
Figure 2.
Putative secondary structures of the tRNA genes identified in this study. Bars indicate Watson-Crick base pairings, and dots between G and U pairs mark canonical base pairings appearing in RNA. Seven tRNA genes identified in this study of the mitochondrial genomes of Aleurodicus dugesii (Ad), Neomaskellia andropogonis (Na) and Tetraleurodes acaciae (Ta) with species name abbreviation below gene name.
2.2. Re-Annotation for Whitefly Mitochondrial Genomes
In the camellia whitefly mt genome, we identified 36 of 37 genes usually found in animal mt genomes, and gene trnI was not found. Gene absence is common in reported Aleyrodidae mt genomes, e.g., A. aceris without trnI, Aleurodicus dugesii without trnS1 and trnQ, T. acaciae without trnI, trnS1 and trnN, and N. andropogonis without trnA, trnR, trnN, trnI and trnS2 [21]. The event of a gene disappearing has also occurred in mt genome of other animals, such as booklice [8,19] and arrow worm [29]. In some extreme cases, Cnidaria and Chaetognatha species lose nearly all their transfer RNA genes [29,30]. Current studies suggest that there are three reasons for gene absence of animal mt genomes. The first, the missing genes have been deleted and functionally replaced by nuclear tRNAs [31]. The second, the mt genome is fragmented and the missing genes are encoded by another unsequenced chromosome, like booklice [8,19], human lice [6], rotifer [32,33] and yellow tea thrips [18]. Thirdly, the disappeared genes actually exist in the circular DNA molecule, but have not been identified due to their rapid evolutionary rates. For example, earlier study indicated that the nad3 gene was lost from the mt genome of Metaseiulus occidentalis [34], and the gene had been identified in a subsequent report [35].
In this study, we have constructed tRNA secondary structure using ARWEN additionally and aligned homologous gene sequences from other whiteflies. Five tRNA genes have been identified firstly in three whiteflies: trnS1 and trnQ for A. dugesii, trnI and trnN for T. acaciae and trnI for N. andropogonis (Figure 2 and Figure 3, Table S1). For the mt genome of N. andropogonis, trnK and trnS2 have been re-annotated and cox2 gene has been reduced to 661 bp by tRNA punctuation model of trnK [36]. In original annotation, cox2 gene (717 bp) is much longer than those of other Aleyrodidae species (661–667 bp), and trnK takes up the usual position of trnS2: thus, trnS2 (14428–14493) huddles together with trnW (14430–14493). Gene trnQ of two whiteflies, A. camelliae and A. dugesii, have almost identical anticodon stem and loops, acceptor stems and TψC stem and loops. However, trnQ of A. dugesii nearly lost the DHU arm. The gene sequence has been much changed in the process of evolution, which increases the difficulty of gene identification. It infers that most gene absences of Aleyrodidae mt genome belong to the third situation of those mentioned above.
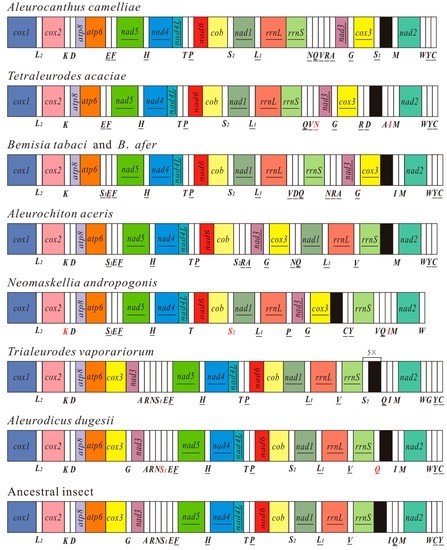
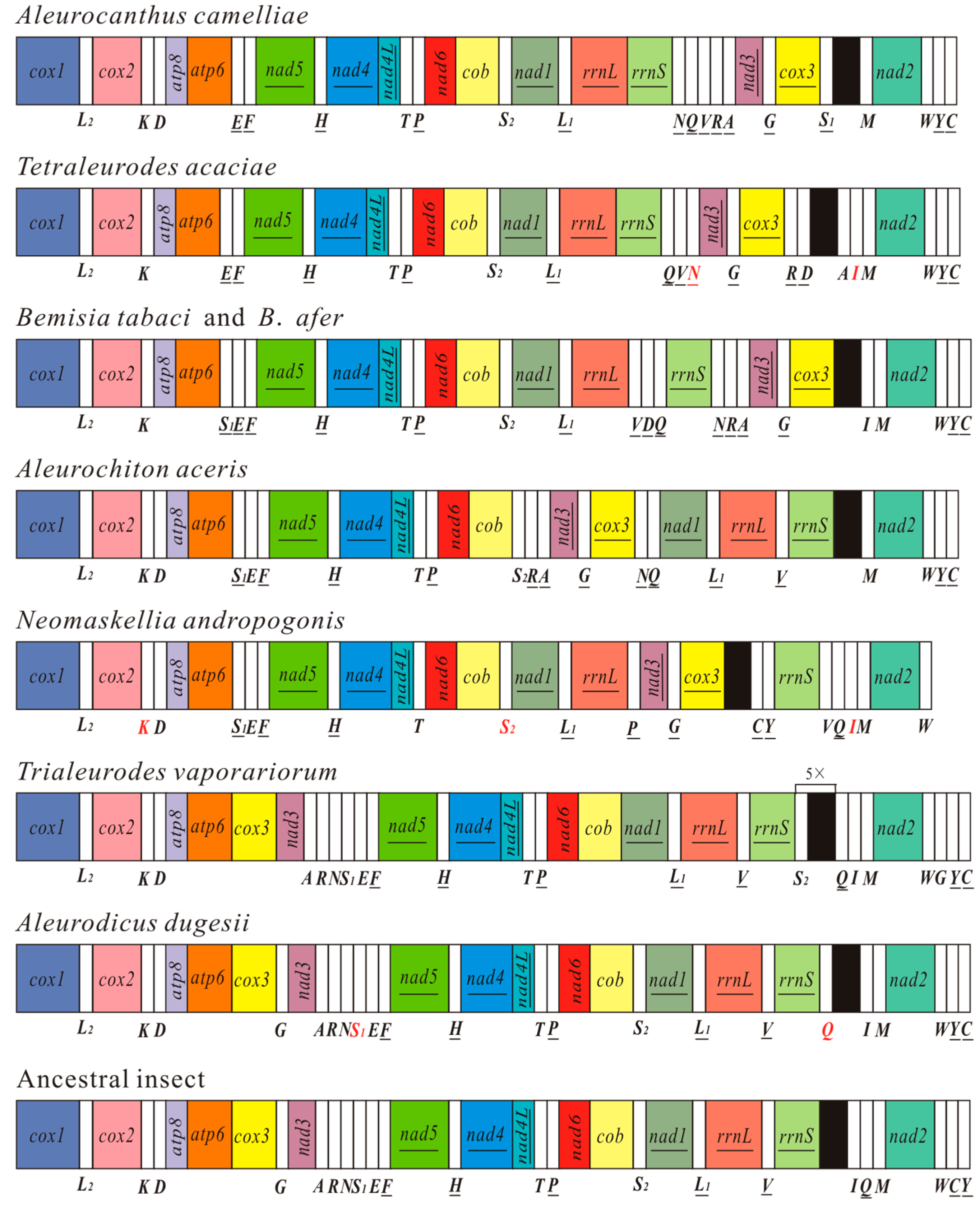
Figure 3.
Arrangement of mitochondrial genes in the family Aleyrodidae and the hypothetical ancestral insect. Circular genomes have been arbitrarily linearized for ease of comparison. Gene names are the standard abbreviations used in the present study. Transfer RNA genes are designated by the single-letter amino acid codes, and red letters represent the tRNAs identified in this study. Genes which are underlined are encoded on the different strand from that of cox1. White boxes represent transfer RNA genes and bright colors represent 13 protein-coding genes and 2 ribosomal RNA genes.
2.3. Gene Arrangement
The mt gene arrangement in A. camelliae is unique and different from that of the ancestral insect and other whiteflies (Figure 3). Mt gene rearrangement is also a common phenomenon for all Aleyrodidae species. As for most whiteflies, A. camelliae has the specific inverse transposition of gene block cox3-trnG-nad3 and the position of trnY and trnC [21]. The gene order of camellia spiny whitefly is similar to that of T. acaciae. The two gene blocks, atp8-atp6-trnE-trnF-nad5-trnH-nad4-nad4L-trnT-trnP-nad6-cob-trnS2-nad1-trnL1-rrnL-rrnS and trnQ-trnV, only exist in the mt genomes of A. camelliae and T. acaciae. For the two whiteflies, gene rrnS is immediately followed by rrnL, with no transfer RNA gene but a long non coding sequence (94 and 53 bp) between them. However, the other whiteflies have one or more transfer genes between the two rRNAs [21,22,23,24,27].
There are numerous gene rearrangements have apparently occurred in the whitefly mt genomes, and the gene order map gives evidences to understand the process of rearrangements. For the major rearrangement, cox3-trnG-nad3-trnA-trnR-trnN firstly inverse transposed as a unit [21], subsequently, trnA, trnR and trnN have undergone transpositions independently. In Figure 3, B. tabaci and B. afer contain the whole inverse unit, A. camelliae and A. aceris with inverse cox3-trnG-nad3-trnA-trnR, and T. acaciae and N. andropogonis just have inverse cox3-trnG-nad3. Additionally, tRNA cluster trnS1-trnE-trnF has been left in the mt genomes of B. tabaci, B. afer, A. aceris and N. andropogonis. With cox3-trnG-nad3-trnA-trnR-trnN rearranged, the two ends of the unit became “hot spots” of tRNA gene transposition and insertion. For the mt genome of A. camellia, trnQ-trnV could be inserted after cox3-trnG-nad3-trnA-trnR-trnN rearranged, while six genes of the rearrangement unit maintain the original relative position. For T. acaciae, A. aceris and N. andropogonis, one or more tRNAs inserted into "hot spots" and trnN or all the three tRNAs (trnA, trnR and trnN) moved away. Thus, trnA, trnR and trnN in N. andropogonis have probably been rearranged several times and the sequences have been changed a lot. Once the tRNA genes lost their anticodons and functions, they would be eliminated. Rearrangement steps of trnS1-trnE-trnF are much clearer. The first step is the inversion of trnS1 (A. aceris and N. andropogonis), the second is inversion of trnE (B. tabaci and B. afer) and the last is the transposition of trnS1 (A. camelliae and T. acaciae). These rearrangement steps could be used to gain information of phylogenies.
To understand the mechanism that causes whitefly mt genome rearrangement, we conduct a comparative analysis among all Aleyrodidae mt genomes. Gene block trnT-trnP is common in all whitefly mt genomes, and a 71 bp non-coding region replaces trnP between trnT and nad6 in N. andropogonis (Table S1). The mt genomes of A. camelliae and T. acaciae have the same situation, and a non-coding region instead of trnV locates between the two rRNA genes. These observations are highly consistent with the tandem duplication/random loss (TDRL) model, which is the widely accepted mechanism for local gene rearrangements in animal mt genomes [37]. However, most of genome rearrangements in Aleyrodidae are consistent with the intramolecular recombination mechanism, because gene inversions cannot be explained without some form of recombination [38,39,40]. The control region (CR) has been considered as a “hot spot” of recombination [41]. For whiteflies, gene block cox3-trnG-nad3 usually translocates close to the control region. Besides cox3-trnG-nad3, many rearrangements with inversions occurred around CR, such as rrnS-trnV of N. andropogonis, trnA and trnD of T. acaciae, and trnQ of A. dugesii. The results indicate that both TDRL model and intramolecular recombination participate in genome rearrangement of whiteflies, and the latter plays a more important role.
2.4. Phylogenetic Analyses
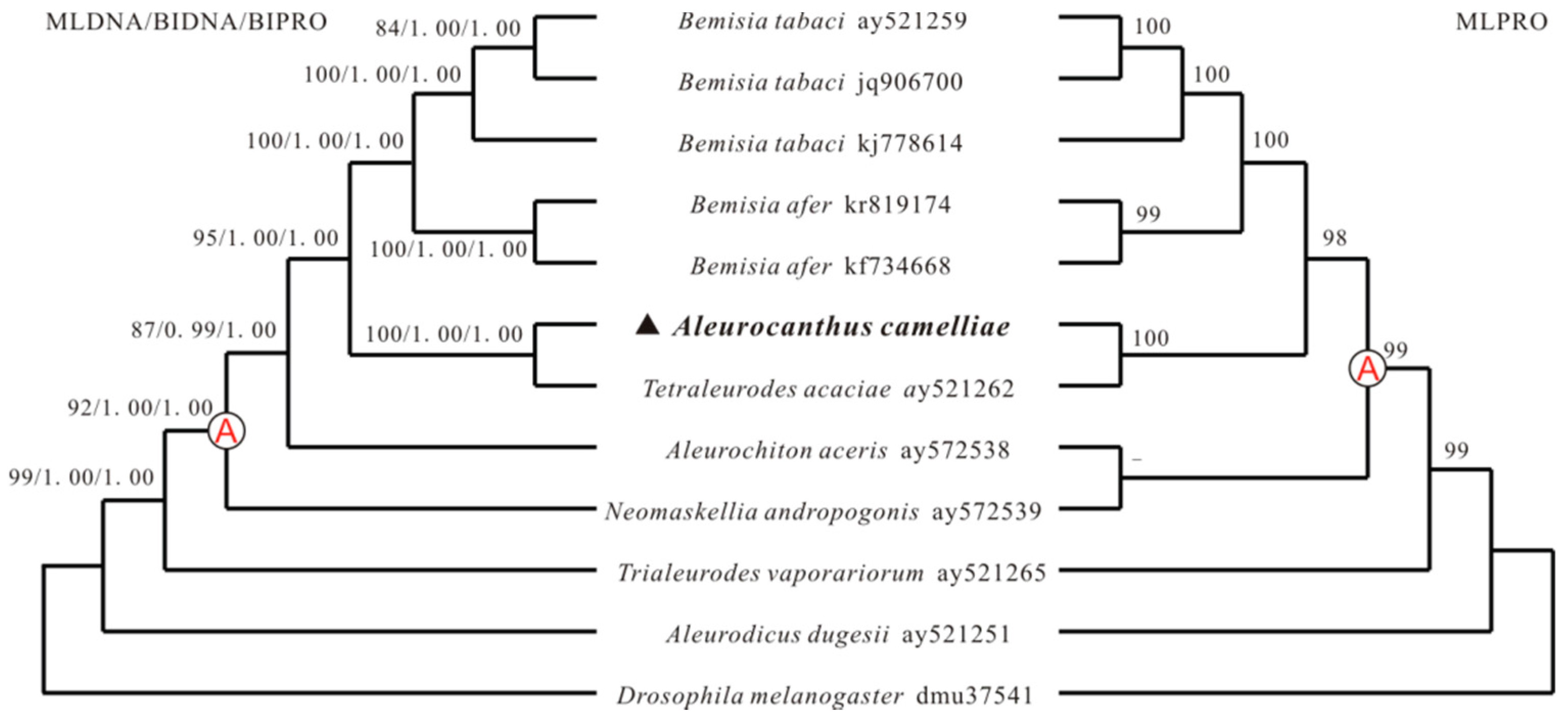
Two methods, maximum likelihood (ML) and Bayesian inference (BI), were used to determine phylogenetic relationships among 11 whiteflies from the family Aleyrodidae, with nucleotide and deducted amino acid sequences of mt genomes (Figure 4). Phylogenetic relationships among 11 whiteflies and D. melanogaster based on mt genome sequence are concordant with that inferred from mt gene order. A. camelliae and T. acaciae share most gene boundaries to each other and they are clustered into a branch of the phylogenetic tree with strong support (100% bootstrap value and 1.0 posterior probabilities). Similarly, three B. tabaci and two B. afer strains gather together and they have the same mitochondrial gene arrangement. In the cluster A, all species contain the insertion position of cox3-trnG-nad3 and differ in tRNAs, that it is possible tRNA rearrangements followed the insertion of the transposed fragment in a common ancestor [21]. Based on rearrangement steps of trnS1-trnE-trnF, A. aceris and N. andropogonis split out firstly with the trnS1 inversion, B. tabaci and B. afer followed them with trnE inversion. The gene order of the mt genome of A. dugesii and T. vaporariorum nearly have the ancestral gene composition and arrangement and contain a few tRNA rearrangements. Meanwhile, these two whiteflies locate more closely than the others to D. melanogaster in the phylogenetic tree, which insect has the same mt gene order as the ancestral insect [3,42]. It can be inferred that gene rearrangement plays an important role in the mt genome evolution in the family Aleyrodidae, and confirmed that the rearrangements are reliable phylogenetic markers [3,7,43].

Figure 4.
Phylogeny from Aleyrodidae mitochondrial genome sequences. Numbers above the branches show maximum likelihood (ML) bootstrap support values and Bayesian (BI) posterior probabilities for the phylogenies. Only support above 50% is shown. All mitochondrial genomes of clade A contain the rearrangement of cox3-trnG-nad3.
3. Materials and Methods
3.1. Ethics Statement and Sample Collection
The camellia spiny whiteflies were collected at tea plantation in Yongchuan, Chongqing, China in 16 March 2015, and identified to species by morphology [2] and sequence of cox1 [2,44] in 30 March 2015. Voucher specimens (#CQNKY-HE-02-01-01) were deposited at the Insect Collection, Tea Research Institute of Chongqing Academy of Agricultural Science, Chongqing, China. The sampling tea plantation belongs to Chongqing Academy of Agricultural Science, and the whitefly is a tea pest and not a protected species.
3.2. DNA Extraction and mt Genome Amplification
Total genomic DNA was extracted using a Dneasy Blood and Tissue Kit (Qiagen, Hilden, Germany) and stored at −20 °C. Referenced gene order of other whiteflies, the mt genome of A. camelliae were amplified in seven overlapping fragments by long-PCR with conserved insect primers [12,45,46] and specific primers designed in this study (Table 4). All fragments were sequenced and assembled into a contig with SeqMan (DNAStar).
Table 4.
PCR primers for amplification of the mitochondrial genome of Aleurocanthus camelliae.
Each long-PCR reaction is 25 μL in total volume, containing 1.0 μL each of forward primer (10 μM) and reverse primer (10 μM), 4.0 μL of dNTPs mix (each 2.5 mM), 1.0 μL of template DNA, 2.5 μL MgCl2 (25 mM), 2.5 μL of 10× LA PCR reaction buffer II, 12.75 μL ddH2O and 0.25 μL LA Taq DNA polymerase (5 U/μL, Takara, Dalian, China). All reactions were carried out using C1000™ thermal cyclers (Bio-RAD, Hercules, CA, USA) with the following conditions: 2 min denaturation at 94 °C, 35 cycles of 94 °C for 30 s, 55 °C for 30 s, 68 °C for 1–5 min (depending on target size, ~1 min/kb), followed by a final extension at 68 °C for 10 min. PCR products were directly sequenced with both forward and reverse PCR primers and internal primers by primer walking. All products in this study were sequenced by Life Technologies in Guangzhou, China.
3.3. Sequence Annotation and Analysis
The protein-coding genes were firstly identified by the ORF Finder (available online: http://www.ncbi.nlm.nih.gov/gorf/gorf.html) and rRNA genes by BLAST searches, then confirmed by alignment with homologous genes from other species of Aleyrodidae (Table 5). The transfer RNA genes were identified by cloverleaf secondary structure using ARWEN [47] with default parameters and tRNAscan-SE 1.21 [48] with the parameters: Search Mode = “EufindtRNA-Cove”, Genetic Code = “Invertebrate Mito” and Cove score cutoff = 1. The base composition was analyzed with BioEdit 7.2.5 (http://www.softpedia.com/get/Science-CAD/BioEdit.shtml). Sequences of mt genomes of other whiteflies were retrieved from GenBank (Table 5).
Table 5.
GenBank accession numbers and mitochondrial genome sizes of the species included in phylogenetic analysis in this study.
3.4. Phylogenetic Analyses
We indicated the relationship of the camellia spiny whitefly to seven other whiteflies (three strains of Bemisia tabaci and two strains of Bemisi aafer) with concatenated mt gene sequences of: (1) nucleotide sequences of 12 protein-coding genes and two rRNA genes (atp8 excluded); (2) amino acid sequence of 12 protein-coding genes (atp8 excluded). Atp8 and tRNA genes were excluded because they are too short to align among 12 mt genomes. All sequences of genes were aligned individually by ClustalW in MEGA 5 [49]. Nucleotide sequences of PCGs were aligned at the amino acid level, then back-translated into nucleotide sequences. Poorly aligned sites of each alignment have been removed using the Gblocks server (available online: http://molevol.cmima.csic.es/castresana/Gblocks_server.html) [50] with all options for a stringent selection were chosen. We using DAMBE 5.3.9 to test substitution saturations of aligned nucleotide sequences [51]. Whole PCG nucleotide sequences would enter the next step if Iss was significantly lower than Iss.c (p < 0.05). Ten protein-coding genes passed this test, and the third codon positions of nad4L and nad6 were excluded then the remainder passed the test. Subsequently, alignments of individual genes were concatenated. According to the Akaike Information Criterion, the best fit models for nucleotide sequence alignment was determined by jModelTest 2.1.4 [52,53], and amino acid sequence alignment by ProtTest 3.2 [54]. Then, GTR+I+G model and MtArt+I+G model were chosen. The two concatenated alignments were used in maximum likelihood (ML) and Bayesian inference (BI) with PhyML3.0 (available online: http://www.atgc-montpellier.fr/phyml/) [55] and MrBayes v3.2.2 [56,57]. ML analyses were performed with substitution models “GTR” or “MtArt”, type of tree improvement “SPR & NNI”, and the shape parameters and the propotions of invariable sites were estimated by jModelTest 2.1.4 and ProtTest 3.2. For BI analyses, four independent Markov chains were simultaneously run for 2,000,000 generations with a heating scheme (temp = 0.2); Trees were sampled every 100 generations (sample-freq = 100) and the first 25% were discarded as burn-in. Stationarity was considered to be reached when the average standard deviation of split frequencies was below 0.01 [58].
4. Conclusions
We sequenced the complete mt genome of camellia spiny whitefly, Aleurocanthus camelliae. This mt genome shares many features with those reported insect mt genomes. It is a typical closed-circular and double stranded DNA molecule in size of 15,188 bp. Distinctively, it has unique gene arrangement and trnI could not be identified. The mt genome encodes 13 protein-coding genes, 2 ribosomal and 22 transfer RNA genes, but no trnI. Gene absence is a common phenomenon of whitefly mt genomes. Using ARWEN and homologous sequence alignment, we have identified five tRNA genes and revised genome annotations of A. dugesii, N. andropogonis and T. acaciae in this study. The gene order of camellia spiny whitefly is similar to that of T. acaciae. Phylogenetic relationships among 11 whiteflies and D. melanogaster based on mt genome sequence are concordant with that inferred from mt gene order. This infers that gene rearrangement plays an important role in the mt genome evolved from whiteflies.
Supplementary Materials
Supplementary materials can be found at www.mdpi.com/1422-0067/17/11/1843/s1.
Acknowledgments
This work was supported by General Fundamental and Advanced Research Project of Chongqing (cstc2015jcyjA80038), National Tea Industry Technology System Research Project of China (CARS-23), Agricultural Development Fund of Chongqing Academy of Agricultural Sciences (NKY-2016AC014) and Natural Science Foundation Project of Yongchuan Science & Technology Commission (Ycstc, 2015nc1005).
Author Contributions
Shi-Chun Chen, Xiao-Qing Wang, Jin-Jun Wang and Ping Peng conceived and designed the experiments; Shi-Chun Chen, Xiao-Qing Wang, Xiang Hu and Pin-Wu Li performed the experiments and analyzed the data; Ping Peng contributed reagents/materials/analysis tools; Shi-Chun Chen wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fu, J.Y.; Han, B.Y.; Xiao, Q. Mitochondrial COI and 16sRNA evidence for a single species hypothesis of E. vitis, J. formosana and E. onukii in East Asia. PLoS ONE 2014, 9, e115259. [Google Scholar] [CrossRef] [PubMed]
- Kanmiya, K.; Ueda, S.; Kasai, A.; Yamashita, K.; Sato, Y.; Yoshiyasu, Y. Proposal of new specific status for tea-infesting populations of the nominal citrus spiny whitefly Aleurocanthus spiniferus (Homoptera: Aleyrodidae). Zootaxa 2011, 2797, 25–44. [Google Scholar]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.C.; Wei, D.D.; Shao, R.; Dou, W.; Wang, J.J. The complete mitochondrial genome of the booklouse, Liposcelis decolor: Insights into gene arrangement and genome organization within the genus Liposcelis. PLoS ONE 2014, 9, e91902. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.; Barker, S.C.; Li, H.; Song, S.; Poudel, S.; Su, Y. Fragmented mitochondrial genomes in two suborders of parasitic lice of eutherian mammals (Anoplura and Rhynchophthirina, Insecta). Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.; Zhu, X.Q.; Barker, S.C.; Herd, K. Evolution of extensively fragmented mitochondrial genomes in the lice of humans. Genome Biol. Evol. 2012, 4, 1088–1101. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.L.; Yoshizawa, K.; Mizukoshi, A.; Whiting, M.F.; Johnson, K.P. Mitochondrial genome deletions and minicircles are common in lice (Insecta: Phthiraptera). BMC Genom. 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.C.; Wei, D.D.; Shao, R.; Shi, J.X.; Dou, W.; Wang, J.J. Evolution of multipartite mitochondrial genomes in the booklice of the genus Liposcelis (Psocoptera). BMC Genom. 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.D.; Yuan, M.L.; Wang, B.J.; Zhou, A.W.; Dou, W.; Wang, J.J. Population genetics of two asexually and sexually reproducing psocids species inferred by the analysis of mitochondrial and nuclear DNA sequences. PLoS ONE 2012, 7, e33883. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.H.; Yuan, Z.J.; Zhang, C.X.; Yin, K.S.; Tang, M.J.; Guo, H.W.; Fu, J.Y.; Xiao, Q. Detecting deep divergence in seventeen populations of tea geometrid (Ectropis obliqua Prout) in China by COI mtDNA and cross-breeding. PLoS ONE 2014, 9, e99373. [Google Scholar] [CrossRef] [PubMed]
- Gissi, C.; Iannelli, F.; Pesole, G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity 2008, 101, 301–320. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.; Buckley, T.R.; Frati, F.; Stewart, J.B.; Beckenbach, A.T. Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 2006, 37, 545–579. [Google Scholar] [CrossRef]
- Bernt, M.; Braband, A.; Schierwater, B.; Stadler, P.F. Genetic aspects of mitochondrial genome evolution. Mol. Phylogenet. Evol. 2013, 69, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- Burger, G.; Gray, M.W.; Lang, B.F. Mitochondrial genomes: Anything goes. Trends Genet. 2003, 19, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Simon, S.; Hadrys, H. A comparative analysis of complete mitochondrial genomes among Hexapoda. Mol. Phylogenet. Evol. 2013, 69, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Wolstenholme, D.R. Animal mitochondrial DNA: structure and evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar] [PubMed]
- Dickey, A.M.; Kumar, V.; Morgan, J.K.; Jara-Cavieres, A.; Shatters, R.G., Jr.; McKenzie, C.L.; Osborne, L.S. A novel mitochondrial genome architecture in thrips (Insecta: Thysanoptera): Extreme size asymmetry among chromosomes and possible recent control region duplication. BMC Genom. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.D.; Shao, R.; Yuan, M.L.; Dou, W.; Barker, S.C.; Wang, J.J. The multipartite mitochondrial genome of Liposcelis bostrychophila: Insights into the evolution of mitochondrial genomes in bilateral animals. PLoS ONE 2012, 7, e33973. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.H.; Mifsud, D.; Rapisarda, C. The whiteflies (Hemiptera: Aleyrodidae) of Europe and the Mediterranean Basin. Bull. Entomol. Res. 2000, 90, 407–448. [Google Scholar] [CrossRef] [PubMed]
- Thao, M.L.; Baumann, L.; Baumann, P. Organization of the mitochondrial genomes of whiteflies, aphids, and psyllids (Hemiptera, Sternorrhyncha). BMC Evol. Biol. 2004, 4. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.L.; Xiao, N.; Yang, J.; Wang, X.W.; Colvin, J.; Liu, S.S. The complete mitochondrial genome of Bemisia afer (Hemiptera: Aleyrodidae). Mitochondr. DNA A 2016, 27, 98–99. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.L.; Yang, J.; Boykin, L.M.; Zhao, Q.Y.; Li, Q.; Wang, X.W.; Liu, S.S. The characteristics and expression profiles of the mitochondrial genome for the Mediterranean species of the Bemisia tabaci complex. BMC Genom. 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.L.; Zhang, Z.; Bing, X.L.; Liu, Y.Q.; Liu, S.S.; Wang, X.W. A complete mitochondrial DNA genome derived from a Chinese population of the Bemisia afer species complex (Hemiptera: Aleyrodidae). Mitochondr. DNA A 2015, 27, 3500–35011. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. Complete mitochondrial genome sequence of the polychaete annelid Platynereis dumerilii. Mol. Biol. Evol. 2001, 18, 1413–1416. [Google Scholar] [CrossRef] [PubMed]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Tay, W.T.; Elfekih, S.; Court, L.; Gordon, K.H.; de Barro, P.J. Complete mitochondrial DNA genome of Bemisia tabaci cryptic pest species complex Asia I (Hemiptera: Aleyrodidae). Mitochondr. DNA 2016, 27, 972–973. [Google Scholar] [CrossRef] [PubMed]
- Lavrov, D.V.; Brown, W.M.; Boore, J.L. A novel type of RNA editing occurs in the mitochondrial tRNAs of the centipede Lithobius forficatus. Proc. Natl. Acad. Sci. USA 2000, 97, 13738–13742. [Google Scholar] [CrossRef] [PubMed]
- Helfenbein, K.G.; Fourcade, H.M.; Vanjani, R.G.; Boore, J.L. The mitochondrial genome of Paraspadella gotoi is highly reduced and reveals that chaetognaths are a sister group to protostomes. Proc. Natl. Acad. Sci. USA 2004, 101, 10639–10643. [Google Scholar] [CrossRef] [PubMed]
- Kayal, E.; Lavrov, D.V. The mitochondrial genome of Hydra oligactis (Cnidaria, Hydrozoa) sheds new light on animal mtDNA evolution and cnidarian phylogeny. Gene 2008, 410, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Duchene, A.M.; Pujol, C.; Marechal-Drouard, L. Import of tRNAs and aminoacyl-tRNA synthetases into mitochondria. Curr. Genet. 2009, 55, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.S.; Suga, K.; Sakakura, Y.; Park, H.G.; Hagiwara, A.; Rhee, J.S.; Lee, J.S. Complete mitochondrial genome of the monogonont rotifer, Brachionus koreanus (Rotifera, Brachionidae). Mitochondr. DNA 2014, 25, 29–30. [Google Scholar] [CrossRef] [PubMed]
- Suga, K.; Welch, D.B.M.; Tanaka, Y.; Sakakura, Y.; Hagiwarak, A. Two circular chromosomes of unequal copy number make up the mitochondrial genome of the rotifer Brachionus plicatilis. Mol. Biol. Evol. 2008, 25, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Jeyaprakash, A.; Hoy, M.A. The mitochondrial genome of the predatory mite Metaseiulus occidentalis (Arthropoda:Chelicerata:Acari:Phytoseiidae) is unexpectedly large and contains several novel features. Gene 2007, 391, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Dermauw, W.; Vanholme, B.; Tirry, L.; van Leeuwen, T. Mitochondrial genome analysis of the predatory mite Phytoseiulus persimilis and a revisit of the Metaseiulus occidentalis mitochondrial genome. Genome 2010, 53, 285–301. [Google Scholar] [PubMed]
- Stewart, J.B.; Beckenbach, A.T. Characterization of mature mitochondrial transcripts in Drosophila, and the implications for the tRNA punctuation model in arthropods. Gene 2009, 445, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. The duplication/random loss model for gene rearrangement exemplified by mitochondrial genomes of deuterostome animals. Comp. Genom. 2000, 1, 133–147. [Google Scholar]
- Mao, M.; Gibson, T.; Dowton, M. Evolutionary dynamics of the mitochondrial genome in the evaniomorpha (hymenoptera)—A group with an intermediate rate of gene rearrangement. Genome Biol. Evol. 2014, 6, 1862–1874. [Google Scholar] [CrossRef] [PubMed]
- Mao, M.; Dowton, M. Complete mitochondrial genomes of Ceratobaeus sp. and Idris sp. (Hymenoptera: Scelionidae): Shared gene rearrangements as potential phylogenetic markers at the tribal level. Mol. Biol. Rep. 2014, 41, 6419–6427. [Google Scholar] [CrossRef] [PubMed]
- Dowton, M.; Campbell, N.J. Intramitochondrial recombination—Is it why some mitochondrial genes sleep around? Trends Ecol. Evol. 2001, 16, 269–271. [Google Scholar] [CrossRef]
- Kurabayashi, A.; Sumida, M.; Yonekawa, H.; Glaw, F.; Vences, M.; Hasegawa, M. Phylogeny, recombination, and mechanisms of stepwise mitochondrial genome reorganization in mantellid frogs from Madagascar. Mol. Biol. Evol. 2008, 25, 874–891. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.L.; Farr, C.L.; Farquhar, A.L.; Kaguni, L.S. Sequence, organization, and evolution of the A + T region of Drosophila melanogaster mitochondrial DNA. Mol. Biol. Evol. 1994, 11, 523–538. [Google Scholar] [PubMed]
- Dowton, M.; Castro, L.R.; Austin, A.D. Mitochondrial gene rearrangements as phylogenetic characters in the invertebrates: The examination of genome “morphology“. Invertebr. Syst. 2002, 16, 345–356. [Google Scholar] [CrossRef]
- Uesugi, K.; Sato, Y.; Han, B.Y.; Huang, Z.-D.; Yara, K.; Furuhashi, K. Molecular evidence for multiple phylogenetic groups within two species of invasive spiny whiteflies and their parasitoid wasp. Bull. Entomol. Res. 2016, 106, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, D.R.; Torres-Jerez, I.; Bedford, I.D.; Markham, P.G.; Brown, J.K. A phylogeographical analysis of the Bemisia tabaci species complex based on mitochondrial DNA markers. Mol. Ecol. 1999, 8, 1683–1691. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.; Frati, F.; Beckenbach, A.; Crespi, B.; Liu, H.; Flook, P. Evolution, weighting, and phylogenetic utility of mitochondrial gene-sequences and a compilation of conserved polymerase chain-reaction primers. Ann. Entomol. Soc. Am. 1994, 87, 651–701. [Google Scholar] [CrossRef]
- Laslett, D.; Canback, B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Lemey, P. Assessing substitution saturation with DAMBE. In Phylogenetic Handbook: A Practical Approach to Phylogenetic Analysis and Hypothesis Testing, 2nd ed.; Cambridge University Press: Cambridge, UK, 2009; pp. 615–630. [Google Scholar]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9. [Google Scholar] [CrossRef] [PubMed]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F.; Nielsen, R.; Bollback, J.P. Bayesian inference of phylogeny and its impact on evolutionary biology. Science 2001, 294, 2310–2314. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).