
Immunomodulatory Function of the Tumor Suppressor p53 in Host Immune Response and the Tumor Microenvironment

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Trp53 Dysfunction and Inflammation—The Non-Canonical and Non-Cell Autonomous Mechanism of Tumorigenesis
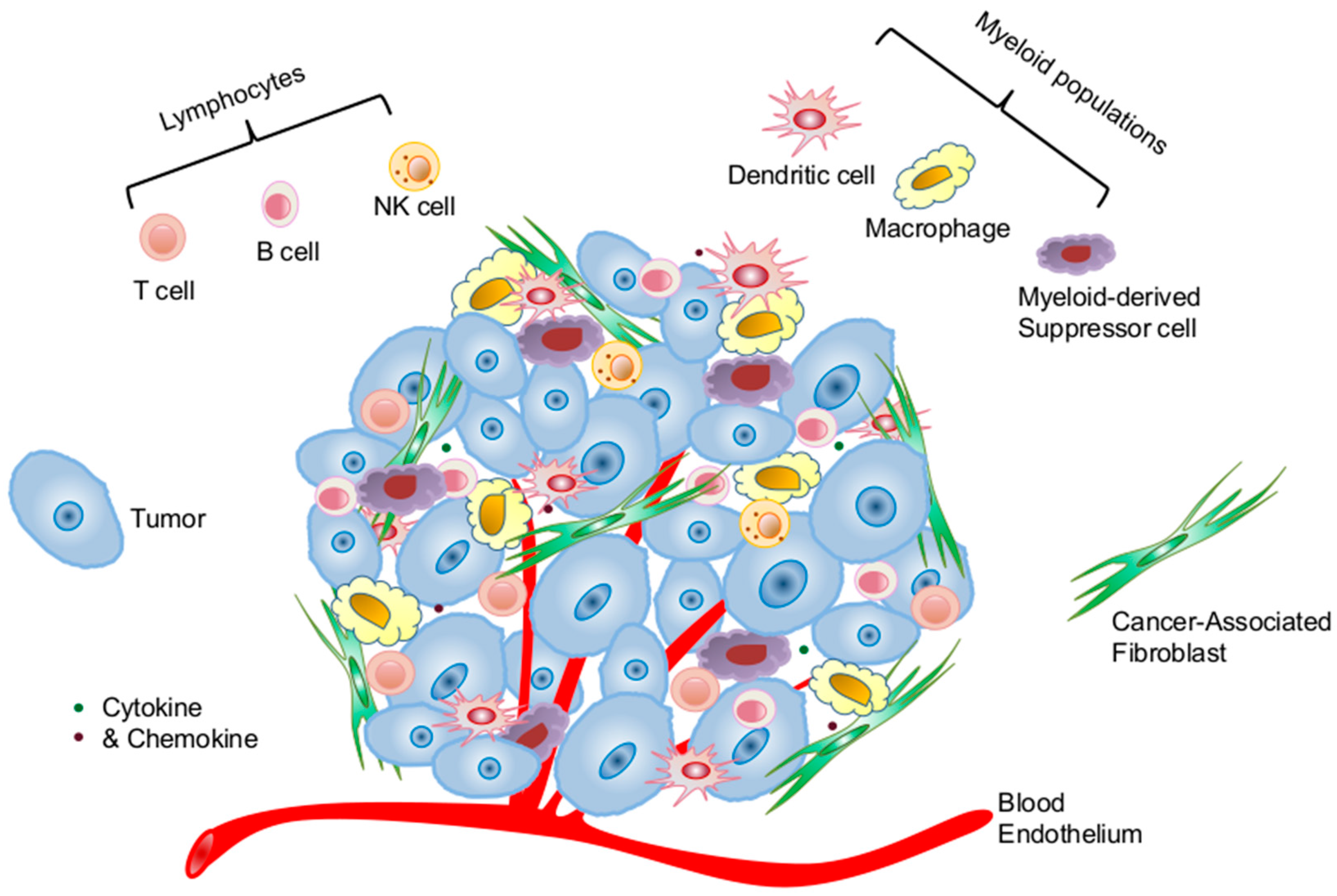
2.1. Cellular Components of the Tumor Microenvironment Involved in p53 Dysfunction-Induced Chronic Inflammation
2.1.1. Cancer Cells of Epithelial Origin
2.1.2. Cancer-Associated Fibroblasts
2.1.3. Immune Cells
2.2. Molecular Pathways that Link p53 Dysfunction and Inflammation
2.2.1. NF-κB Hyperactivity
2.2.2. Signal Transducer and Activators of Transcription Pathways
2.2.3. Other Immune Regulatory Molecules and Pathways
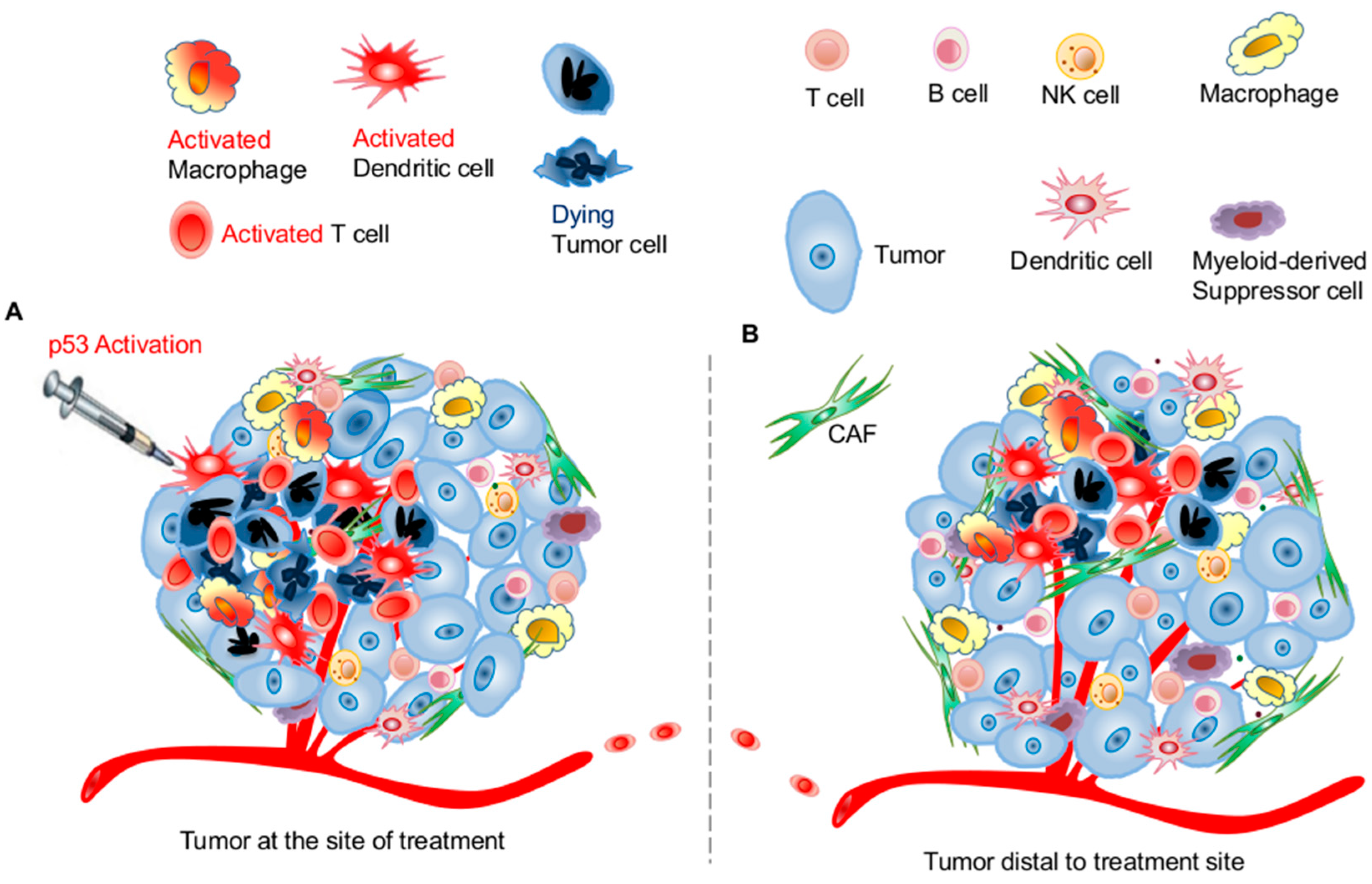
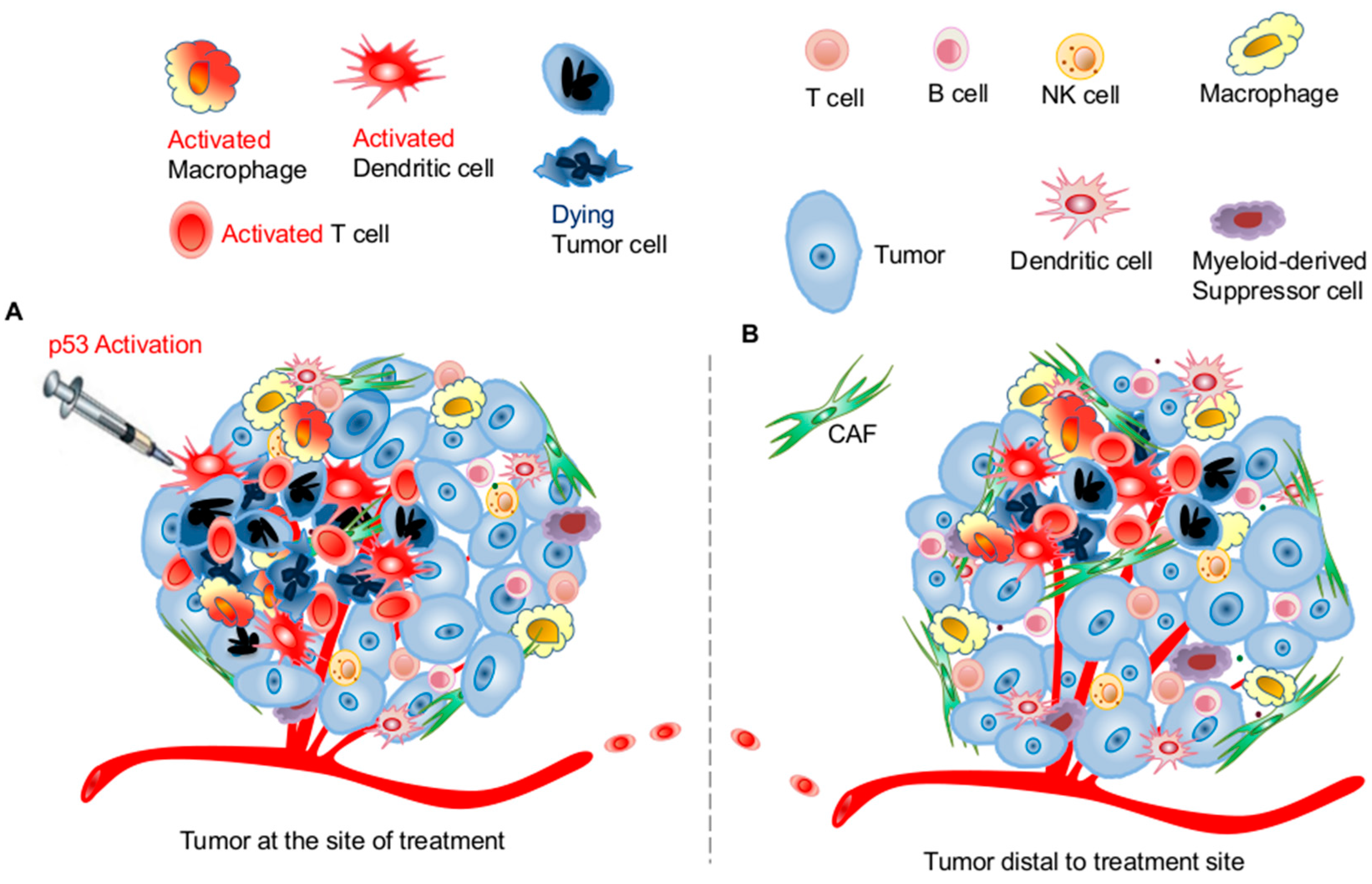
3. Targeting the p53 Pathway in the Tumor Microenvironment for Improving Systemic Antitumor Immunity
3.1. Effects of p53 Activation on Innate Immunity
3.2. Effects of p53 Activation on Adaptive Immunity
3.3. Potential Application of Targeting the p53 Pathway for Harnessing Host Antitumor Immunity
3.3.1. Small Molecule-Based Therapy Targeting the p53–MDM2 Axis
3.3.2. Restoration of Mutant p53 to Wild-Type Configuration and Function
3.3.3. Combining p53-Activation Therapy with Other Active Immunotherapy to Improve Therapeutic Efficacy
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. 2007, 8, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.; Levine, A. p53 Research: The past thirty years and the next thirty years. Cold Spring Harb. Perspect. Biol. 2010, 2, a000893. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the light: The growing complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Menendez, D.; Inga, A.; Resnick, M.A. The expanding universe of p53 targets. Nat. Rev. Cancer 2009, 9, 724–737. [Google Scholar] [CrossRef] [PubMed]
- Aylon, Y.; Oren, M. New plays in the p53 theater. Curr. Opin. Genet. Dev. 2011, 21, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Lu, C.; Hu, W.; Sun, Y.; Levine, A.J. Multiple roles of p53-related pathways in somatic cell reprogramming and stem cell differentiation. Cancer Res. 2012, 72, 5635–5645. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 2009, 139, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Rivlin, N.; Koifman, G.; Rotter, V. p53 orchestrates between normal differentiation and cancer. Semin. Cancer Biol. 2015, 32, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.P.; Hollstein, M.H.; Harris, C.C. p53 tumor suppressor gene: At the crossroads of molecular carcinogenesis, molecular epidemiology, and human risk assessment. Ann. N. Y. Acad. Sci. 2000, 919, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.P.; Amstad, P.; Raja, K.; Ambs, S.; Nagashima, M.; Bennett, W.P.; Shields, P.G.; Ham, A.J.; Swenberg, J.A.; Marrogi, A.J.; et al. Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: A cancer-prone chronic inflammatory disease. Cancer Res. 2000, 60, 3333–3337. [Google Scholar] [PubMed]
- Schetter, A.J.; Heegaard, N.H.; Harris, C.C. Inflammation and cancer: Interweaving microRNA, free radical, cytokine and p53 pathways. Carcinogenesis 2010, 31, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Gudkov, A.V.; Gurova, K.V.; Komarova, E.A. Inflammation and p53: A tale of two stresses. Genes Cancer 2011, 2, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- DeLeo, A.B.; Whiteside, T.L. Development of multi-epitope vaccines targeting wild-type sequence p53 peptides. Expert Rev. Vaccines 2008, 7, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Vermeij, R.; Leffers, N.; van der Burg, S.H.; Melief, C.J.; Daemen, T.; Nijman, H.W. Immunological and clinical effects of vaccines targeting p53-overexpressing malignancies. J. Biomed. Biotechnol. 2011, 2011, 702146. [Google Scholar] [CrossRef] [PubMed]
- Chiappori, A.A.; Soliman, H.; Janssen, W.E.; Antonia, S.J.; Gabrilovich, D.I. INGN-225: A dendritic cell-based p53 vaccine (Ad.p53-DC) in small cell lung cancer: Observed association between immune response and enhanced chemotherapy effect. Expert Opin. Biol. Ther. 2010, 10, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Soussi, T. p53 alterations in human cancer: More questions than answers. Oncogene 2007, 26, 2145–2156. [Google Scholar] [CrossRef] [PubMed]
- Soussi, T.; Hamroun, D.; Hjortsberg, L.; Rubio-Nevado, J.M.; Fournier, J.L.; Beroud, C. MUT-TP53 2.0: A novel versatile matrix for statistical analysis of TP53 mutations in human cancer. Hum. Mutat. 2010, 31, 1020–1025. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Marrero, L.; Rodriguez, P.; Del Valle, L.; Ochoa, A.; Cui, Y. Trp53 inactivation in the tumor microenvironment promotes tumor progression by expanding the immunosuppressive lymphoid-like stromal network. Cancer Res. 2013, 73, 1668–1675. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zheng, M.; Kibe, R.; Huang, Y.; Marrero, L.; Warren, S.; Zieske, A.W.; Iwakuma, T.; Kolls, J.K.; Cui, Y. Trp53 negatively regulates autoimmunity via the STAT3-Th17 axis. FASEB J. 2011, 25, 2387–2398. [Google Scholar] [CrossRef] [PubMed]
- Menendez, D.; Shatz, M.; Resnick, M.A. Interactions between the tumor suppressor p53 and immune responses. Curr. Opin. Oncol. 2013, 25, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Komarova, E.A.; Krivokrysenko, V.; Wang, K.; Neznanov, N.; Chernov, M.V.; Komarov, P.G.; Brennan, M.L.; Golovkina, T.V.; Rokhlin, O.W.; Kuprash, D.V.; et al. p53 is a suppressor of inflammatory response in mice. FASEB J. 2005, 19, 1030–1032. [Google Scholar] [CrossRef] [PubMed]
- Martins, C.P.; Brown-Swigart, L.; Evan, G.I. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell 2006, 127, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Kiaris, H.; Chatzistamou, I.; Trimis, G.; Frangou-Plemmenou, M.; Pafiti-Kondi, A.; Kalofoutis, A. Evidence for nonautonomous effect of p53 tumor suppressor in carcinogenesis. Cancer Res. 2005, 65, 1627–1630. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Akkari, L.; Simon, J.; Grace, D.; Tschaharganeh, D.F.; Bolden, J.E.; Zhao, Z.; Thapar, V.; Joyce, J.A.; Krizhanovsky, V.; et al. Non-cell-autonomous tumor suppression by p53. Cell 2013, 153, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef] [PubMed]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Meng, Y.; Blank, C.; Brown, I.; Kacha, A.; Kline, J.; Harlin, H. Immune resistance orchestrated by the tumor microenvironment. Immunol. Rev. 2006, 213, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Kerkar, S.P.; Restifo, N.P. Cellular constituents of immune escape within the tumor microenvironment. Cancer Res. 2012, 72, 3125–3130. [Google Scholar] [CrossRef] [PubMed]
- Swartz, M.A.; Iida, N.; Roberts, E.W.; Sangaletti, S.; Wong, M.H.; Yull, F.E.; Coussens, L.M.; DeClerck, Y.A. Tumor microenvironment complexity: Emerging roles in cancer therapy. Cancer Res. 2012, 72, 2473–2480. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G. Cancer and inflammation: An old intuition with rapidly evolving new concepts. Annu. Rev. Immunol. 2012, 30, 677–706. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Lawrence, T.; Nizet, V. Innate immunity gone awry: Linking microbial infections to chronic inflammation and cancer. Cell 2006, 124, 823–835. [Google Scholar] [CrossRef]
- Cooks, T.; Harris, C.C.; Oren, M. Caught in the cross fire: p53 in inflammation. Carcinogenesis 2014, 35, 1680–1690. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.P.; Zvaifler, N.J.; Green, D.R.; Firestein, G.S. Rheumatoid arthritis and p53: How oxidative stress might alter the course of inflammatory diseases. Immunol. Today 2000, 21, 78–82. [Google Scholar] [CrossRef]
- Yamanishi, Y.; Boyle, D.L.; Rosengren, S.; Green, D.R.; Zvaifler, N.J.; Firestein, G.S. Regional analysis of p53 mutations in rheumatoid arthritis synovium. Proc. Natl. Acad. Sci. USA 2002, 99, 10025–10030. [Google Scholar] [CrossRef] [PubMed]
- Tapinos, N.I.; Polihronis, M.; Moutsopoulos, H.M. Lymphoma development in Sjogren’s syndrome: Novel p53 mutations. Arthritis Rheum. 1999, 42, 1466–1472. [Google Scholar] [CrossRef]
- Staib, F.; Robles, A.I.; Varticovski, L.; Wang, X.W.; Zeeberg, B.R.; Sirotin, M.; Zhurkin, V.B.; Hofseth, L.J.; Hussain, S.P.; Weinstein, J.N.; et al. The p53 tumor suppressor network is a key responder to microenvironmental components of chronic inflammatory stress. Cancer Res. 2005, 65, 10255–10264. [Google Scholar] [CrossRef] [PubMed]
- Yamanishi, Y.; Boyle, D.L.; Pinkoski, M.J.; Mahboubi, A.; Lin, T.; Han, Z.; Zvaifler, N.J.; Green, D.R.; Firestein, G.S. Regulation of joint destruction and inflammation by p53 in collagen-induced arthritis. Am. J. Pathol. 2002, 160, 123–130. [Google Scholar] [CrossRef]
- Zheng, S.J.; Lamhamedi-Cherradi, S.E.; Wang, P.; Xu, L.; Chen, Y.H. Tumor suppressor p53 inhibits autoimmune inflammation and macrophage function. Diabetes 2005, 54, 1423–1428. [Google Scholar] [CrossRef] [PubMed]
- Okuda, Y.; Okuda, M.; Bernard, C.C. Regulatory role of p53 in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2003, 135, 29–37. [Google Scholar] [CrossRef]
- Yao, Q.; Wang, S.; Glorioso, J.C.; Evans, C.H.; Robbins, P.D.; Ghivizzani, S.C.; Oligino, T.J. Gene transfer of p53 to arthritic joints stimulates synovial apoptosis and inhibits inflammation. Mol. Ther 2001, 3, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Di Minin, G.; Bellazzo, A.; Dal Ferro, M.; Chiaruttini, G.; Nuzzo, S.; Bicciato, S.; Piazza, S.; Rami, D.; Bulla, R.; Sommaggio, R.; et al. Mutant p53 reprograms TNF signaling in cancer cells through interaction with the tumor suppressor DAB2IP. Mol. Cell 2014, 56, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Dibra, D.; Xia, X.; Mitra, A.; Cutrera, J.J.; Lozano, G.; Li, S. Mutant p53 in concert with an interleukin-27 receptor alpha deficiency causes spontaneous liver inflammation, fibrosis, and steatosis in mice. Hepatology 2016, 63, 1000–1012. [Google Scholar] [CrossRef] [PubMed]
- Schwitalla, S.; Ziegler, P.K.; Horst, D.; Becker, V.; Kerle, I.; Begus-Nahrmann, Y.; Lechel, A.; Rudolph, K.L.; Langer, R.; Slotta-Huspenina, J.; et al. Loss of p53 in enterocytes generates an inflammatory microenvironment enabling invasion and lymph node metastasis of carcinogen-induced colorectal tumors. Cancer Cell 2013, 23, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Weinberg, R.A. Stromal fibroblasts in cancer: A novel tumor-promoting cell type. Cell Cycle 2006, 5, 1597–1601. [Google Scholar] [CrossRef] [PubMed]
- Shields, J.D.; Kourtis, I.C.; Tomei, A.A.; Roberts, J.M.; Swartz, M.A. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science 2010, 328, 749–752. [Google Scholar] [CrossRef] [PubMed]
- Katakai, T.; Hara, T.; Sugai, M.; Gonda, H.; Shimizu, A. Lymph node fibroblastic reticular cells construct the stromal reticulum via contact with lymphocytes. J. Exp. Med. 2004, 200, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, A.L.; Malhotra, D.; Turley, S.J. Lymph node stroma broaden the peripheral tolerance paradigm. Trends Immunol. 2011, 32, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Epardaud, M.; Sun, J.; Becker, J.E.; Cheng, A.C.; Yonekura, A.R.; Heath, J.K.; Turley, S.J. Peripheral antigen display by lymph node stroma promotes T cell tolerance to intestinal self. Nat. Immunol. 2007, 8, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Turley, S.J.; Fletcher, A.L.; Elpek, K.G. The stromal and haematopoietic antigen-presenting cells that reside in secondary lymphoid organs. Nat. Rev. Immunol. 2010, 10, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Peduto, L.; Dulauroy, S.; Lochner, M.; Spath, G.F.; Morales, M.A.; Cumano, A.; Eberl, G. Inflammation recapitulates the ontogeny of lymphoid stromal cells. J. Immunol. 2009, 182, 5789–5799. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.; Song, Y.; Cardiff, R.D.; van Dyke, T. Selective evolution of stromal mesenchyme with p53 loss in response to epithelial tumorigenesis. Cell 2005, 123, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Patocs, A.; Zhang, L.; Xu, Y.; Weber, F.; Caldes, T.; Mutter, G.L.; Platzer, P.; Eng, C. Breast-cancer stromal cells with TP53 mutations and nodal metastases. N. Engl. J. Med. 2007, 357, 2543–2551. [Google Scholar] [CrossRef] [PubMed]
- Yamanishi, Y.; Boyle, D.L.; Green, D.R.; Keystone, E.C.; Connor, A.; Zollman, S.; Firestein, G.S. p53 tumor suppressor gene mutations in fibroblast-like synoviocytes from erosion synovium and non-erosion synovium in rheumatoid arthritis. Arthritis Res. Ther. 2005, 7, R12–R18. [Google Scholar] [CrossRef] [PubMed]
- Fukino, K.; Shen, L.; Patocs, A.; Mutter, G.L.; Eng, C. Genomic instability within tumor stroma and clinicopathological characteristics of sporadic primary invasive breast carcinoma. JAMA 2007, 297, 2103–2111. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Weinberg, R.A. Heterogeneity of stromal fibroblasts in tumors. Cancer Biol. Ther. 2007, 6, 618–619. [Google Scholar] [CrossRef] [PubMed]
- Addadi, Y.; Moskovits, N.; Granot, D.; Lozano, G.; Carmi, Y.; Apte, R.N.; Neeman, M.; Oren, M. p53 status in stromal fibroblasts modulates tumor growth in an SDF1-dependent manner. Cancer Res. 2010, 70, 9650–9658. [Google Scholar] [CrossRef] [PubMed]
- Moskovits, N.; Kalinkovich, A.; Bar, J.; Lapidot, T.; Oren, M. p53 Attenuates cancer cell migration and invasion through repression of SDF-1/CXCL12 expression in stromal fibroblasts. Cancer Res. 2006, 66, 10671–10676. [Google Scholar] [CrossRef] [PubMed]
- Mlecnik, B.; Bindea, G.; Kirilovsky, A.; Angell, H.K.; Obenauf, A.C.; Tosolini, M.; Church, S.E.; Maby, P.; Vasaturo, A.; Angelova, M.; et al. The tumor microenvironment and Immunoscore are critical determinants of dissemination to distant metastasis. Sci. Transl. Med. 2016, 8, 327ra26. [Google Scholar] [CrossRef] [PubMed]
- Angell, H.; Galon, J. From the immune contexture to the Immunoscore: The role of prognostic and predictive immune markers in cancer. Curr. Opin. Immunol. 2013, 25, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Mlecnik, B.; Bindea, G.; Angell, H.K.; Maby, P.; Angelova, M.; Tougeron, D.; Church, S.E.; Lafontaine, L.; Fischer, M.; Fredriksen, T.; et al. Integrative analyses of colorectal cancer show Immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity 2016, 44, 698–711. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Xu, J.; Song, J.; Liu, C.; Wang, J.; Weng, C.; Sun, H.; Wei, H.; Xiao, W.; Sun, R.; et al. The predictive value of centre tumour CD8+ T cells in patients with hepatocellular carcinoma: Comparison with Immunoscore. Oncotarget 2015, 6, 35602–35615. [Google Scholar] [PubMed]
- Park, J.S.; Lim, M.A.; Cho, M.L.; Ryu, J.G.; Moon, Y.M.; Jhun, J.Y.; Byun, J.K.; Kim, E.K.; Hwang, S.Y.; Ju, J.H.; et al. p53 controls autoimmune arthritis via STAT-mediated regulation of the Th17 cell/Treg cell balance in mice. Arthritis Rheuma. 2013, 65, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, H.; Takatori, H.; Suzuki, K.; Iwata, A.; Yokota, M.; Suto, A.; Minamino, T.; Hirose, K.; Nakajima, H. Tumor suppressor p53 inhibits systemic autoimmune diseases by inducing regulatory T cells. J. Immunol. 2013, 191, 3614–3623. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Li, H.; Yusuf, N.; Elmets, C.A.; Li, J.; Mountz, J.D.; Xu, H. IL-17 promotes tumor development through the induction of tumor promoting microenvironments at tumor sites and myeloid-derived suppressor cells. J. Immunol. 2010, 184, 2281–2288. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yi, T.; Kortylewski, M.; Pardoll, D.M.; Zeng, D.; Yu, H. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J. Exp. Med. 2009, 206, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yi, T.; Zhang, W.; Pardoll, D.M.; Yu, H. IL-17 enhances tumor development in carcinogen-induced skin cancer. Cancer Res. 2010, 70, 10112–10120. [Google Scholar] [CrossRef] [PubMed]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. 2008, 9, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. The common mechanisms of transformation by the small DNA tumor viruses: The inactivation of tumor suppressor gene products: p53. Virology 2009, 384, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Havre, P.A.; Yuan, J.; Hedrick, L.; Cho, K.R.; Glazer, P.M. p53 inactivation by HPV16 E6 results in increased mutagenesis in human cells. Cancer Res. 1995, 55, 4420–4424. [Google Scholar] [PubMed]
- Jiang, D.; Srinivasan, A.; Lozano, G.; Robbins, P.D. SV40 T antigen abrogates p53-mediated transcriptional activity. Oncogene 1993, 8, 2805–2812. [Google Scholar] [PubMed]
- Quaresma, J.A.; Yoshikawa, G.T.; Koyama, R.V.; Dias, G.A.; Fujihara, S.; Fuzii, H.T. HTLV-1, immune response and autoimmunity. Viruses 2015, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Ak, P.; Levine, A.J. p53 and NF-κB: Different strategies for responding to stress lead to a functional antagonism. FASEB J. 2010, 24, 3643–3652. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. Activated p53 induces NF-κB DNA binding but suppresses its transcriptional activation. Biochem. Biophys. Res Commun. 2008, 372, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. p53 regulates glucose metabolism through an IKK-NF-κB pathway and inhibits cell transformation. Nat. Cell Biol. 2008, 10, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Son, D.S.; Kabir, S.M.; Dong, Y.L.; Lee, E.; Adunyah, S.E. Inhibitory effect of tumor suppressor p53 on proinflammatory chemokine expression in ovarian cancer cells by reducing proteasomal degradation of IκB. PLoS ONE 2012, 7, e51116. [Google Scholar] [CrossRef] [PubMed]
- Lowe, J.M.; Menendez, D.; Bushel, P.R.; Shatz, M.; Kirk, E.L.; Troester, M.A.; Garantziotis, S.; Fessler, M.B.; Resnick, M.A. p53 and NF-κB coregulate proinflammatory gene responses in human macrophages. Cancer Res. 2014, 74, 2182–2192. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Davalos, A.R.; Kawahara, M.; Malhotra, G.K.; Schaum, N.; Huang, J.; Ved, U.; Beausejour, C.M.; Coppe, J.P.; Rodier, F.; Campisi, J. p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J. Cell Biol. 2013, 201, 613–629. [Google Scholar] [CrossRef] [PubMed]
- Bollrath, J.; Greten, F.R. IKK/NF-κB and STAT3 pathways: Central signalling hubs in inflammation-mediated tumour promotion and metastasis. EMBO Rep. 2009, 10, 1314–1319. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Grivennikov, S.I.; Karin, M. The unholy trinity: Inflammation, cytokines, and STAT3 shape the cancer microenvironment. Cancer Cell 2011, 19, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Niu, G.; Wright, K.L.; Ma, Y.; Wright, G.M.; Huang, M.; Irby, R.; Briggs, J.; Karras, J.; Cress, W.D.; Pardoll, D.; et al. Role of Stat3 in regulating p53 expression and function. Mol. Cell. Biol. 2005, 25, 7432–7440. [Google Scholar] [CrossRef] [PubMed]
- Hudson, J.D.; Shoaibi, M.A.; Maestro, R.; Carnero, A.; Hannon, G.J.; Beach, D.H. A proinflammatory cytokine inhibits p53 tumor suppressor activity. J. Exp. Med. 1999, 190, 1375–1382. [Google Scholar] [CrossRef] [PubMed]
- Bucala, R.; Donnelly, S.C. Macrophage migration inhibitory factor: A probable link between inflammation and cancer. Immunity 2007, 26, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.A.; Liao, H.; Chesney, J.; Fingerle-Rowson, G.; Baugh, J.; David, J.; Bucala, R. Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: Regulatory role in the innate immune response. Proc. Natl. Acad. Sci. USA 2002, 99, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.M.; Wilkinson, J.E.; Miller, R.A. Macrophage migration inhibitory factor-knockout mice are long lived and respond to caloric restriction. FASEB J. 2010, 24, 2436–2442. [Google Scholar] [CrossRef] [PubMed]
- Cortez, M.A.; Ivan, C.; Valdecanas, D.; Wang, X.; Peltier, H.J.; Ye, Y.; Araujo, L.; Carbone, D.P.; Shilo, K.; Giri, D.K.; et al. PDL1 Regulation by p53 via miR-34. J. Natl. Cancer Inst. 2016, 108, djv303. [Google Scholar] [CrossRef] [PubMed]
- Aloisi, F.; Pujol-Borrell, R. Lymphoid neogenesis in chronic inflammatory diseases. Nat. Rev. Immunol. 2006, 6, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D. Cancer and the immune system: Basic concepts and targets for intervention. Semin. Oncol. 2015, 42, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.M.; Marabelle, A.; Eggermont, A.; Soria, J.C.; Kroemer, G.; Zitvogel, L. Targeting the tumor microenvironment: Removing obstruction to anticancer immune responses and immunotherapy. Ann. Oncol. 2016, 27, 1482–1492. [Google Scholar] [CrossRef] [PubMed]
- Burnette, B.C.; Liang, H.; Lee, Y.; Chlewicki, L.; Khodarev, N.N.; Weichselbaum, R.R.; Fu, Y.X.; Auh, S.L. The efficacy of radiotherapy relies upon induction of type I interferon-dependent innate and adaptive immunity. Cancer Res. 2011, 71, 2488–2496. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; Andre, F.; Tesniere, A.; Kroemer, G. The anticancer immune response: Indispensable for therapeutic success? J. Clin. Investig. 2008, 118, 1991–2001. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Galluzzi, L.; Smyth, M.J.; Kroemer, G. Mechanism of action of conventional and targeted anticancer therapies: Reinstating immunosurveillance. Immunity 2013, 39, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Adjemian, S.; Mattarollo, S.R.; Yamazaki, T.; Aymeric, L.; Yang, H.; Portela Catani, J.P.; Hannani, D.; Duret, H.; Steegh, K.; et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity 2013, 38, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Cekic, C.; Linden, J. Purinergic regulation of the immune system. Nat. Rev. Immunol. 2016, 16, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Shatz, M.; Menendez, D.; Resnick, M.A. The human TLR innate immune gene family is differentially influenced by DNA stress and p53 status in cancer cells. Cancer Res. 2012, 72, 3948–3957. [Google Scholar] [CrossRef] [PubMed]
- Taura, M.; Eguma, A.; Suico, M.A.; Shuto, T.; Koga, T.; Komatsu, K.; Komune, T.; Sato, T.; Saya, H.; Li, J.D.; et al. p53 regulates Toll-like receptor 3 expression and function in human epithelial cell lines. Mol. Cell. Biol. 2008, 28, 6557–6567. [Google Scholar] [CrossRef] [PubMed]
- Slatter, T.L.; Wilson, M.; Tang, C.; Campbell, H.G.; Ward, V.K.; Young, V.L.; Van Ly, D.; Fleming, N.I.; Braithwaite, A.W.; Baird, M.A. Antitumor cytotoxicity induced by bone-marrow-derived antigen-presenting cells is facilitated by the tumor suppressor protein p53 via regulation of IL-12. Oncoimmunology 2016, 5, e1112941. [Google Scholar] [CrossRef] [PubMed]
- Gasser, S.; Orsulic, S.; Brown, E.J.; Raulet, D.H. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 2005, 436, 1186–1190. [Google Scholar] [CrossRef] [PubMed]
- Gasser, S.; Raulet, D.H. The DNA damage response arouses the immune system. Cancer Res. 2006, 66, 3959–3962. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Lakshmikanth, T.; Garofalo, C.; Enge, M.; Spinnler, C.; Anichini, A.; Szekely, L.; Karre, K.; Carbone, E.; Selivanova, G. Pharmacological activation of p53 triggers anticancer innate immune response through induction of ULBP2. Cell Cycle 2011, 10, 3346–3358. [Google Scholar] [CrossRef] [PubMed]
- Textor, S.; Fiegler, N.; Arnold, A.; Porgador, A.; Hofmann, T.G.; Cerwenka, A. Human NK cells are alerted to induction of p53 in cancer cells by upregulation of the NKG2D ligands ULBP1 and ULBP2. Cancer Res. 2011, 71, 5998–6009. [Google Scholar] [CrossRef] [PubMed]
- Iannello, A.; Thompson, T.W.; Ardolino, M.; Lowe, S.W.; Raulet, D.H. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J. Exp. Med. 2013, 210, 2057–2069. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C.; Alpers, J.D.; Nowell, P.C.; Hoover, R.G. Sequential expression of protooncogenes during lectin-stimulated mitogenesis of normal human lymphocytes. Proc. Natl. Acad. Sci. USA 1986, 83, 3982–3986. [Google Scholar] [CrossRef] [PubMed]
- Terada, N.; Lucas, J.J.; Gelfand, E.W. Differential regulation of the tumor suppressor molecules, retinoblastoma susceptibility gene product (Rb) and p53, during cell cycle progression of normal human T cells. J. Immunol. 1991, 147, 698–704. [Google Scholar] [PubMed]
- Boehme, S.A.; Lenardo, M.J. TCR-mediated death of mature T lymphocytes occurs in the absence of p53. J. Immunol. 1996, 156, 4075–4078. [Google Scholar] [PubMed]
- Watanabe, M.; Moon, K.D.; Vacchio, M.S.; Hathcock, K.S.; Hodes, R.J. Downmodulation of tumor suppressor p53 by T cell receptor signaling is critical for antigen-specific CD4+ T cell responses. Immunity 2014, 40, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Cheok, C.F.; Verma, C.S.; Baselga, J.; Lane, D.P. Translating p53 into the clinic. Nat. Rev. Clin. Oncol. 2011, 8, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Khoo, K.H.; Verma, C.S.; Lane, D.P. Drugging the p53 pathway: Understanding the route to clinical efficacy. Nat. Rev. Drug Discov. 2014, 13, 217–236. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.M.; Goren, E.M.; Taylor, P.A.; Mueller, S.N.; Stefanski, H.E.; Osborn, M.J.; Scott, H.S.; Komarova, E.A.; Gudkov, A.V.; Hollander, G.A.; et al. Short-term inhibition of p53 combined with keratinocyte growth factor improves thymic epithelial cell recovery and enhances T-cell reconstitution after murine bone marrow transplantation. Blood 2010, 115, 1088–1097. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Saha, M.N.; Micallef, J.; Qiu, L.; Chang, H. Pharmacological activation of the p53 pathway in haematological malignancies. J. Clin. Pathol. 2010, 63, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Issaeva, N.; Bozko, P.; Enge, M.; Protopopova, M.; Verhoef, L.G.; Masucci, M.; Pramanik, A.; Selivanova, G. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat. Med. 2004, 10, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Pflaum, J.; Schlosser, S.; Muller, M. p53 family and cellular stress responses in cancer. Front. Oncol. 2014, 4, 285. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, C.; Tommasini, A.; Zauli, G. The MDM2 inhibitor Nutlin-3 modulates dendritic cell-induced T cell proliferation. Hum. Immunol. 2012, 73, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Secchiero, P.; Toffoli, B.; Melloni, E.; Agnoletto, C.; Monasta, L.; Zauli, G. The MDM2 inhibitor Nutlin-3 attenuates streptozotocin-induced diabetes mellitus and increases serum level of IL-12p40. Acta Diabetol. 2013, 50, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Stegh, A.H. Targeting the p53 signaling pathway in cancer therapy—The promises, challenges and perils. Expert Opin. Ther. Targets 2012, 16, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; Demaria, S. Combining radiotherapy and cancer immunotherapy: A paradigm shift. J. Natl. Cancer Inst. 2013, 105, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; Kroemer, G. Immunological aspects of cancer chemotherapy. Nat. Rev. Immunol. 2008, 8, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Melero, I.; Arina, A.; Murillo, O.; Dubrot, J.; Alfaro, C.; Pérez-Gracia, J.L.; Bendandi, M.; Hervás-Stubbs, S. Immunogenic cell death and cross-priming are reaching the clinical immunotherapy arena. Clin. Cancer Res. 2006, 12, 2385–2389. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, Y.; Guo, G. Immunomodulatory Function of the Tumor Suppressor p53 in Host Immune Response and the Tumor Microenvironment. Int. J. Mol. Sci. 2016, 17, 1942. https://doi.org/10.3390/ijms17111942
Cui Y, Guo G. Immunomodulatory Function of the Tumor Suppressor p53 in Host Immune Response and the Tumor Microenvironment. International Journal of Molecular Sciences. 2016; 17(11):1942. https://doi.org/10.3390/ijms17111942
Chicago/Turabian StyleCui, Yan, and Gang Guo. 2016. "Immunomodulatory Function of the Tumor Suppressor p53 in Host Immune Response and the Tumor Microenvironment" International Journal of Molecular Sciences 17, no. 11: 1942. https://doi.org/10.3390/ijms17111942
APA StyleCui, Y., & Guo, G. (2016). Immunomodulatory Function of the Tumor Suppressor p53 in Host Immune Response and the Tumor Microenvironment. International Journal of Molecular Sciences, 17(11), 1942. https://doi.org/10.3390/ijms17111942