
Glucose-6-Phosphate Dehydrogenase: Update and Analysis of New Mutations around the World

 , , , , ,
, , , , ,
Abstract
: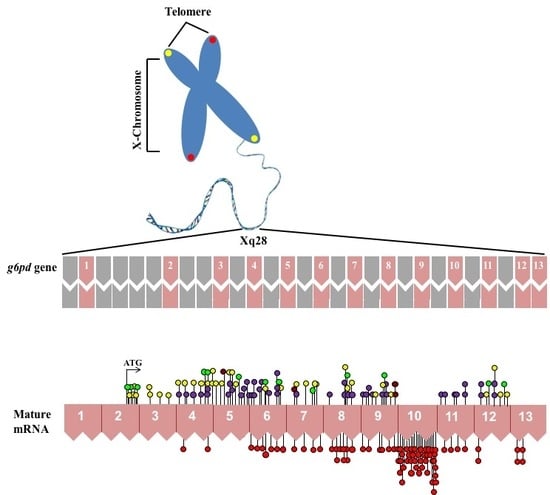
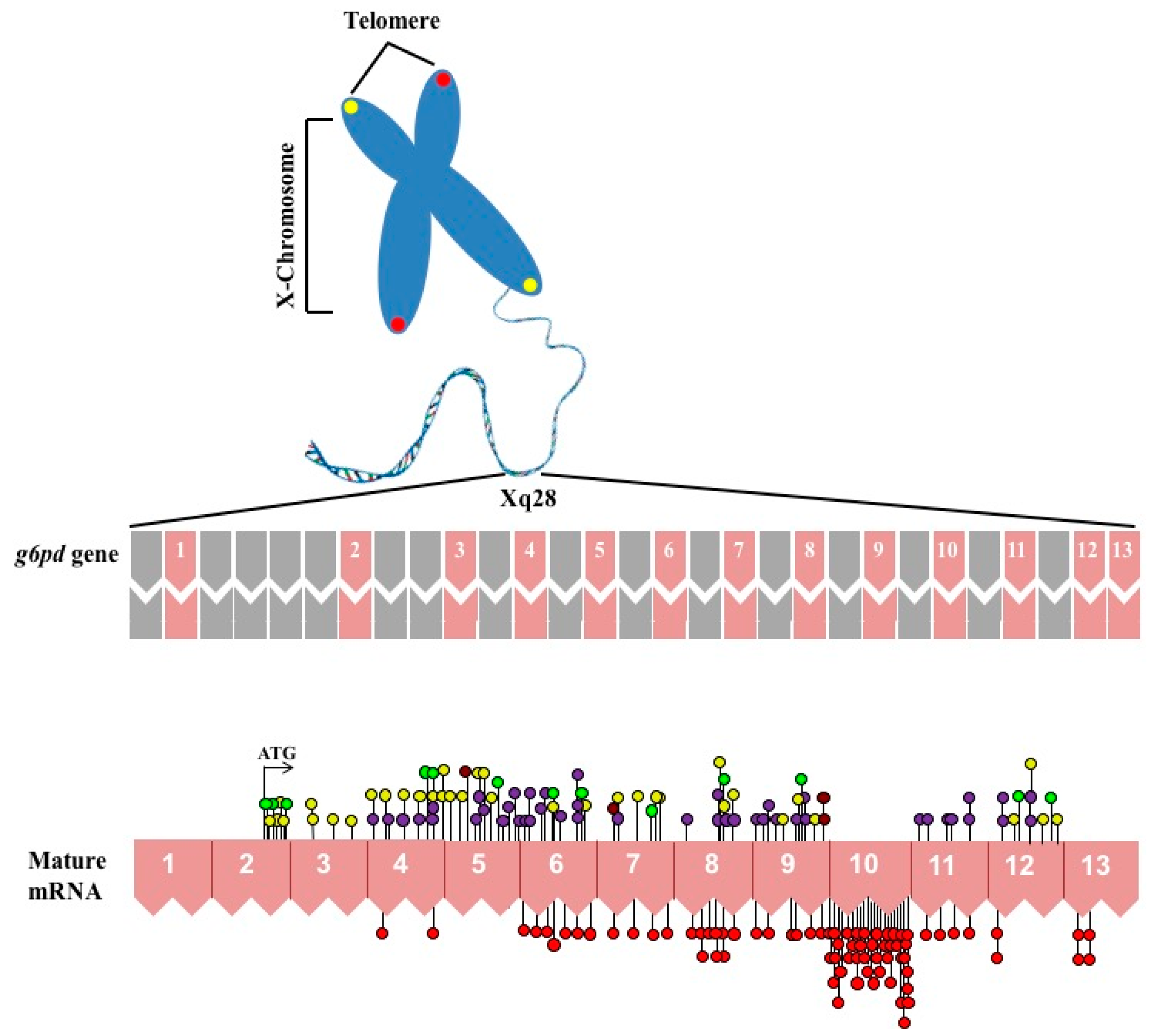
1. Gene Structure of Glucose-6-Phosphate Dehydrogenase (G6PD)
2. Epidemiology G6PD Deficiency
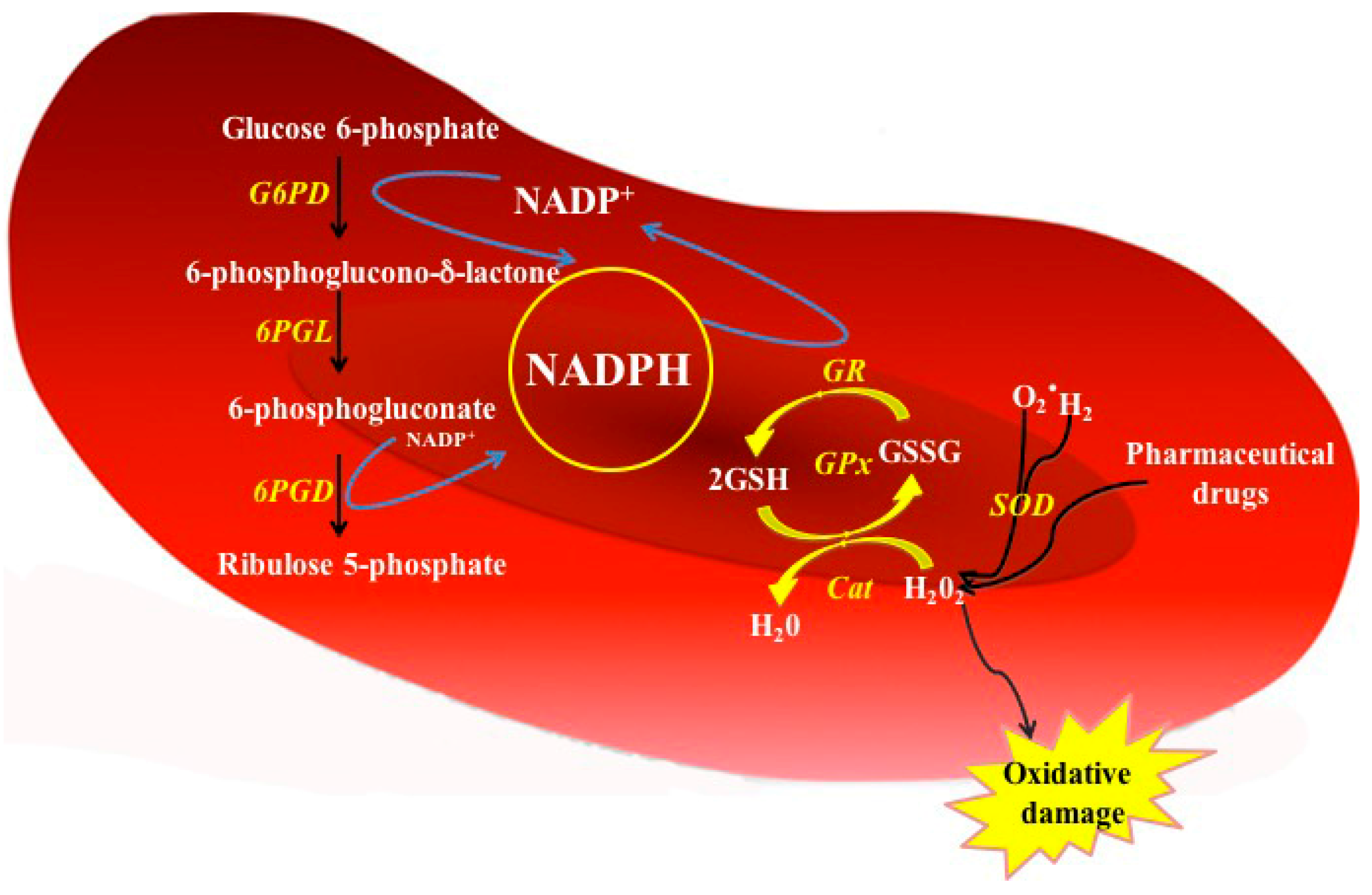
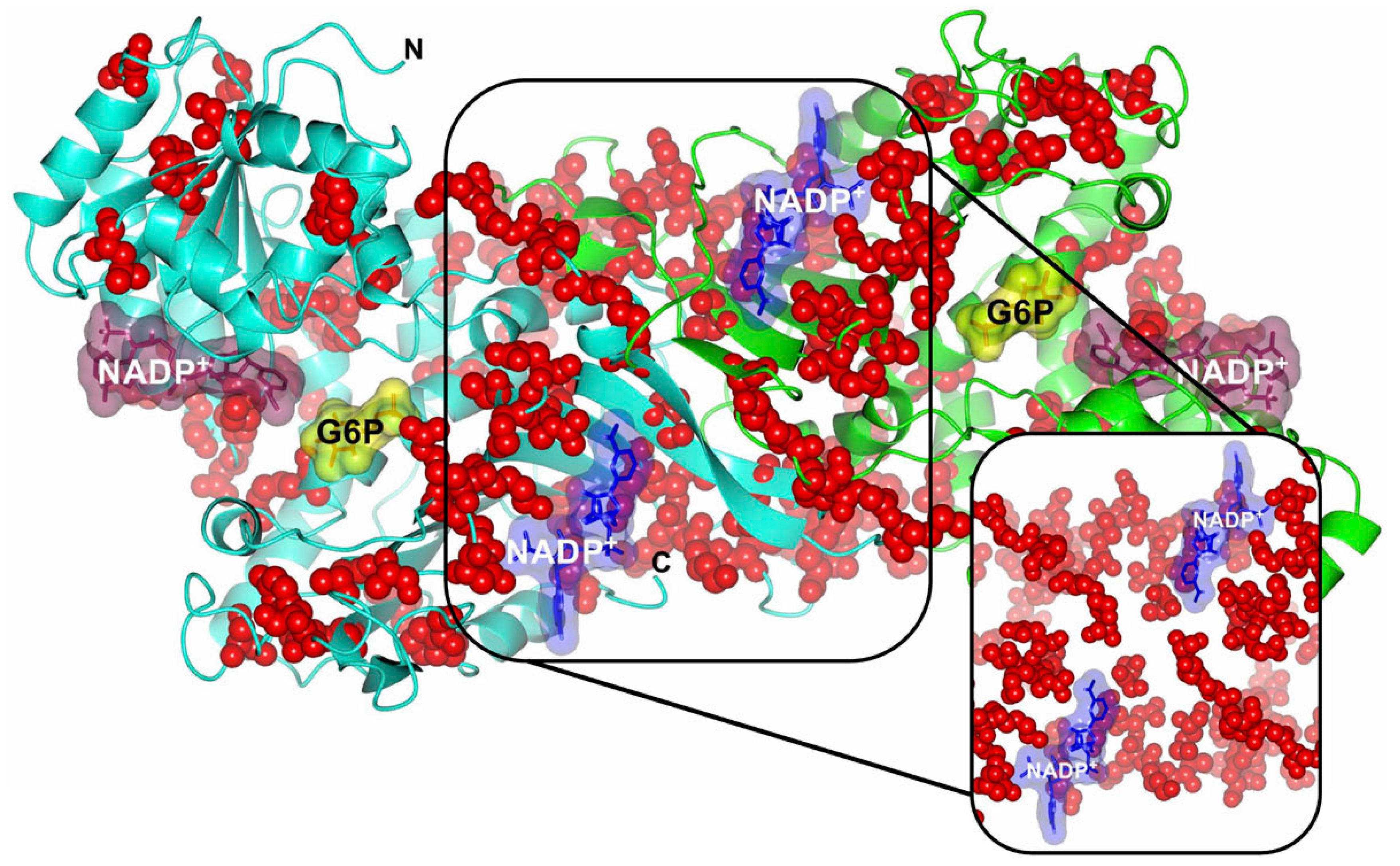
3. Biochemical Aspects of the G6PD (Metabolic Function and Deficiency)
4. Molecular Characterization of G6PD Variants
5. Clinical G6PD Deficiency
5.1. Neonatal Jaundice
5.2. Hemolytic Anemia
5.3. Chronic Nonspherocytic Hemolytic Anemia (CNSHA)
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pai, G.S.; Sprenkle, J.A.; Do, T.T.; Mareni, C.E.; Migeon, B.R. Localization of loci for hypoxanthine phosphoribosyltransferase and glucose-6-phosphate dehydrogenase 202 and biochemical evidence of nonrandom X chromosome expression from studies of a human X-autosome translocation. Proc. Natl. Acad. Sci. USA 1980, 77, 2810–2813. [Google Scholar] [CrossRef] [PubMed]
- Luzzatto, L.; Battistuzzi, G. Glucose-6-phosphate dehydrogenase. Adv. Hum. Genet. 1985, 14, 217–386. [Google Scholar] [PubMed]
- Allahverdiyev, A.M.; Bagirova, M.; Elcicek, S.; Koc, R.C.; Ates, S.C.; Baydar, S.Y.; Yaman, S.; Abamor, E.S.; Oztel, O.N. Glucose-6-Phosphate Dehydrogenase Deficiency and Malaria: A Method to Detect Primaquine-Induced Hemolysis In Vitro. In Biochemistry, Genetics and Molecular Biology; Canuto, R.A., Ed.; In Tech: Rijeka, Croatia, 2012. [Google Scholar]
- Szabo, P.; Purrello, M.; Rocchi, M.; Archidiacono, N.; Alhadeff, B.; Filippi, G.; Toniolo, D.; Martini, G.; Luzzatto, L.; Siniscalco, M. Cytological mapping of the human glucose-6-phosphate dehydrogenase gene distal to the fragile-X site suggests a high rate of meiotic recombination across this site. Proc. Natl. Acad. Sci. USA 1984, 81, 7855–7859. [Google Scholar] [CrossRef] [PubMed]
- Patterson, M.; Schwartz, C.; Bell, M.; Hofker, M.; Trask, B.; van den Engh, G.; Davies, K.E. Physical mapping studies on the human X chromosome in the region Xq27-Xqter. Genomics 1987, 1, 297–306. [Google Scholar] [CrossRef]
- Oberle, I.; Camerino, G.; Wrogemann, K.; Arveiler, B.; Hanauer, A.; Raimondi, E.; Mandel, J.L. Multipoint genetic mapping of the Xq26-q28 region in families with fragile X mental retardation and in normal families reveals tight linkage of markers in q26-q27. Hum. Genet. 1987, 77, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Motulsky, A.G. Normal and abnormal color-vision genes. Am. J. Hum. Genet. 1988, 42, 405–407. [Google Scholar] [PubMed]
- Filosa, S.; Calabro, V.; Lania, G.; Vulliamy, T.J.; Brancati, C.; Tagarelli, A.; Luzzatto, L.; Martini, G.G. G6PD haplotypes spanning Xq28 from F8C to red/green color vision. Genomics 1993, 17, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Boyer, S.H.; Graham, J.B. Linkage between the X chromosome loci for glucose-6-phosphate dehydrogenase electrophoretic variation and hemophilia A. Am. J. Hum. Genet. 1965, 17, 320–324. [Google Scholar] [PubMed]
- Arngrimsson, R.; Dokal, I.; Luzzatto, L.; Connor, J. Dyskeratosis congenita: Three additional families show linkage to a locus in Xq28. J. Med. Genet. 1993, 30, 618–619. [Google Scholar] [CrossRef] [PubMed]
- Persico, M.G.; Viglietto, G.; Martini, G.; Toniolo, D.; Paonessa, G.; Moscatelli, C.; Dono, R.; Vulliamy, T.; Luzzatto, L.; D’Urso, M. Isolation of human glucose-6-phosphate dehydrogenase (G6PD) cDNA clones: Primary structure of the protein and unusual 5′ non-coding region. Nucleic Acids Res. 1986, 14, 2511–2522. [Google Scholar] [CrossRef] [PubMed]
- Rattazzi, M.C. Glucose 6-phosphate dehydrogenase from human erythrocytes: Molecular weight determination by gel filtration. Biochem. Biophys. Res. Commun. 1968, 31, 16–24. [Google Scholar] [CrossRef]
- Vulliamy, T.J.; D’Urso, M.; Battistuzzi, G.; Estrada, M.; Foulkes, N.S.; Martini, G.; Calabrof, V.; Poggi, V.; Giordano, R.; Town, M.; et al. Diverse point mutations in the human glucose-6-phosphate dehydrogenase gene cause enzyme deficiency and mild or severe hemolytic anemia. Proc. Natl. Acad. Sci. USA 1998, 85, 5171–5175. [Google Scholar] [CrossRef]
- Ruwende, C.; Hill, A. Glucose-6-phosphate dehydrogenase deficiency and malaria. J. Mol. Med. 1998, 76, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Janney, S.K.; Joist, J.J.; Fitch, C.D. Excess Release of ferriheme in G6PD-deficient erythrocytes: Possible cause of hemolysis and resistance to Malaria. Blood 1986, 67, 331–333. [Google Scholar] [PubMed]
- Martini, G.; Toniolo, D.; Vulliamy, T.; Luzzatto, L.; Dono, R.; Viglietto, G.; Paonessa, G.; D’Urso, M.; Persico, M.G. Structural analysis of the X-linked gene encoding human glucose 6-phosphate dehydrogenase. EMBO J. 1986, 5, 1849–1855. [Google Scholar] [PubMed]
- Nkhoma, E.T.; Poole, C.; Vannappagari, V.; Hall, S.A.; Beutler, E. The global prevalence of glucose-6-phosphate dehydrogenase deficiency: A systematic review and meta-analysis. Blood Cells Mol. Dis. 2009, 42, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Frank, J.E. Diagnosis and management of G6PD deficiency. Am. Fam. Phys. 2005, 7, 1277–1282. [Google Scholar]
- Peters, A.L.; Van Noorden, C.J. Glucose-6-phosphate dehydrogenase deficiency and malaria: Cytochemical detection of heterozygous G6PD deficiency in women. J. Histochem. Cytochem. 2009, 57, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Manzo, S.; Terrón-Hernández, J.; De la Mora-De la Mora, I.; González-Valdez, A.; Marcial-Quino, J.; García-Torres, I.; Vanoye-Carlo, A.; López-Velázquez, G.; Hernández-Alcantara, G.; Oria-Hernández, J.; et al. The stability of G6PD is affected by mutations with different clinical phenotypes. Int. J. Mol. Sci. 2014, 15, 21179–21201. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Manzo, S.; Marcial-Quino, J.; Vanoye-Carlo, A.; Enríquez-Flores, S.; De la Mora-De la Mora, I.; González-Valdez, A.; García-Torres, A.; Martínez-Rosas, V.; Sierra-Palacios, E.; Lazcano-Pérez, F.; et al. Mutations of Glucose-6-Phosphate Dehydrogenase Durham, Santa-Maria and A+ Variants Are Associated with Loss Functional and Structural Stability of the Protein. Int. J. Mol. Sci. 2015, 16, 28657–28668. [Google Scholar] [CrossRef] [PubMed]
- Howes, R.E.; Piel, F.B.; Patil, A.P.; Nyangiri, O.A.; Gething, P.W.; Dewi, M.; Hogg, M.M.; Battle, K.E.; Padilla, C.D.; Baird, J.K.; et al. G6PD deficiency prevalence and estimates of affected populations in malaria endemic countries: A geostatistical model-based map. PLoS Med. 2012, 9, 1001339. [Google Scholar] [CrossRef] [PubMed]
- Lopez, R.; Cooperman, J.M. Glucose-6-phosphate dehydrogenase deficiency and hyperbilirubinaemia in the newborn. Am. J. Dis. Child. 1971, 122, 66–70. [Google Scholar] [PubMed]
- Au, S.W.; Naylor, C.E.; Gover, S.; Vandeputte-Rutten, L.; Scopes, D.A.; Mason, P.J.; Luzzatto, L.; Lam, V.M.; Adams, M.J. Solution of the structure of tetrameric human glucose 6-phosphate dehydrogenase by molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 1999, 55, 826–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howes, R.E.; Battle, K.E.; Satyagraha, A.W.; Baird, J.K.; Hay, S.I. G6PD Deficiency: Global Distribution, Genetic Variants and Primaquine Therapy. Adv. Parasitol. 2013, 81, 134–199. [Google Scholar]
- Wrigley, N.G.; Heather, J.V.; Bonsignore, A.; de Flora, A. Human erythrocyte glucose 6-phosphate dehydrogenase: Electron microscope studies on structure and interconversion of tetramers, dimers and monomers. J. Mol. Biol. 1972, 68, 483–499. [Google Scholar] [CrossRef]
- Turner, N.J. Applications of transketolases in organic synthesis. Curr. Opin. Biotechnol. 2000, 11, 527–531. [Google Scholar] [CrossRef]
- McNicholas, S.; Potterton, E.; Wilson, K.S.; Noble, M.E.M. Presenting your structures: The CCP4mg molecular-graphics software. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Manganelli, G.; Fico, A.; Martini, G.; Filosa, S. Discussion on pharmacogenetic interaction in G6PD deficiency and methods to identify potential hemolytic drugs. Cardiovasc. Hematol. Disord. Drug Targets 2010, 10, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Cappellini, M.D.; Fiorelli, G. Glucose-6-phosphate dehydrogenase deficiency. Lancet 2008, 9606, 64–74. [Google Scholar] [CrossRef]
- Luzzatto, L.; Nannelli, C.; Notaro, R. Glucose-6-Phosphate Dehydrogenase Deficiency. Hematol. Oncol. Clin. N. Am. 2016, 30, 373–393. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Cheng, A.; Lee, A.; Kuang, W.J.; Hillier, L.; Green, P.; Schlessinger, D.; Ciccodicola, A.; D’Urso, M. Sequence of human glucose-6-phosphate dehydrogenase cloned in plasmids and a yeast artificial chromosome. Genomics 1991, 10, 792–800. [Google Scholar] [CrossRef]
- Greene, L.S. G6PD deficiency as protection against falciparum malaria: An epidemiologic critique of population and experimental studies. Am. J. Phys. Anthropol. 1993, 36, 153–178. [Google Scholar] [CrossRef]
- Minucci, A.; Moradkhani, K.; Hwang, M.; Zuppi, C.; Giardina, B.; Capoluongo, P. Glucose-6-phosphate dehydrogenase (G6PD) mutations database: Review of the “old” and update of the new mutations. Blood Cells Mol. Dis. 2012, 48, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Minucci, A.; Giardina, B.; Zuppi, C.; Capoluongo, E. Glucose-6-phosphate dehydrogenase laboratory assay: How, when, and why? IUBMB Life 2009, 61, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Kwok, C.J.; Martin, A.C.; Au, S.W.; Lam, V.M. G6PDdb, an integrated database of glucose-6-phosphate dehydrogenase (G6PD) mutations. Hum. Mutat. 2002, 19, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Vaca, G.; Arámbula, E.; Monsalvo, A.; Medina, C.; Nuñez, C.; Sandoval, L.; López-Guido, B. Glucose-6-phosphate dehydrogenase (G6PD) mutations in Mexico: Four new G6PD variants. Blood Cells Mol. Dis. 2003, 31, 112–120. [Google Scholar] [CrossRef]
- Rigano, P.; Fabiano, C.; Pojero, F.; Niceta, M.; Pecoraro, A.; Maggio, A.; Sammarco, P. Glucose 6-phosphate dehydrogenase Palermo R257M: A novel variant associated with chronic non-spherocytic haemolytic anaemia. Br. J. Haematol. 2010, 149, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Kordes, U.; Richter, A.; Santer, R.; Schäfer, H.; Singer, D.; Sonntag, J.; Janka, G. Neonatal cholestasis and glucose-6-P-dehydrogenase deficiency. Pediatr. Blood Cancer 2010, 54, 758–760. [Google Scholar] [CrossRef] [PubMed]
- McDade, J.; Abramova, T.; Mortier, N.; Howard, T.; Ware, R.E. A novel G6PD mutation leading to chronic hemolytic anemia. Pediatr. Blood Cancer 2008, 51, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.M.; El-Humiany, A.U.-R. Molecular Characterization of New Variants of Glucose-6-phosphate Dehydrogenase Deficiency Gene Isolated in Western Province of Saudi Arabia Causing Hemolytic Anemia. Pak. J. Biol. Sci. 2006, 9, 1605–1616. [Google Scholar]
- Gómez-Manzo, S.; Marcial-Quino, J.; Vanoye-Carlo, A.; Serrano-Posada, H.; González-Valdez, A.; Martínez-Rosas, V.; Hernández-Ochoa, B.; Sierra-Palacios, E.; Castillo-Rodríguez, R.A.; Cuevas-Cruz, M.; et al. Functional and biochemical characterization of three recombinant human Glucose-6-Phosphate Dehydrogenase mutants: Zacatecas, Vanua-Lava and Viangchan. Int. J. Mol. Sci. 2016, 17, 787. [Google Scholar] [CrossRef] [PubMed]
- Kiani, F.; Schwarzl, S.; Fischer, S.; Efferth, T. Three-dimensional modeling of glucose-6-phosphate dehydrogenase-deficient variants from German ancestry. PLoS ONE 2007, 7, e625. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, W.M.; Val, F.F.A.; Siqueira, A.M.; Franca, G.P.; Sampaio, V.S.; Melo, G.C.; Almeida, A.C.G.; Brito, M.A.M.; Peixoto, H.M.; Fuller, D.; et al. G6PD deficiency in Latin America: Systematic review on prevalence and variants. Mem. Inst. Oswaldo Cruz 2014, 109, 553–568. [Google Scholar] [CrossRef] [PubMed]
- Chalvam, R.; Kedar, P.S.; Colah, R.B.; Ghosh, K.; Mukherjee, M.B. A novel R198H mutation in the glucose-6-phosphate dehydrogenase gene in the tribal groups of the Nilgiris in Southern India. J. Hum. Genet. 2008, 53, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Jalloh, A.; Jalloh, M.; Gamanga, I.; Baion, D.; Sahr, F.; Gbakima, A.; Matsuoka, H. G6PD deficiency assessment in Freetown, Sierra Leone, reveals further insight into the molecular heterogeneity of G6PD A. J. Hum. Genet. 2008, 53, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, R.A.; Oshiro, M.; Hirata, M.H.; Hirata, R.D.; Ribeiro, G.S.; Medeiros, T.; Barretto, O.C. A novel point mutation in a class IV glucose-6-phosphate dehydrogenase variant (G6PD São Paulo) and polymorphic G6PD variants in Sõ Paulo State, Brazil. Genet. Mol. Biol. 2009, 32, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.F.; Xia, W.Q.; Ni, P.H.; Hu, Y.Q.; Jiang, X.C. Analysis of glucose-6-phosphate dehydrogenase gene mutations: A novel missense mutation. J. Shanghai Jiaotong Univ. 2010, 30, 698–702. [Google Scholar]
- Moiz, B.; Nasir, A.; Moatter, T.; Naqvi, Z.A.; Khurshid, M. Molecular characterization of glucose-6-phosphate dehydrogenase deficiency in Pakistani population. Int. J. Lab. Hematol. 2011, 33, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Hue, N.T.; Charlieu, J.P.; Chau, T.T.; Day, N.; Farrar, J.J.; Hien, T.T.; Dunstan, S.J. Glucose-6-phosphate dehydrogenase (G6PD) mutations and haemoglobinuria syndrome in the Vietnamese population. Malar. J. 2009, 8, 152. [Google Scholar] [CrossRef] [PubMed]
- Moura Neto, J.P.; Dourado, M.V.; Reis, M.G.; Gonçalves, M.S. A novel c.197T → A variant among Brazilian neonates with glucose-6-phosphate dehydrogenase deficiency. Genet. Mol. Biol. 2008, 31, 33–35. [Google Scholar] [CrossRef]
- Chaves, A.; Eandi, S.; Defelipe, L.; Pepe, C.; Milanesio, B.; Aguirre, F.; Feliú-torres, A. Two novel DNA variants associated with glucose-6-phosphate dehydrogenase deficiency found in Argentine pediatric patients. Clin. Biochem. 2016, 49, 808–810. [Google Scholar] [CrossRef] [PubMed]
- Warny, M.; Lausen, B.; Birgens, H.; Knabe, N.; Petersen, J. Severe G6PD Deficiency Due to a New Missense Mutation in an Infant of Northern European Descent. J. Pediatr. Hematol. Oncol. 2015, 37, 497–499. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.A.; Kim, J.Y.; Lee, K.O.; Kim, S.H.; Koo, H.H.; Kim, H.J. A novel de novo mutation in the G6PD gene in a korean boy with glucose-6-phosphate dehydrogenase deficiency: Case report. Annal. Clin. Lab. Sci. 2015, 45, 446–448. [Google Scholar]
- Chen, X.; Lv, R.; Wen, F.; Chen, Y.; Liu, F. A Novel A1088T Mutation in the Glucose-6-Phosphate Dehydrogenase Gene Detected by RT-PCR Combined with DNA Sequencing. Ind. J. Hemat. Blood Transf. 2016, 32, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Benmansour, I.; Moradkhani, K.; Moumni, I.; Wajcman, H.; Hafsia, R.; Ghanem, A.; Préhu, C. Two new class III G6PD variants (G6PD Tunis (c.920A > C: p.307Gln > Pro) and G6PD Nefza (c.968T > C: p.323 Leu > Pro)) and overview of the spectrum of mutations in Tunisia. Blood Cells Mol. Dis. 2013, 50, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Nantakomol, R.P.; Attakorn, P.; Day, N.P.J.; White, N.J.; Imwong, M. Evaluation of the phenotypic test and genetic analysis in the detection of glucose-6-phosphate dehydrogenase deficiency Duangdao. Malar. J. 2013, 12. [Google Scholar] [CrossRef] [PubMed]
- García-Magallanes, N.; Luque-Ortega, F.; Aguilar-Medina, E.M.; Ramos-Payán, R.; Galaviz-Hernández, C.; Romero-Quintana, J.G.; Arámbula-Meraz, E. Glucose-6-phosphate dehydrogenase deficiency in Northern Mexico and description of a novel mutation. J. Genet. 2014, 93, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Sirdah, M.; Reading, N.S.; Vankayalapati, H.; Perkins, S.L.; Shubair, M.E.; Aboud, L.; Prchal, J.T. Molecular heterogeneity of glucose-6-phosphate dehydrogenase deficiency in Gaza Strip Palestinians. Blood Cells Mol. Dis. 2012, 49, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Wei-Liang, L.; Fang, L. Glucose-6-Phosphate Dehydrogenase Qingzhen: Identification of a Novel Splice Mutation (IVS5–1G > A). Pediatr. Blood Cancer 2012, 58, 825–826. [Google Scholar]
- Bendaoud, B.; Hosni, I.; Mosbahi, I.; Hafsia, R.; Prehu, C.; Abbes, S. Three new mutations account for the prevalence of glucose 6 phosphate deshydrogenase (G6PD) deficiency in Tunisia. Pathol. Biol. 2013, 61, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Beutler, E.; Vulliamy, T.J. Hematologically important mutations: Glucose-6-phosphate dehydrogenase. Blood Cells Mol. Dis. 2002, 28, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Mason, P.J.; Vulliamy, T.J. Glucose-6-phosphate dehydrogenase deficiency. Baillieres Best Pract. Res. Clin. Haematol. 2000, 13, 21–38. [Google Scholar] [CrossRef] [PubMed]
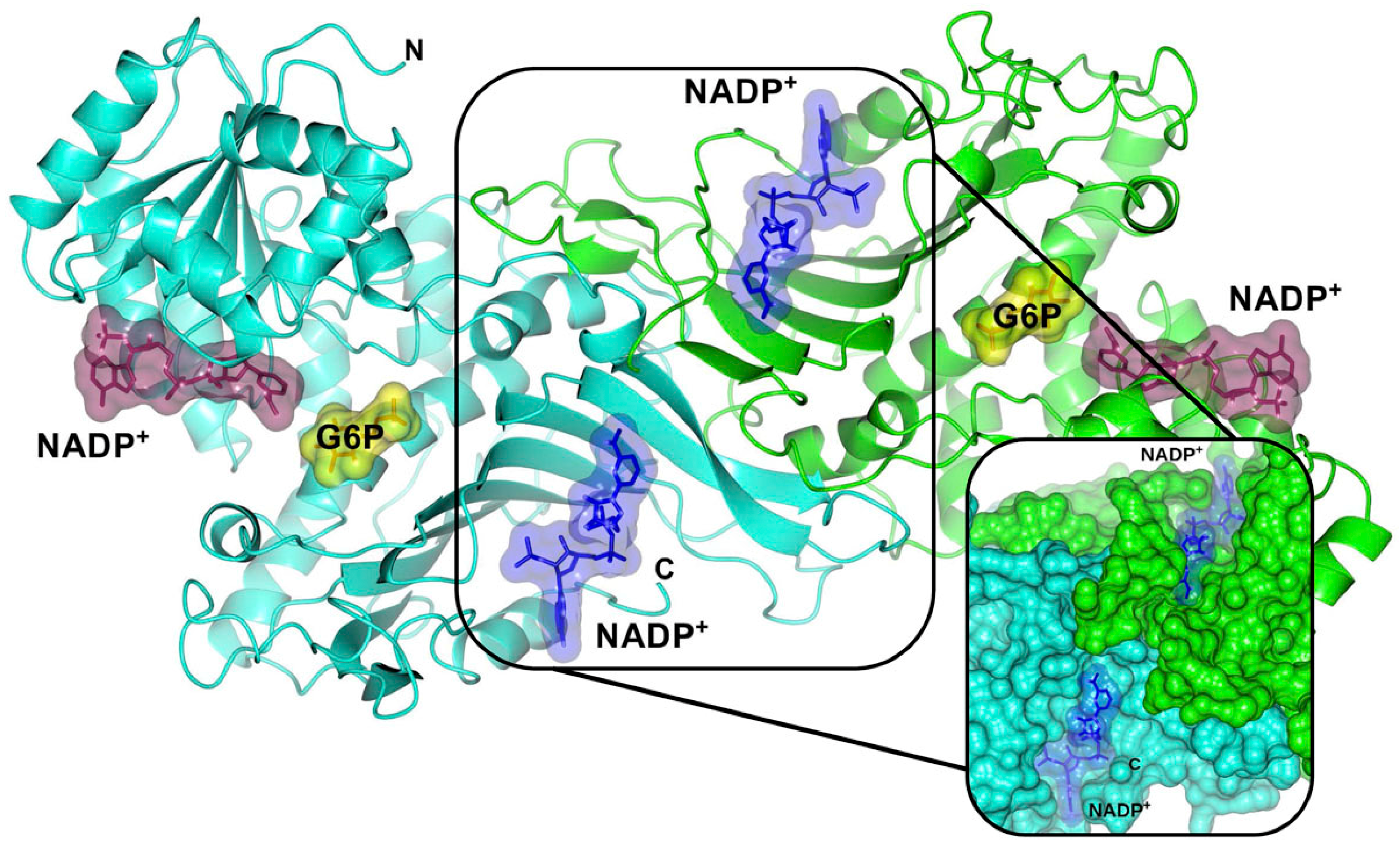
- Wang, X.T.; Chan, T.F.; Lam, V.; Engel, P. What is the role of the second “structural” NADP+-binding site in human glucose-6-phosphate dehydrogenase? Protein Sci. 2008, 17, 1403–1411. [Google Scholar] [CrossRef] [PubMed]
- Au, S.W.N.; Gover, S.; Lam, V.; Adams, M. Human glucose-6-phosphate dehydrogenase: The cristal structure reveals a structural NADP+ molecule and provides. Structure 2000, 8, 293–303. [Google Scholar] [CrossRef]
- Mason, P.J.; Bautista, J.M.; Gilsanz, F. G6PD deficiency: The genotype-phenotype association. Blood Rev. 2007, 5, 267–283. [Google Scholar] [CrossRef] [PubMed]
- Filosa, S.; Calabrò, V.; Vallone, D.; Poggi, V.; Mason, P.; Pagnini, D.; Alfinito, F.; Rotoli, B.; Martini, G.; Luzzatto, L.; et al. Molecular basis of chronic non-spherocytic haemolytic anaemia: A new G6PD variant (393 Arg–His) with abnormal KmG6P and marked in vivo instability. Br. J. Haematol. 1992, 80, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Filosa, S.; Cai, W.; Galanello, R.; Cao, A.; de Mattia, D.; Schettini, F.; Martini, G. A novel single-base mutation in the glucose 6-phosphate dehydrogenase gene is associated with chronic non-spherocytic haemolytic anaemia. Hum. Genet. 1994, 94, 560–562. [Google Scholar] [CrossRef] [PubMed]
- Luzzatto, L.; Mehta, A.; Vulliamy, T. Glucose 6-phosphate dehydrogenase deficiency. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; Chapter 179; pp. 4517–4553. [Google Scholar]
- Beutler, E. Glucose-6-phosphate dehydrogenase deficiency: A historical perspective. Blood 2008, 111, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Luzzatto, L. Glucose 6-phosphate dehydrogenase deficiency: From genotype to phenotype. Haematologica 2006, 91, 1303–1306. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.H.; Bhutani, V.K. System-based approach to management of neonatal jaundice and prevention of kernicterus. J. Pediatr. 2002, 140, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Luzzatto, L.; Poggi, V.E. Glucose 6-phosphate dehydrogenase deficiency. In Nathan and Oski’s Hematology of Infancy and Childhood; Orkin, S.H., Nathan, D.G., Ginsburg, D., Look, A.T., Fisher, D.E., Lux, S., IV, Eds.; Saunders: Philadelphia, PA, USA, 2009; pp. 883–907. [Google Scholar]
- Maisels, M.J. Neonatal jaundice. Pediatr. Rev. 2006, 27, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Carette, C.; Dubois-Laforgue, D.; Gautier, J.F.; Timsit, J. Diabetes mellitus and glucose-6-phosphate dehydrogenase deficiency: From one crisis to another. Diabetes Metab. 2011, 37, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Beutler, E. Red Cell Metabolism: A Manual of Biochemical Methods, 3rd ed.; Grune and Stratton: New York, NY, USA, 1984; p. 105. [Google Scholar]
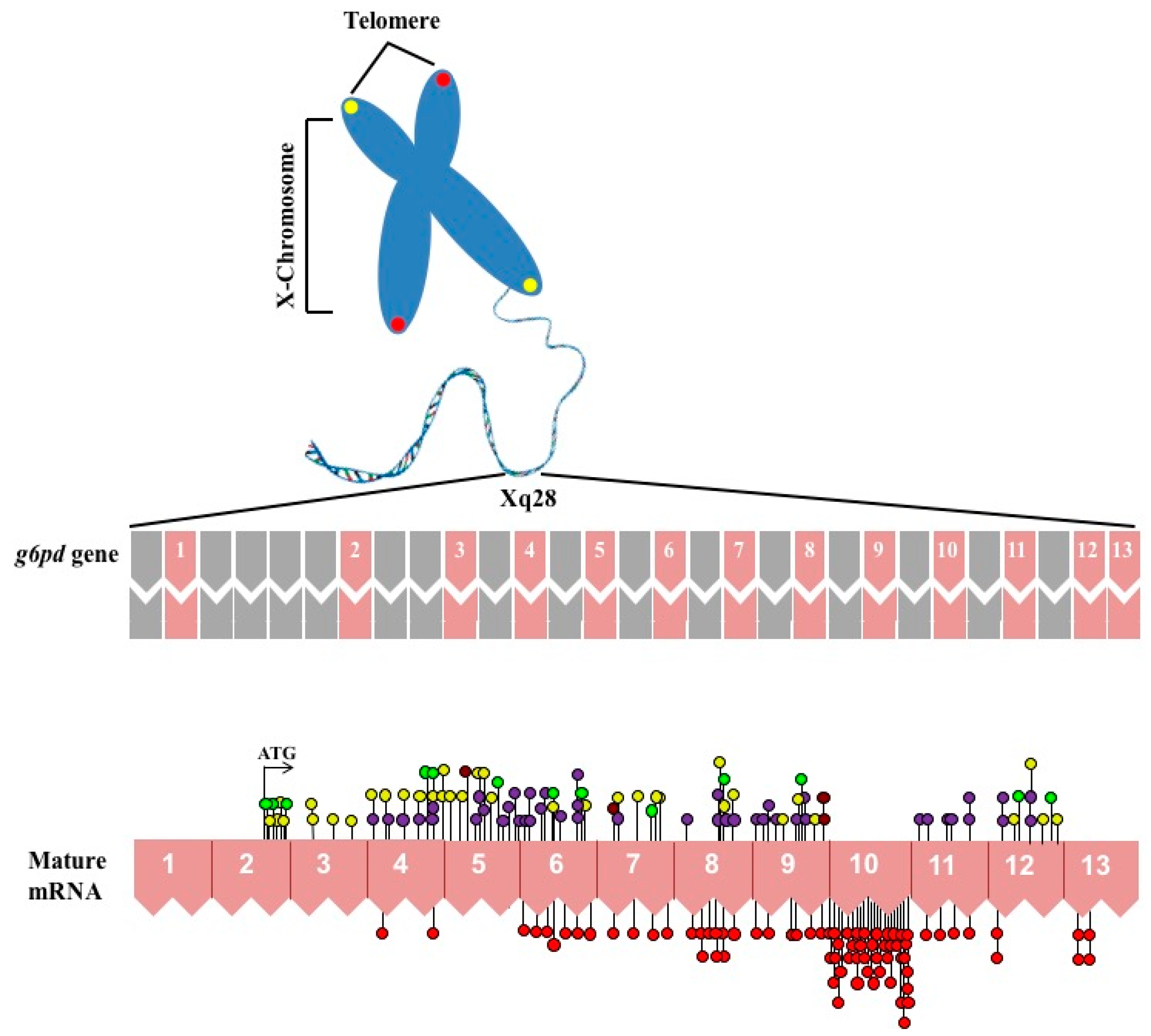






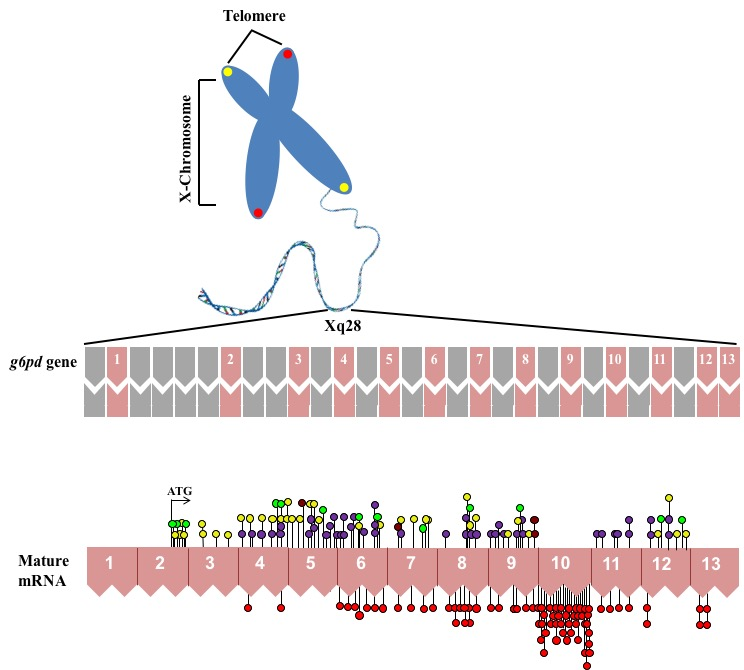{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
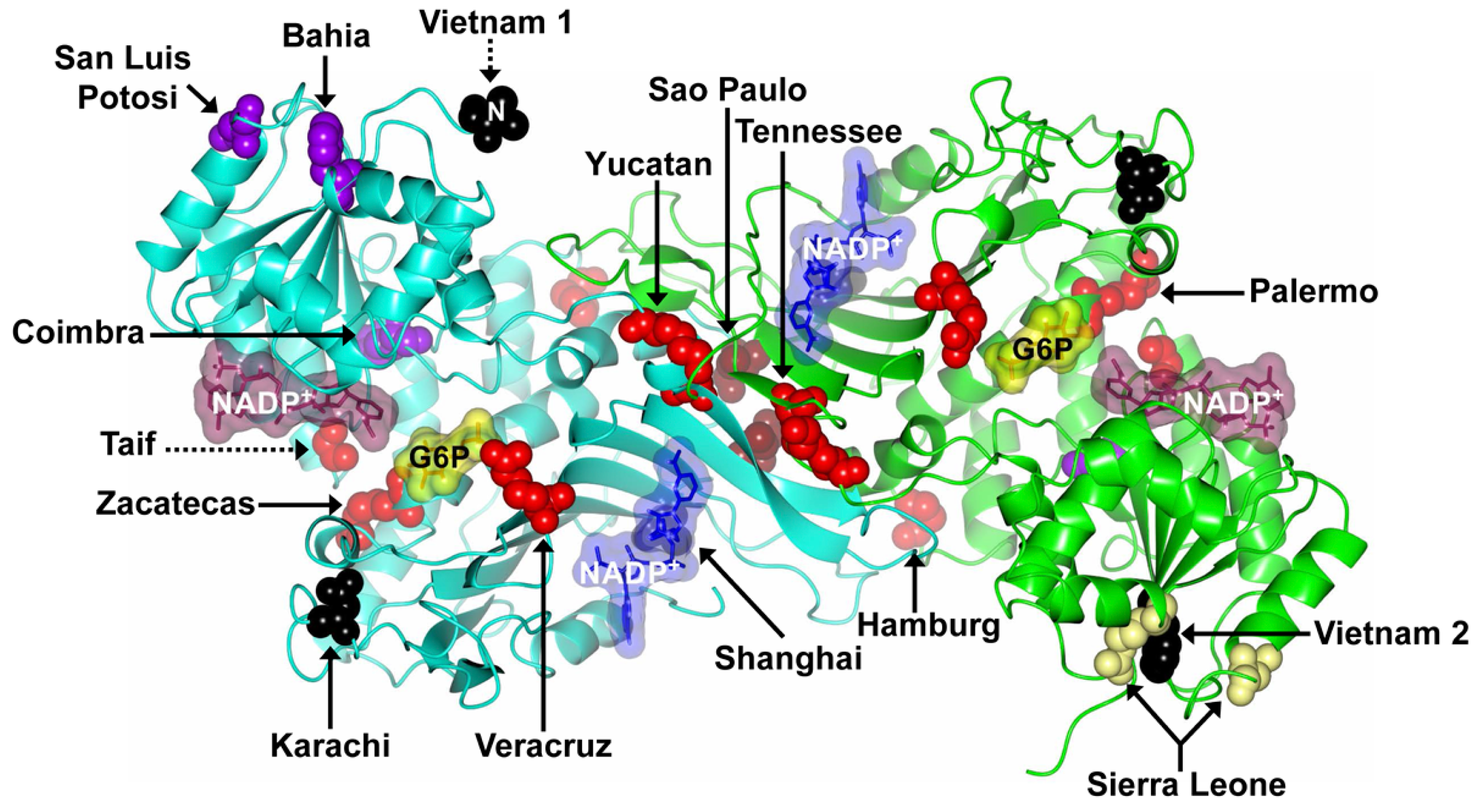
| Mutation Name | cDNA Nucleotide Substitution | Codon | Amino Acid Substitution | Exon | Class | Origin’s Country | Reference |
|---|---|---|---|---|---|---|---|
| Single Missense Mutations | |||||||
| Vietnam 1 | 7G > A | 3 | Glu → Lys | 2 | NR | Vietnam | [50] |
| México DF | 193A > G | 65 | Thr → Ala | 4 | NR | Mexico | [58] |
| Bahia | 197T > A | 66 | Phe → Thr | 4 | II | Brazil | [51] |
| Vietnam 2 | 197T > G | 66 | Phe → Cys | 4 | NR | Vietnam | [50] |
| San Luis Potosi | 376A > T | 126 | Asn → Tyr | 5 | II | Mexico | [37] |
| Gaza | 536G > A | 179 | Ser → Asn | 6 | NR | Palestine | [59] |
| Herlev | 592C > A | 198 | Arg → Ser | 6 | I/II | Denmark | [53] |
| Coimbra | 593G > A | 198 | Arg → His | 6 | II | India | [45] |
| San Paulo | 660C > G | 220 | Ile → Met | 7 | IV | Brazil | [47] |
| Shanghai | 691G > C | 231 | Ala → Pro | 7 | NR | Chinese | [48] |
| Tunisia | 737T > C | 246 | Arg → Leu | 7 | III | Tunisia | [61] |
| Zacatecas | 770G > T | 257 | Arg → Leu | 7 | I | Mexico | [37] |
| Hamburg | 827C > T | 276 | Pro → Leu | 8 | I | Germany | [39] |
| Tunis | 920A > C | 307 | Gln → Pro | 9 | II | Tunisia | [56] |
| Nefza | 968T > C | 323 | Leu → Pro | 9 | III | Tunisia | [56] |
| Karachi | 973G > A | 325 | Asp → Asn | 9 | NR | Pakistan | [49] |
| Quilmes | 995C > T | 332 | Ser → Phe | 9 | I | Argentine | [52] |
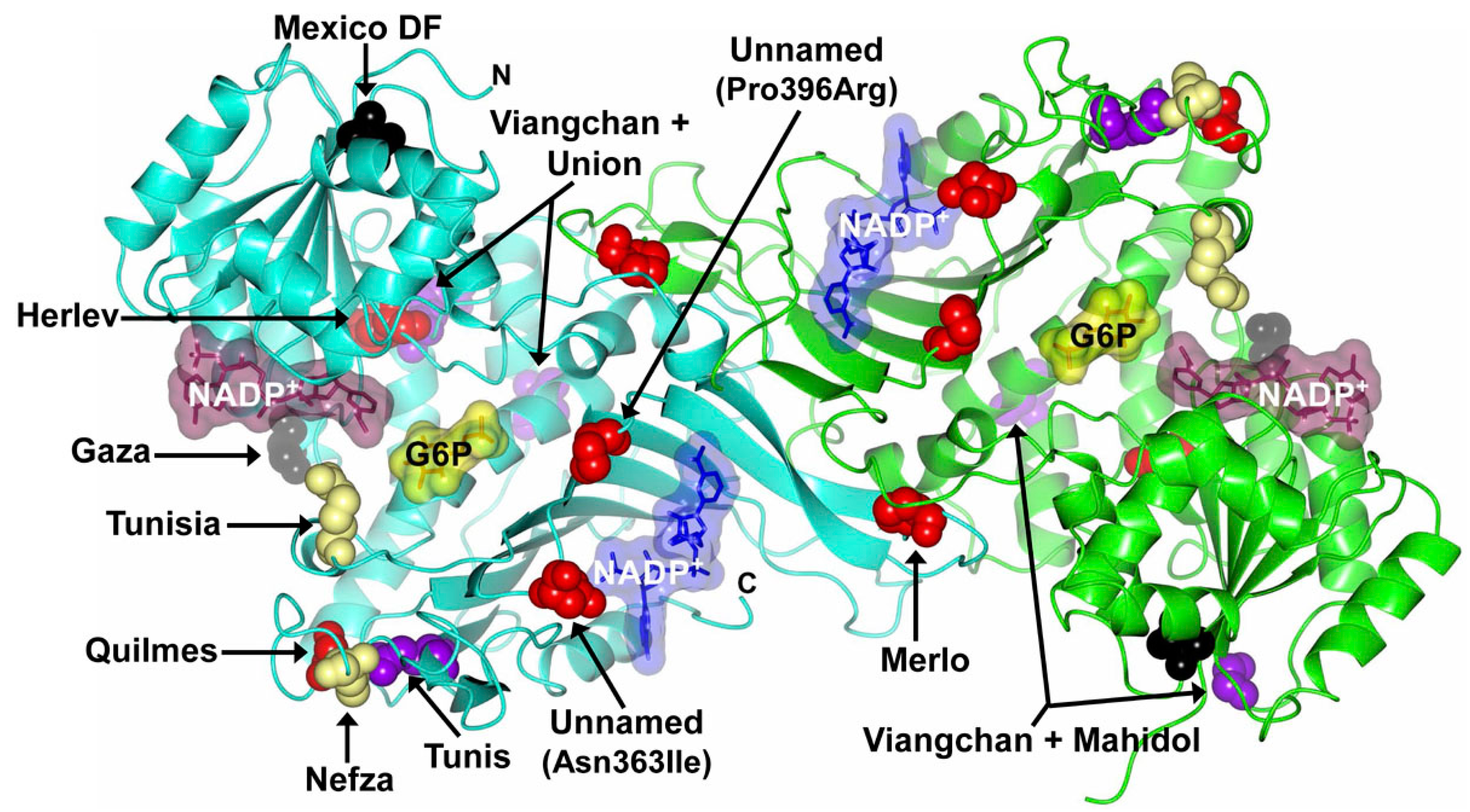
| Unnamed | 1088A > T | 363 | Asn → Ile | 10 | I | China | [55] |
| Veracruz | 1094G > A | 365 | Arg → His | 10 | I | Mexico | [37] |
| Unnamed | 1187C > G | 396 | Pro → Arg | 10 | I | Korea | [54] |
| Merlo | 1226C > A | 409 | Pro → Gln | 10 | I | Argentine | [52] |
| Yucatan | 1285A > G | 429 | Lys → Glu | 10 | I | Mexico | [37] |
| Tennessee | 1465C > G | 422 | Leu → Val | 10 | I | USA | [40] |
| Multiple Missense Mutations | |||||||
| Taif | 516–518 del | 174 | Gly | 6 | I | Saudi Arabia | [41] |
| Sierra Leona | 311G > A | 104 | Arg → His | 5 | III | Sierra Leone | [46] |
| 376A > G | 126 | Asn → Asp | |||||
| Palermo | 769C > A | 257 | Arg → Met | 7 | I | Italy | [38] |
| 770G > T | |||||||
| Viangchan + Mahidol | 871G > A | 291 | Val → Met | 9, 6 | II/III | Thailand | [57] |
| 487G > A | 163 | Gly → Ser | |||||
| Viangchan + Union | 871G > A | 291 | Val → Met | 9, 11 | II/III | Thailand | [57] |
| 1360C > T | 454 | Arg → Cys | |||||
| Non-Coding Region Substitutions | |||||||
| Qingzhen | IVS5-1G > A | - | - | - | NR | - | [60] |
| Unnamed | IVS-VIII 43G > A | - | - | - | III | Tunisia | [61] |
| Unnamed | IVS-V 655C > T | - | - | - | III | Tunisia | [61] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Manzo, S.; Marcial-Quino, J.; Vanoye-Carlo, A.; Serrano-Posada, H.; Ortega-Cuellar, D.; González-Valdez, A.; Castillo-Rodríguez, R.A.; Hernández-Ochoa, B.; Sierra-Palacios, E.; Rodríguez-Bustamante, E.; et al. Glucose-6-Phosphate Dehydrogenase: Update and Analysis of New Mutations around the World. Int. J. Mol. Sci. 2016, 17, 2069. https://doi.org/10.3390/ijms17122069
Gómez-Manzo S, Marcial-Quino J, Vanoye-Carlo A, Serrano-Posada H, Ortega-Cuellar D, González-Valdez A, Castillo-Rodríguez RA, Hernández-Ochoa B, Sierra-Palacios E, Rodríguez-Bustamante E, et al. Glucose-6-Phosphate Dehydrogenase: Update and Analysis of New Mutations around the World. International Journal of Molecular Sciences. 2016; 17(12):2069. https://doi.org/10.3390/ijms17122069
Chicago/Turabian StyleGómez-Manzo, Saúl, Jaime Marcial-Quino, America Vanoye-Carlo, Hugo Serrano-Posada, Daniel Ortega-Cuellar, Abigail González-Valdez, Rosa Angélica Castillo-Rodríguez, Beatriz Hernández-Ochoa, Edgar Sierra-Palacios, Eduardo Rodríguez-Bustamante, and et al. 2016. "Glucose-6-Phosphate Dehydrogenase: Update and Analysis of New Mutations around the World" International Journal of Molecular Sciences 17, no. 12: 2069. https://doi.org/10.3390/ijms17122069
APA StyleGómez-Manzo, S., Marcial-Quino, J., Vanoye-Carlo, A., Serrano-Posada, H., Ortega-Cuellar, D., González-Valdez, A., Castillo-Rodríguez, R. A., Hernández-Ochoa, B., Sierra-Palacios, E., Rodríguez-Bustamante, E., & Arreguin-Espinosa, R. (2016). Glucose-6-Phosphate Dehydrogenase: Update and Analysis of New Mutations around the World. International Journal of Molecular Sciences, 17(12), 2069. https://doi.org/10.3390/ijms17122069