
Temporal Genetic Modifications after Controlled Cortical Impact—Understanding Traumatic Brain Injury through a Systematic Network Approach

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
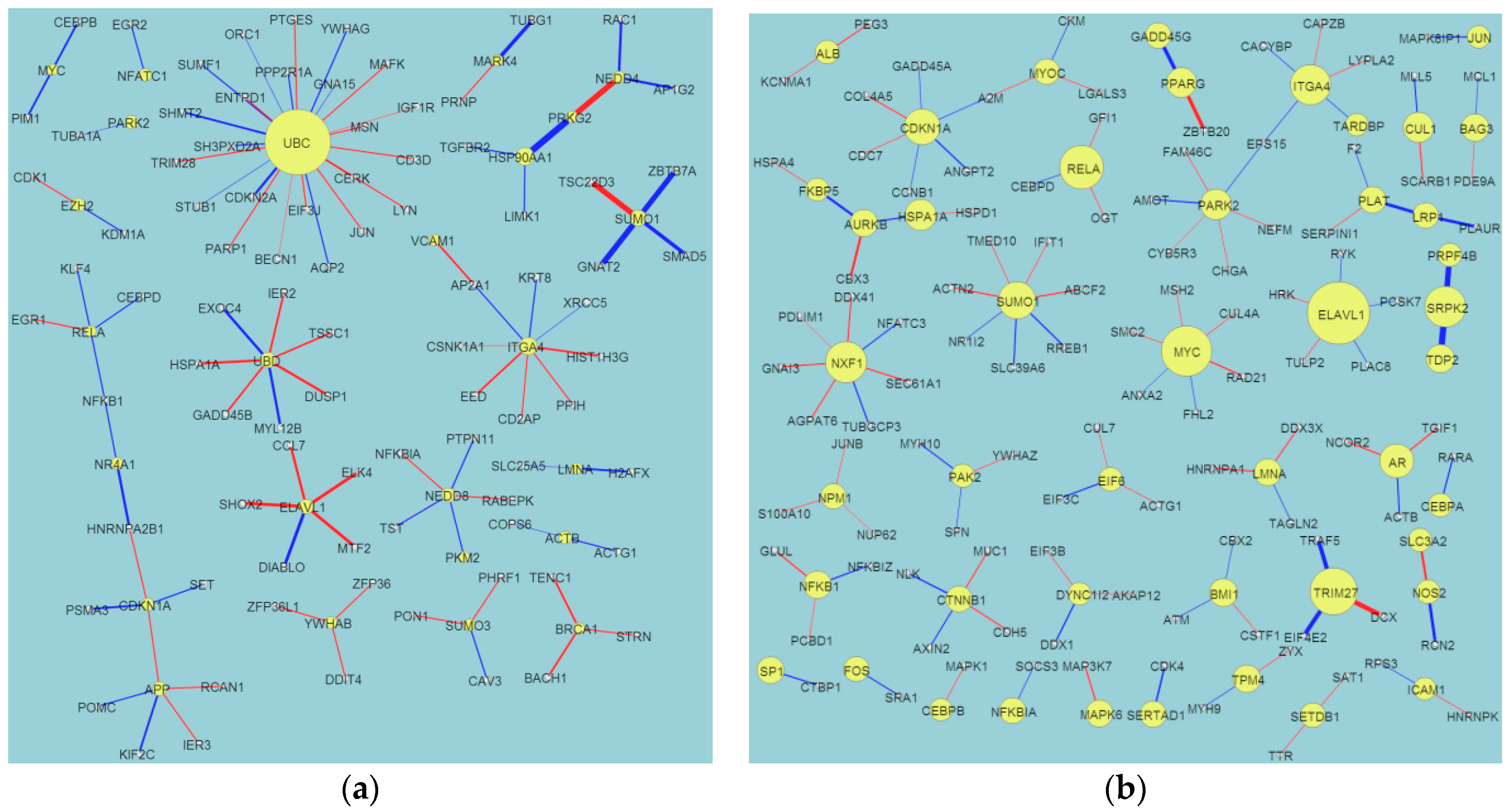
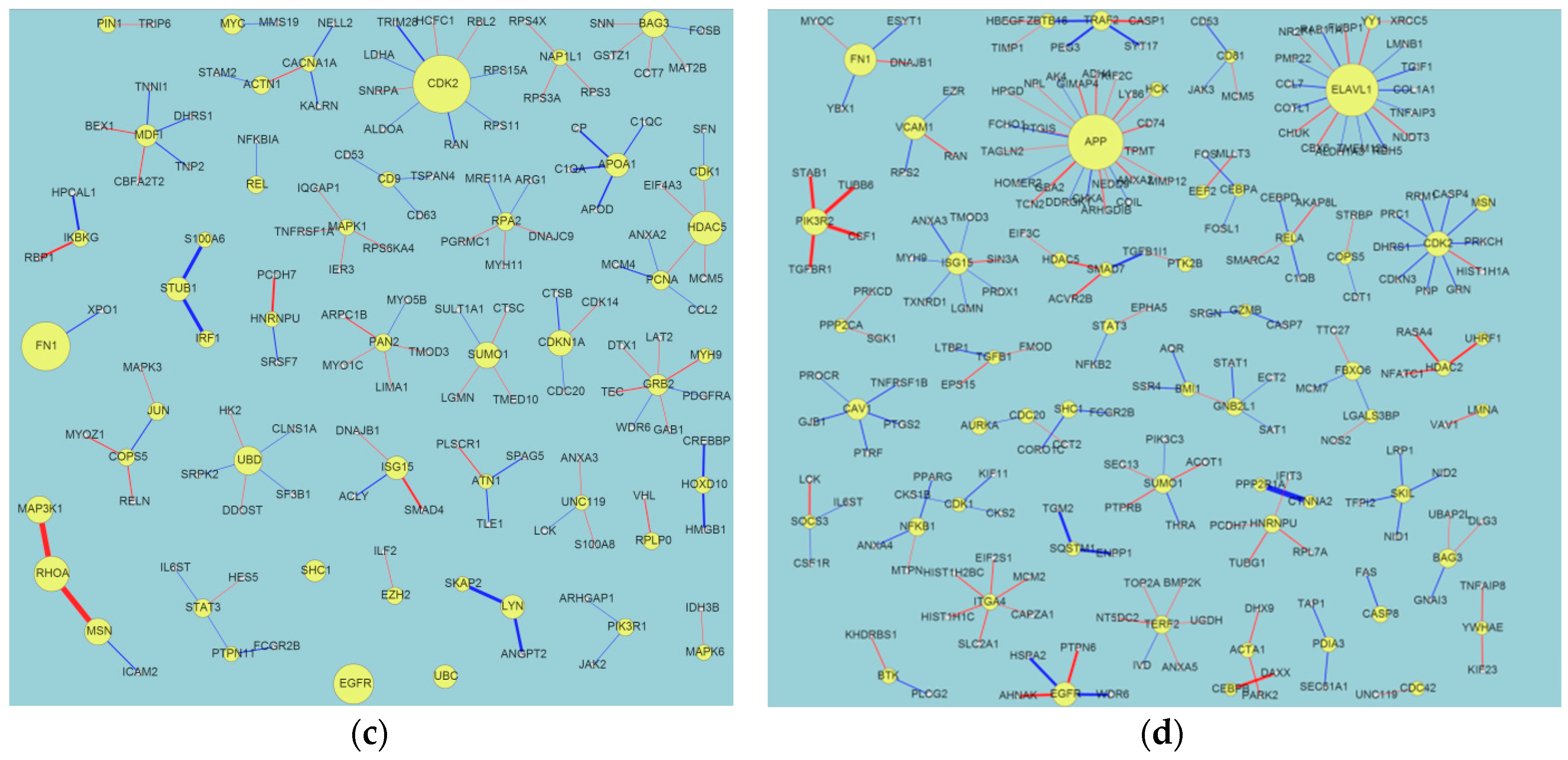
2.1. Evolution of Network Biomarkers at the Four Post-Traumatic Brain Injury (TBI) Time Points

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | TRV | p-Value | TBI_AvgExp | Normal_AvgExp | Log2 FC |
|---|---|---|---|---|---|
| 4 h | |||||
| UBC | 64.77 | <10−9 | 9545 | 20,349 | −1.09 |
| HSP90AA1 | 12.45 | 0.000167 | 11,942 | 22,341 | −0.9 |
| ITGA4 | 11.92 | 0.000167 | 172 | 280 | −0.7 |
| SUMO1 | 10.22 | 0.000167 | 8999 | 9258 | −0.04 |
| UBD | 9.73 | 0.000333 | 126 | 209 | −0.73 |
| ELAVL1 | 8.33 | 0.000667 | 1549 | 1785 | −0.2 |
| APP | 8.29 | 0.000667 | 9608 | 19,299 | −1.01 |
| NEDD8 | 8.1 | 0.000667 | 4903 | 5672 | −0.21 |
| PARK2 | 7.45 | 0.001833 | 47 | 167 | −1.83 |
| EZH2 | 7.42 | 0.002 | 633 | 362 | 0.8 |
| CDKN1A | 6.63 | 0.0045 | 3046 | 1920 | 0.67 |
| ACTB | 6.53 | 0.005 | 15,663 | 26,523 | −0.76 |
| NFATC1 | 6.53 | 0.005167 | 273 | 77 | 1.84 |
| NEDD4 | 6.22 | 0.006833 | 4030 | 4698 | −0.22 |
| YWHAB | 5.81 | 0.011333 | 2014 | 3474 | −0.79 |
| SUMO3 | 5.77 | 0.012333 | 16,117 | 13,002 | 0.31 |
| NR4A1 | 5.75 | 0.0125 | 8539 | 3969 | 1.11 |
| BRCA1 | 5.46 | 0.016667 | 82 | 216 | −1.4 |
| PRKG2 | 5.43 | 0.017667 | 29 | 60 | −1.06 |
| LMNA | 5.25 | 0.021333 | 10,185 | 5878 | 0.79 |
| 8 h | |||||
| ELAVL1 | 16.97 | 0.000868 | 1609 | 1082 | 0.57 |
| MYC | 13.35 | 0.001157 | 356 | 96 | 1.9 |
| TRIM27 | 12.28 | 0.00135 | 3114 | 2773 | 0.17 |
| RELA | 11.34 | 0.001446 | 6282 | 3408 | 0.88 |
| MDM2 | 10.9 | 0.001543 | 2615 | 2317 | 0.17 |
| NXF1 | 10.48 | 0.001543 | 1086 | 1240 | −0.19 |
| SRPK2 | 10.4 | 0.001639 | 1183 | 1232 | −0.06 |
| ITGA4 | 10.26 | 0.001639 | 138 | 231 | −0.74 |
| SUMO1 | 9.35 | 0.002314 | 9180 | 10,837 | −0.24 |
| CDKN1A | 8.92 | 0.002893 | 3417 | 661 | 2.37 |
| NEDD8 | 8.43 | 0.003857 | 4659 | 5525 | −0.25 |
| AR | 8.35 | 0.003857 | 824 | 1585 | −0.94 |
| HSPA1A | 7.5 | 0.005593 | 5663 | 2112 | 1.42 |
| PARK2 | 7.34 | 0.006268 | 28 | 51 | −0.87 |
| CUL1 | 7.22 | 0.006847 | 4406 | 4936 | −0.16 |
| PPARG | 7.17 | 0.007329 | 169 | 126 | 0.42 |
| UBC | 6.93 | 0.0081 | 26,887 | 29,005 | −0.11 |
| TDP2 | 6.69 | 0.00974 | 719 | 1230 | −0.78 |
| BMI1 | 6.58 | 0.010511 | 2465 | 3576 | −0.54 |
| NFKB1 | 6.49 | 0.011765 | 1896 | 1398 | 0.44 |
| 24 h | |||||
| CDK2 | 30.4 | 0.000563 | 612 | 302 | 1.02 |
| FN1 | 25.07 | 0.000805 | 2652 | 1701 | 0.64 |
| EGFR | 20.02 | 0.001689 | 315 | 197 | 0.68 |
| RHOA | 17.25 | 0.002816 | 4082 | 4140 | −0.02 |
| HDAC5 | 16.31 | 0.003057 | 4914 | 7484 | −0.61 |
| UBD | 13.24 | 0.004023 | 97 | 219 | −1.17 |
| MAP3K1 | 12.48 | 0.004586 | 876 | 364 | 1.27 |
| MSN | 12.27 | 0.004666 | 4631 | 1331 | 1.8 |
| BAG3 | 12.13 | 0.004747 | 1206 | 419 | 1.52 |
| SUMO1 | 12 | 0.004747 | 9074 | 10,206 | −0.17 |
| CDKN1A | 11.59 | 0.004988 | 4037 | 740 | 2.45 |
| STUB1 | 10.71 | 0.005632 | 6156 | 5743 | 0.1 |
| LYN | 10.59 | 0.006034 | 350 | 312 | 0.17 |
| APOA1 | 10.52 | 0.006195 | 102 | 63 | 0.71 |
| UBC | 10.27 | 0.006436 | 30,215 | 27,824 | 0.12 |
| ISG15 | 10.26 | 0.006436 | 1137 | 469 | 1.28 |
| MDFI | 9.46 | 0.007723 | 659 | 547 | 0.27 |
| SHC1 | 9.31 | 0.008045 | 788 | 441 | 0.84 |
| GRB2 | 8.91 | 0.008286 | 3458 | 3862 | −0.16 |
| MYC | 8.41 | 0.00901 | 251 | 120 | 1.06 |
| 72 h | |||||
| APP | 34.02 | <10−9 | 31,743 | 38,874 | −0.29 |
| ELAVL1 | 31.92 | <10−9 | 1937 | 1169 | 0.73 |
| FN1 | 18.2 | 0.000309 | 3441 | 1562 | 1.14 |
| PIK3R2 | 14.17 | 0.000386 | 1495 | 1625 | −0.12 |
| CDK2 | 13.29 | 0.000464 | 529 | 383 | 0.47 |
| EGFR | 12.62 | 0.000464 | 325 | 384 | −0.24 |
| VCAM1 | 12.3 | 0.000541 | 1691 | 994 | 0.77 |
| ISG15 | 11.52 | 0.000696 | 1651 | 628 | 1.4 |
| UBC | 11.33 | 0.00085 | 31,436 | 28,733 | 0.13 |
| CAV1 | 10.69 | 0.001005 | 2024 | 802 | 1.34 |
| TRAF2 | 9.53 | 0.001468 | 1479 | 1636 | −0.15 |
| BAG3 | 8.94 | 0.002473 | 1205 | 506 | 1.25 |
| SUMO1 | 8.94 | 0.002473 | 7368 | 9430 | −0.36 |
| NFKB1 | 8.7 | 0.002782 | 2636 | 1476 | 0.84 |
| FBXO6 | 8.6 | 0.003014 | 5920 | 4444 | 0.41 |
| TERF2 | 8.52 | 0.003091 | 878 | 1464 | −0.74 |
| HNRNPU | 8.43 | 0.003246 | 150 | 291 | −0.95 |
| GNB2L1 | 8.26 | 0.003632 | 17,568 | 10,465 | 0.75 |
| SKIL | 7.99 | 0.004405 | 149 | 43 | 1.79 |
| AURKA | 7.89 | 0.004637 | 518 | 175 | 1.56 |


| Gene | Time Point (h) | CRV_Value | p-Value | Case_AvgExp | Control_AvgExp | Log2 FC |
|---|---|---|---|---|---|---|
| UBC | 4 | 64.77 | <10−9 | 9545 | 20,349 | −1.09 |
| UBC | 8 | 6.93 | 0.0081 | 26,887 | 29,005 | −0.11 |
| UBC | 24 | 10.27 | 0.006436 | 30,215 | 27,824 | 0.12 |
| UBC | 72 | 11.33 | 0.00085 | 31,436 | 28,733 | 0.13 |
| SUMO1 | 4 | 10.22 | 0.000167 | 8999 | 9258 | −0.04 |
| SUMO1 | 8 | 9.35 | 0.002314 | 9180 | 10,837 | −0.24 |
| SUMO1 | 24 | 12 | 0.004747 | 9074 | 10,206 | −0.17 |
| SUMO1 | 72 | 8.94 | 0.002473 | 7368 | 9430 | −0.36 |
| CDKN1A | 4 | 6.63 | 0.0045 | 3046 | 1920 | 0.67 |
| CDKN1A | 8 | 8.92 | 0.002893 | 3417 | 661 | 2.37 |
| CDKN1A | 24 | 11.59 | 0.004988 | 4037 | 740 | 2.45 |
| CDKN1A | 72 | 6.68 | 0.010974 | 2257 | 757 | 1.58 |
| MYC | 4 | 4.84 | 0.035667 | 281 | 144 | 0.96 |
| MYC | 8 | 13.35 | 0.001157 | 356 | 96 | 1.9 |
| MYC | 24 | 8.41 | 0.00901 | 251 | 120 | 1.06 |
| MYC | 72 | 5.93 | 0.022411 | 268 | 63 | 2.08 |
2.2. Network Structure Interpretation of the Four Post-TBI Time Points
- (i)
- HSP90AA1: White et al. [19] discussed the relationship between gene expression patterns post-TBI and the inflammatory response. They identified the following significant genes: HSP90AA1, ERAP1 (endoplasmic reticulum aminopeptidase 1), PSMB9 (proteasome subunit beta type-9), CBL (calcineurin B-like), BTK (Bruton’s tyrosine kinase), RORA (retinoic acid receptor-related orphan receptor alpha), THRA (thyroid hormone receptor alpha), and ITGA5 (Integrin, alpha 5, fibronectin receptor).
- (ii)
- (iii)
- CDK2: Zhang discussed the relationships between the expression of BAD, CDK2, and STAT3, and brain function after a TBI in rats [21].
- (iv)
- FN1: White et al. discussed the complex behaviors of FN1, such as its extracellular matrix/cell adhesion (FN1, matrix metalloproteinases (MMPs), and ICAM1) module, its fold changes in a gene interaction hierarchy (GIH) analysis, and others [19].
- (v)
- EGFR: This is a well-known cancer oncogene and the vascular EGFR (VEGFR) receptor is always reported in brain injuries [22]. It is a novel clue for us to identify the relationship between EGFR and TBI.
2.3. Comparison with Our Previous Results for Stroke
2.4. MetaCore™ Results
| No. | Processes | S | T | P | G |
|---|---|---|---|---|---|
| Common to All Four Networks | |||||
| 1 | Viral transcription (97.1%), viral genome expression (97.1%), translational termination (97.1%), cellular protein complex disassembly (97.1%), SRP (signal recognition particle)-dependent cotranslational protein targeting to membrane (97.1%). | 38 | 1 | 0 | 11.39 |
| Common to Two or Three of the Networks | |||||
| 1 | Positive regulation of nucleobase-containing compound metabolic process (75.5%), positive regulation of biosynthetic process (77.6%), enzyme-linked receptor protein signaling pathway (65.3%), positive regulation of nitrogen compound metabolic process (75.5%), positive regulation of macromolecule biosynthetic process (73.5%). | 50 | 16 | 0 | 69.15 |
| 2 | Positive regulation of response to stimulus (60.4%), regulation of response to stimulus (75.0%), response to organic substance (72.9%), transmembrane receptor protein tyrosine kinase signaling pathway (45.8%), response to hormone stimulus (56.2%). | 50 | 14 | 0 | 56.71 |
| 3 | Cell surface receptor signaling pathway (96.0%), signal transduction (96.0%), signaling (96.0%), single organism signaling (96.0%), cell communication (96.0%). | 50 | 2 | 30 | 45.94 |
| Unique to the 4 h Post-TBI Network | |||||
| 1 | Cell activation (54.0%), signal transduction (94.0%), signaling (96.0%), single organism signaling (96.0%), response to wounding (62.0%). | 50 | 6 | 0 | 43.25 |
| 2 | G-protein coupled receptor signaling pathway (71.4%), neuropeptide signaling pathway (30.6%), G-protein coupled receptor signaling pathway, coupled to cyclic nucleotide second messenger (32.7%), cell surface receptor signaling pathway (75.5%), chemokine-mediated signaling pathway (20.4%). | 50 | 5 | 0 | 36.02 |
| 3 | Axis specification (38.0%), canonical Wnt receptor signaling pathway (36.0%), pattern specification process (46.0%), Wnt receptor signaling pathway (38.0%), anterior/posterior pattern specification (36.0%). | 50 | 4 | 0 | 28.78 |
| Unique to the 8 h Post-TBI Network | |||||
| 1 | Response to abiotic stimulus (61.7%), regulation of apoptotic process (63.8%), regulation of programmed cell death (63.8%), positive regulation of cellular process (85.1%), positive regulation of metabolic process (74.5%). | 50 | 9 | 0 | 41.50 |
| 2 | Positive regulation of immune response (43.8%), positive regulation of response to stimulus (58.3%), regulation of response to stress (52.1%), regulation of immune response (47.9%), TRIF (Toll/IL-1 receptor domain-containing adapter inducing interferon-β)-dependent toll-like receptor signaling pathway (27.1%). | 50 | 8 | 0 | 36.87 |
| 3 | Immune response-activating signal transduction (25.0%), immune response-regulating signaling pathway (25.0%), T cell costimulation (17.5%), lymphocyte costimulation (17.5%), cellular defense response (17.5%). | 50 | 5 | 2 | 25.40 |
| Unique to the 24 h Post-TBI Network | |||||
| 1 | Double-strand break repair via synthesis-dependent strand annealing (100.0%), DNA recombinase assembly (100.0%), DNA excision (100.0%), telomere maintenance via semi-conservative replication (100.0%), nucleotide-excision repair, DNA gap filling (100.0%). | 4 | 2 | 0 | 33.30 |
| 2 | Lipoprotein metabolic process (46.2%), protein-lipid complex assembly (34.6%), plasma lipoprotein particle assembly (34.6%), lipid transport (50.0%), lipid localization (50.0%). | 49 | 6 | 0 | 30.34 |
| 3 | Immune response (56.5%), immune system process (67.4%), positive regulation of response to stimulus (58.7%), activation of immune response (39.1%), positive regulation of immune system process (47.8%). | 50 | 6 | 0 | 28.11 |
| Unique to the 72 h Post-TBI Network | |||||
| 1 | Positive regulation of biological process (98.0%), positive regulation of cellular process (90.0%), enzyme linked receptor protein signaling pathway (56.0%), regulation of immune system process (60.0%), regulation of response to stimulus (76.0%). | 50 | 13 | 0 | 65.57 |
| 2 | Positive regulation of apoptotic process (58.0%), positive regulation of programmed cell death (58.0%), positive regulation of cell death (58.0%), positive regulation of biological process (90.0%), regulation of apoptotic process (66.0%). | 50 | 11 | 0 | 55.45 |
| 3 | Enzyme linked receptor protein signaling pathway (63.8%), membrane protein intracellular domain proteolysis (25.5%), nerve growth factor receptor signaling pathway (40.4%), membrane protein proteolysis (25.5%), transmembrane receptor protein tyrosine kinase signaling pathway (46.8%). | 50 | 10 | 0 | 50.91 |
2.4.1. Statistical Interpretation of MetaCore™
2.4.2. Three Statistical Results for the Four Time Points Post-TBI


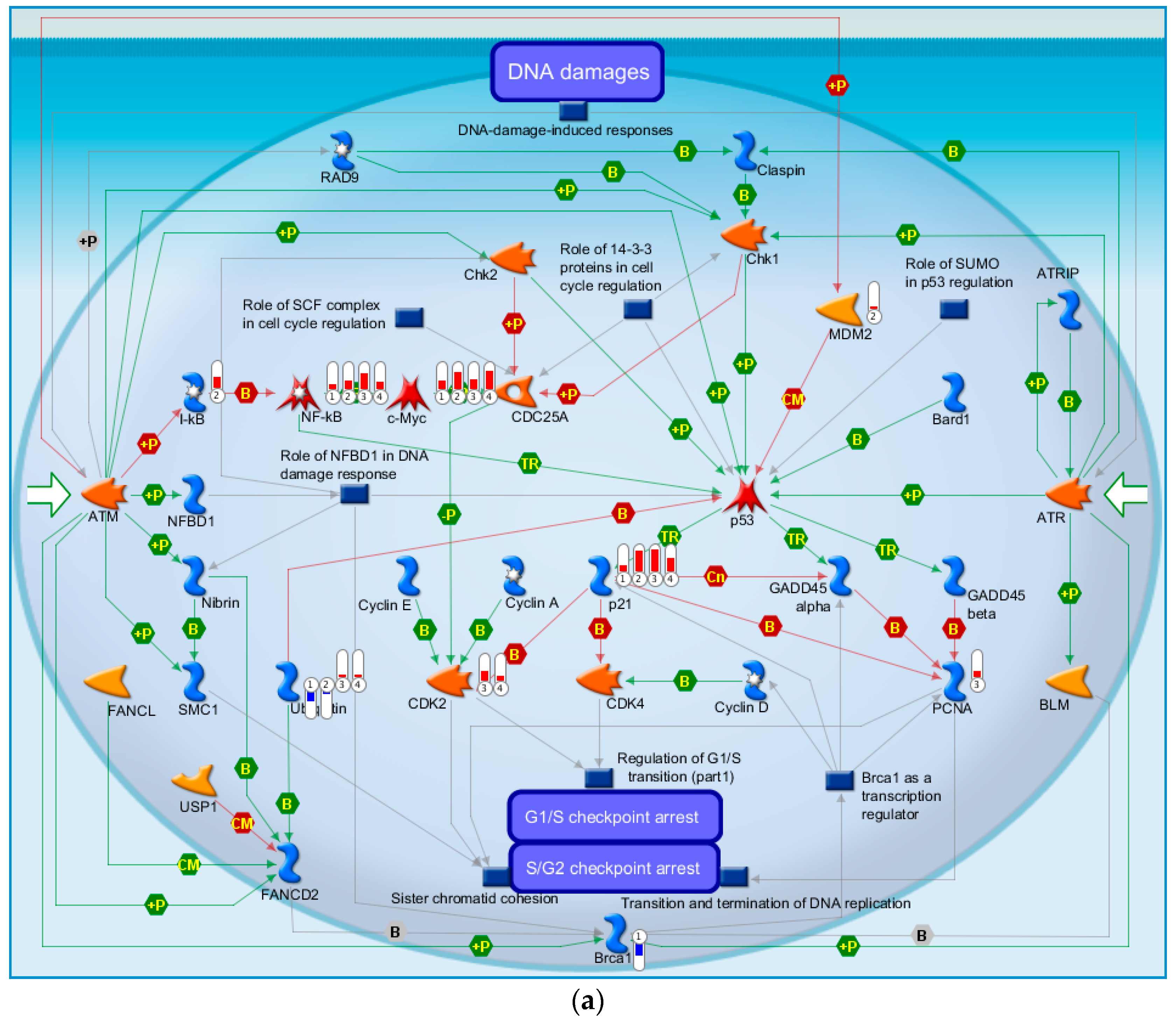
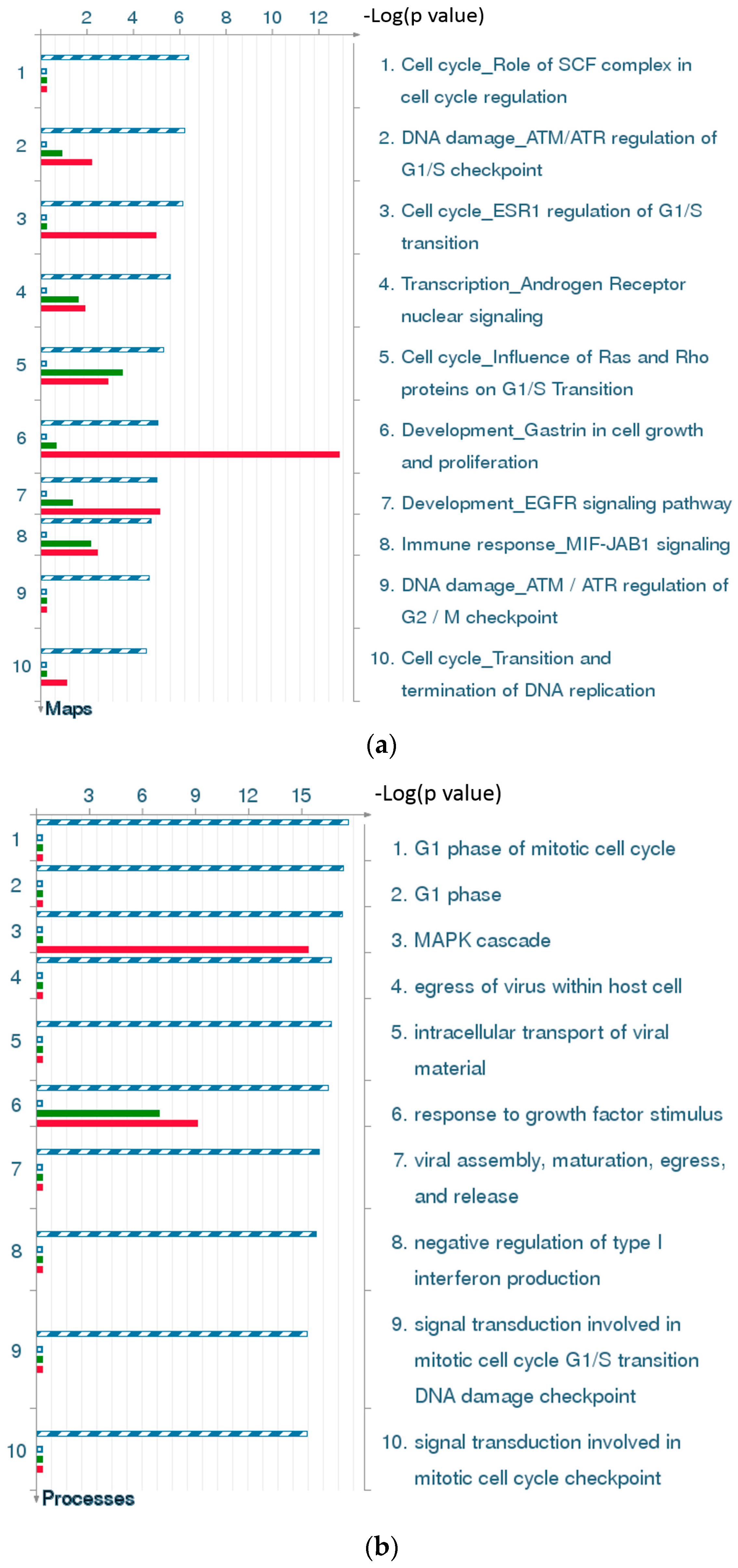
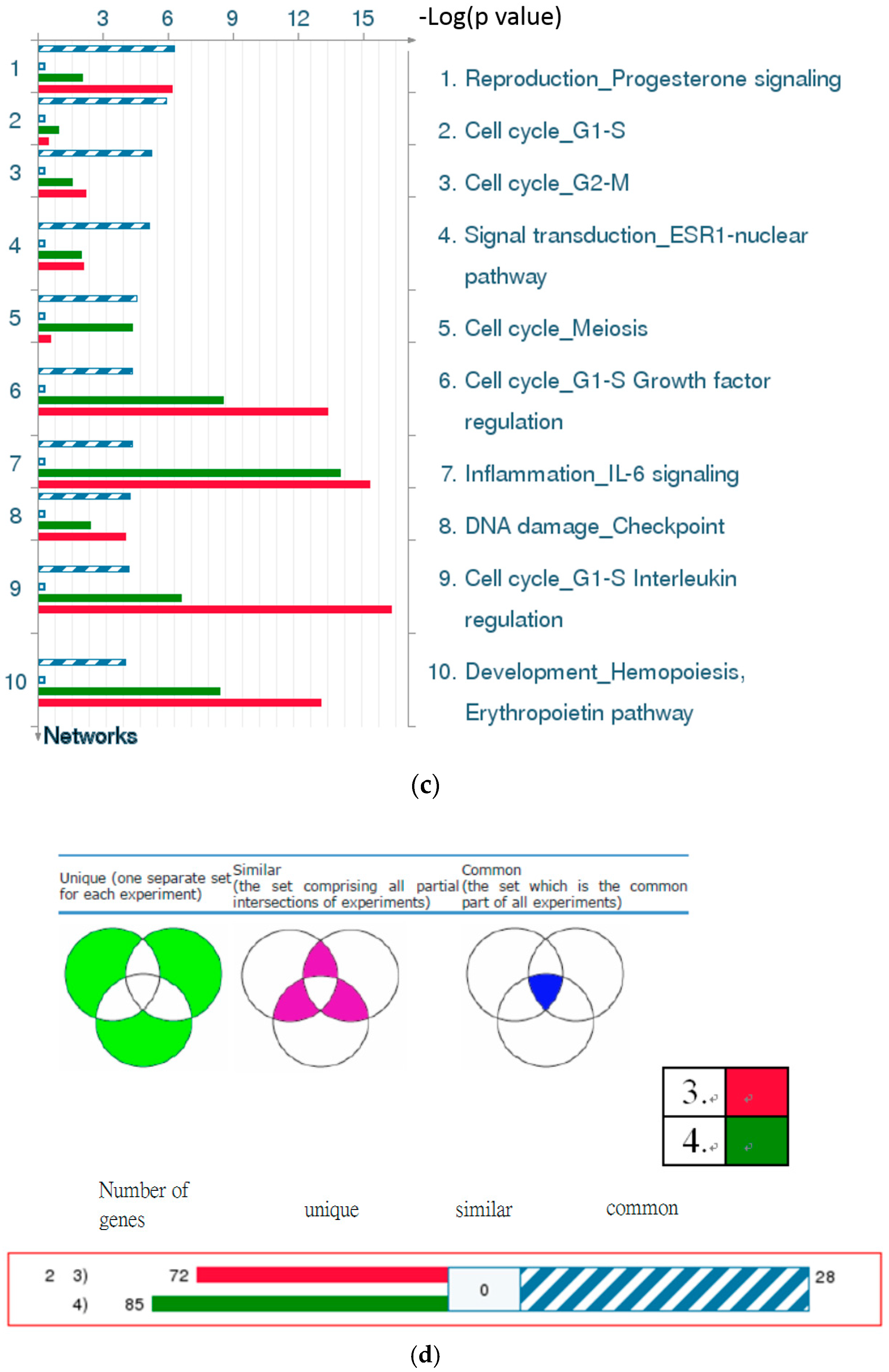
2.4.3. The Highest-Scoring Pathway Map of Post-TBI-Related Biomarker Genes at the Four Time Points


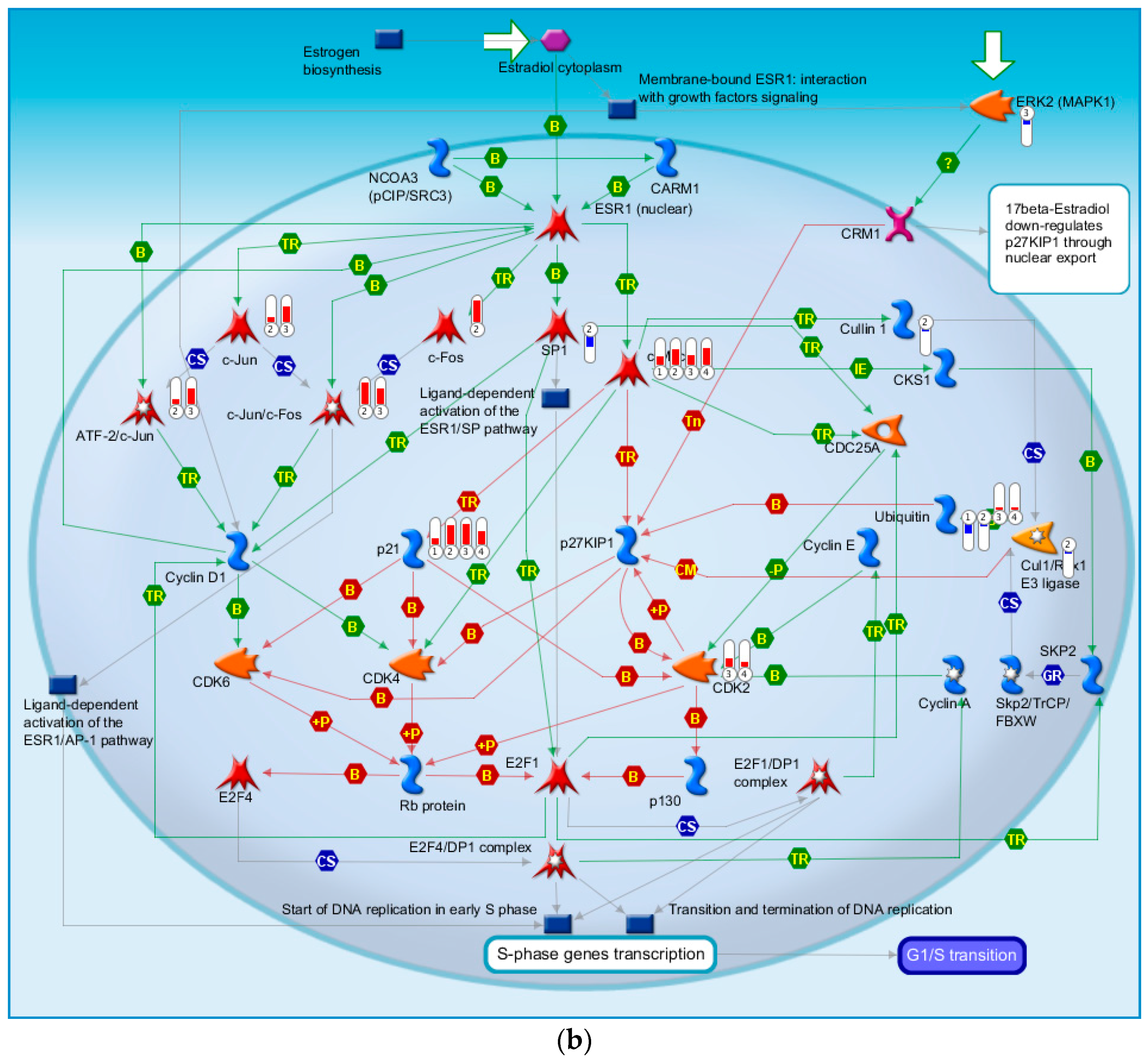
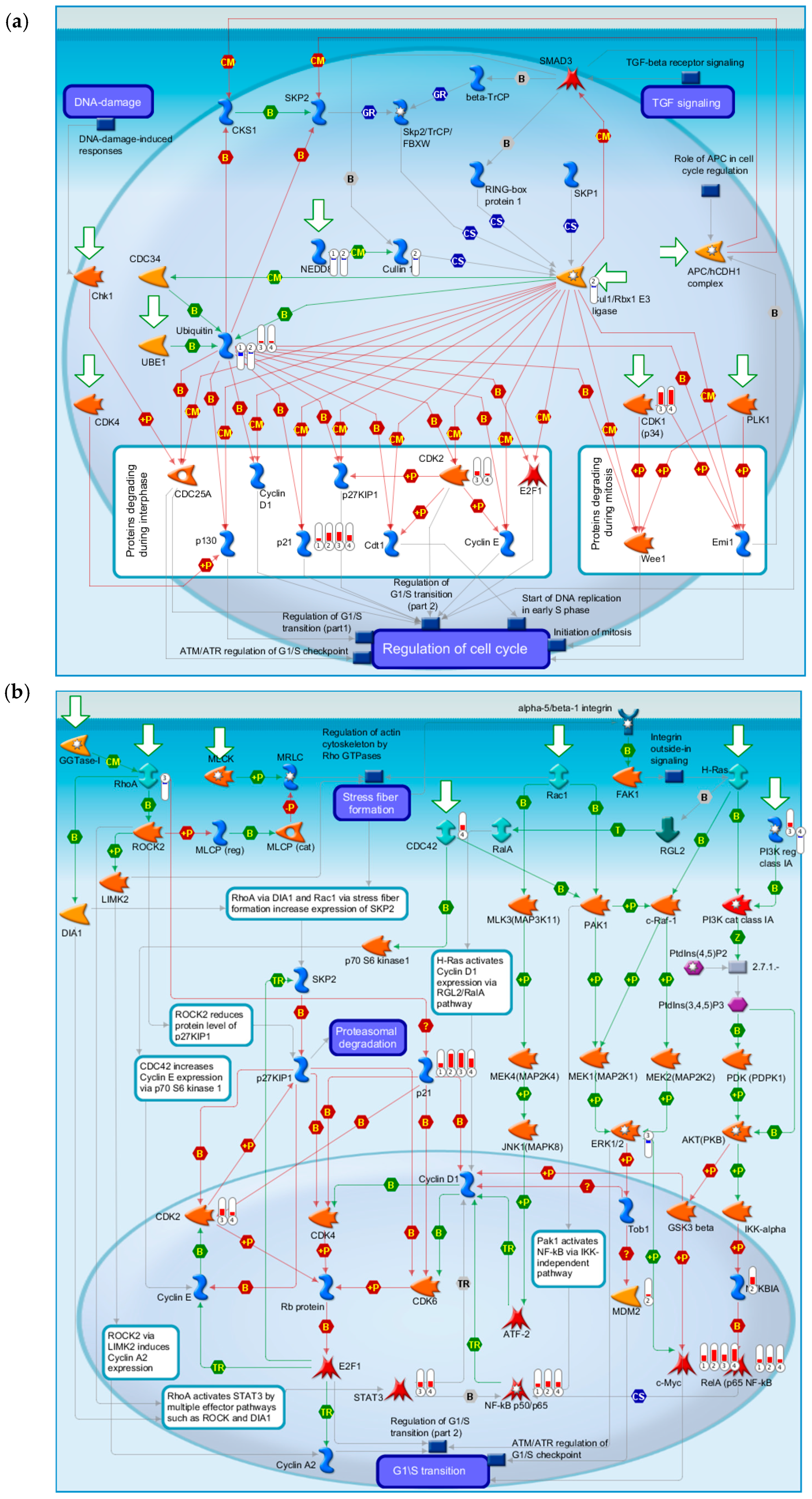
2.4.4. Three Statistical Results for the Last Two Time Points Post-TBI
2.4.5. The Highest-Scoring Pathway Map of Post-TBI-Related Biomarker Genes at 24 and 72 h



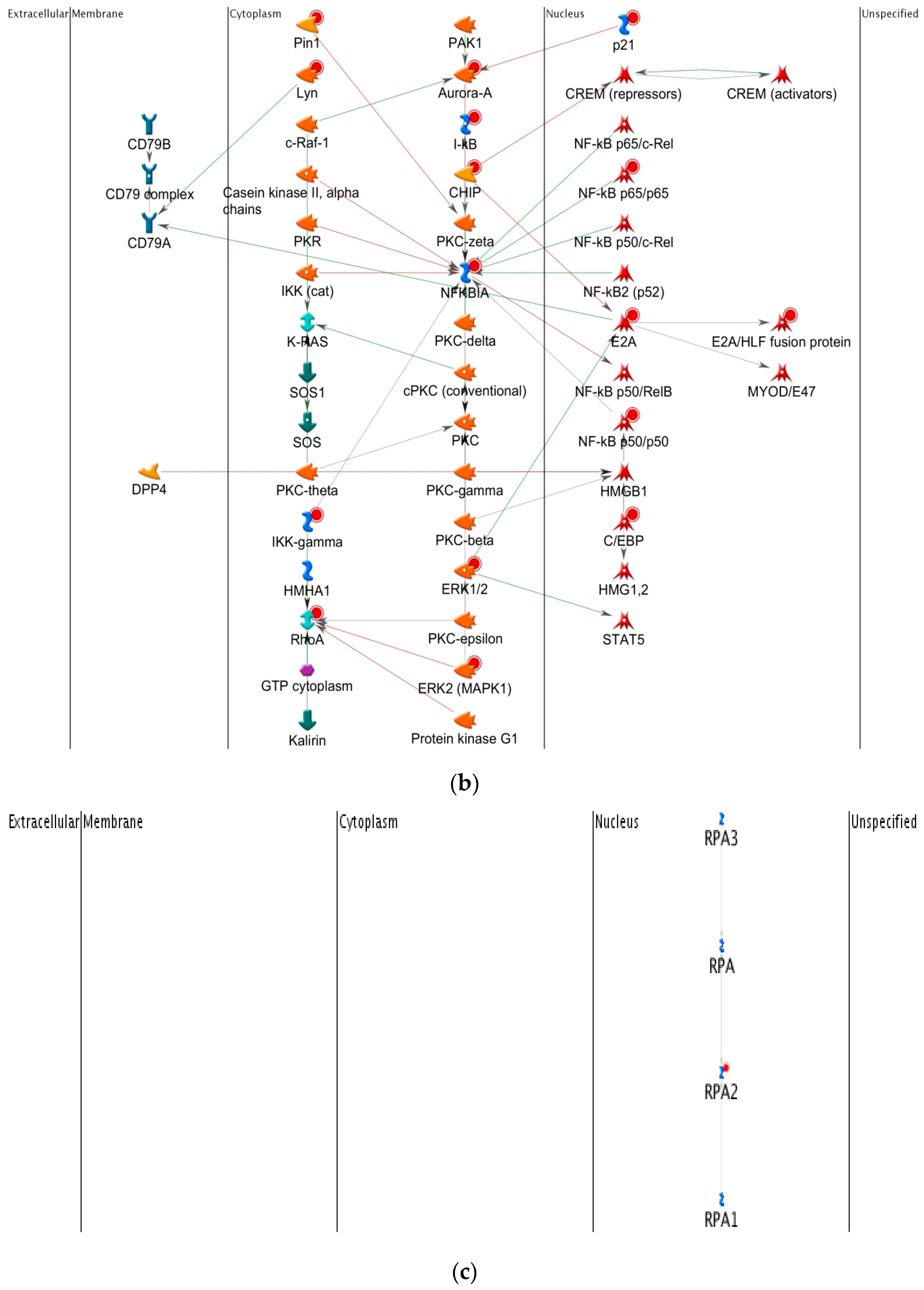
2.4.6. Top-Scoring AN (Analyze Networks Algorithm) Results for the Four Time Points Post-TBI
2.4.7. Discussion of the Cell Cycle Behavior of TBI
2.5. Network Ontology Analysis (NOA) Results



2.6. Summary of the Results in Table 4
| GO: Term | p-Value | Corrected p-Value | R | T | G | O | Term Name |
|---|---|---|---|---|---|---|---|
| 4 h Post-TBI | |||||||
| (1) Biological Processes | |||||||
| GO:0032446 | 1.1 × 10−8 | 1.0 × 10−5 | 14,791 | 26 | 168 | 7 | protein modification by small protein conjugation |
| GO:0070647 | 4.5 × 10−8 | 3.8 × 10−5 | 14,791 | 26 | 204 | 7 | protein modification by small protein conjugation or removal |
| GO:0043687 | 3.7 × 10−7 | 3.1 × 10−4 | 14,791 | 26 | 1243 | 12 | post-translational protein modification |
| GO:0048518 | 5.1 × 10−7 | 4.4 × 10−4 | 14,791 | 26 | 2188 | 15 | positive regulation of biological process |
| GO:0043067 | 8.5 × 10−7 | 7.2 × 10−4 | 14,791 | 26 | 850 | 10 | regulation of programmed cell death |
| GO:0010941 | 9.0 × 10−7 | 7.7 × 10−4 | 14,791 | 26 | 856 | 10 | regulation of cell death |
| GO:0048522 | 1.1 × 10−6 | 9.8 × 10−4 | 14,791 | 26 | 1984 | 14 | positive regulation of cellular process |
| GO:0042221 | 1.3 × 10−6 | 0.0011 | 14,791 | 26 | 1402 | 12 | response to chemical stimulus |
| GO:0009314 | 1.5 × 10−6 | 0.0013 | 14,791 | 26 | 215 | 6 | response to radiation |
| GO:0006464 | 2.6 × 10−6 | 0.0022 | 14,791 | 26 | 1490 | 12 | protein modification process |
| (2) Cellular Components | |||||||
| GO:0043234 | 7.1 × 10−6 | 9.3 × 10−4 | 16,768 | 25 | 2748 | 14 | protein complex |
| GO:0044451 | 1.8 × 10−5 | 0.0024 | 16,768 | 25 | 594 | 7 | nucleoplasm part |
| GO:0005737 | 2.3 × 10−5 | 0.0030 | 16,768 | 25 | 4549 | 17 | cytoplasm |
| GO:0017053 | 3.4 × 10−5 | 0.0045 | 16,768 | 25 | 43 | 3 | transcriptional repressor complex |
| GO:0044428 | 4.4 × 10−5 | 0.0057 | 16,768 | 25 | 1932 | 11 | nuclear part |
| GO:0016235 | 4.4 × 10−5 | 0.0058 | 16,768 | 25 | 7 | 2 | aggresome |
| GO:0032991 | 6.5 × 10−5 | 0.0085 | 16,768 | 25 | 3312 | 14 | macromolecular complex |
| GO:0035098 | 7.6 × 10−5 | 0.0100 | 16,768 | 25 | 9 | 2 | ESC/E(Z) complex (Extra Sex Combs/Enhancer of Zeste complex) |
| GO:0034708 | 7.7 × 10−5 | 0.0100 | 16,768 | 25 | 56 | 3 | methyltransferase complex |
| GO:0035097 | 7.7 × 10−5 | 0.0100 | 16,768 | 25 | 56 | 3 | histone methyltransferase complex |
| (3) Molecular Functions | |||||||
| GO:0005515 | 7.5 × 10−7 | 1.2 × 10−4 | 15,767 | 26 | 8097 | 25 | protein binding |
| GO:0019899 | 2.9 × 10−6 | 4.8 × 10−4 | 15,767 | 26 | 584 | 8 | enzyme binding |
| GO:0050815 | 1.5 × 10−5 | 0.0025 | 15,767 | 26 | 4 | 2 | phosphoserine binding |
| GO:0032403 | 4.2 × 10−5 | 0.0069 | 15,767 | 26 | 243 | 5 | protein complex binding |
| GO:0016563 | 4.9 × 10−5 | 0.0082 | 15,767 | 26 | 419 | 6 | transcription activator activity |
| GO:0042802 | 8.3 × 10−5 | 0.0139 | 15,767 | 26 | 677 | 7 | identical protein binding |
| GO:0016564 | 1.7 × 10−4 | 0.0292 | 15,767 | 26 | 329 | 5 | transcription repressor activity |
| GO:0045309 | 2.7 × 10−4 | 0.0449 | 15,767 | 26 | 15 | 2 | protein phosphorylated amino acid binding |
| GO:0004407 | 3.0 × 10−4 | 0.0513 | 15,767 | 26 | 16 | 2 | histone deacetylase activity |
| GO:0033558 | 3.5 × 10−4 | 0.0581 | 15,767 | 26 | 17 | 2 | protein deacetylase activity |
| 8 h Post-TBI | |||||||
| (1) Biological Processes | |||||||
| GO:0031325 | 4.7 × 10−11 | 5.9 × 10−8 | 14,791 | 48 | 965 | 19 | positive regulation of cellular metabolic process |
| GO:0009893 | 1.1 × 10−10 | 1.4 × 10−7 | 14,791 | 48 | 1015 | 19 | positive regulation of metabolic process |
| GO:0048523 | 1.9 × 10−10 | 2.4 × 10−7 | 14,791 | 48 | 1815 | 24 | negative regulation of cellular process |
| GO:0042127 | 4.4 × 10−10 | 5.6 × 10−7 | 14,791 | 48 | 839 | 17 | regulation of cell proliferation |
| GO:0031328 | 5.0 × 10−10 | 6.3 × 10−7 | 14,791 | 48 | 727 | 16 | positive regulation of cellular biosynthetic process |
| GO:0009891 | 6.5 × 10−10 | 8.2 × 10−7 | 14,791 | 48 | 740 | 16 | positive regulation of biosynthetic process |
| GO:0048519 | 1.2 × 10−9 | 1.5 × 10−6 | 14,791 | 48 | 1983 | 24 | negative regulation of biological process |
| GO:0048518 | 1.5 × 10−9 | 1.8 × 10−6 | 14,791 | 48 | 2188 | 25 | positive regulation of biological process |
| GO:0051173 | 2.0 × 10−9 | 2.6 × 10−6 | 14,791 | 48 | 683 | 15 | positive regulation of nitrogen compound metabolic process |
| GO:0031323 | 2.2 × 10−9 | 2.8 × 10−6 | 14,791 | 48 | 3768 | 32 | regulation of cellular metabolic process |
| (2) Cellular Components | |||||||
| GO:0005634 | 4.9 × 10−11 | 8.3 × 10−9 | 16768 | 45 | 5037 | 35 | nucleus |
| GO:0044428 | 7.5 × 10−11 | 1.2 × 10−8 | 16,768 | 45 | 1932 | 23 | nuclear part |
| GO:0032991 | 1.1 × 10−7 | 1.8 × 10−5 | 16,768 | 45 | 3312 | 25 | macromolecular complex |
| GO:0005737 | 1.8 × 10−7 | 3.0 × 10−5 | 16,768 | 45 | 4549 | 29 | cytoplasm |
| GO:0043231 | 3.3 × 10−7 | 5.6 × 10−5 | 16,768 | 45 | 7996 | 38 | intracellular membrane-bounded organelle |
| GO:0043227 | 3.3 × 10−7 | 5.6 × 10−5 | 16,768 | 45 | 7998 | 38 | membrane-bounded organelle |
| GO:0005829 | 5.5 × 10−7 | 9.2 × 10−5 | 16,768 | 45 | 1269 | 15 | cytosol |
| GO:0043229 | 1.1 × 10−6 | 1.8 × 10−4 | 16,768 | 45 | 8759 | 39 | intracellular organelle |
| GO:0043226 | 1.1 × 10−6 | 1.9 × 10−4 | 16,768 | 45 | 8773 | 39 | organelle |
| GO:0044424 | 1.6 × 10−6 | 2.8 × 10−4 | 16,768 | 45 | 11,001 | 43 | intracellular part |
| (3) Molecular Functions | |||||||
| GO:0005515 | 1.2 × 10−11 | 3.1 × 10−9 | 15,767 | 48 | 8097 | 46 | protein binding |
| GO:0010843 | 4.0 × 10−11 | 9.9 × 10−9 | 15,767 | 48 | 111 | 9 | promoter binding |
| GO:0044212 | 5.6 × 10−11 | 1.3 × 10−8 | 15,767 | 48 | 115 | 9 | DNA regulatory region binding |
| GO:0016563 | 4.1 × 10−7 | 1.0 × 10−4 | 15,767 | 48 | 419 | 10 | transcription activator activity |
| GO:0042802 | 5.9 × 10−7 | 1.4 × 10−4 | 15,767 | 48 | 677 | 12 | identical protein binding |
| GO:0003690 | 6.2 × 10−7 | 1.5 × 10−4 | 15,767 | 48 | 102 | 6 | double-stranded DNA binding |
| GO:0035326 | 5.7 × 10−6 | 0.0014 | 15,767 | 48 | 39 | 4 | enhancer binding |
| GO:0003705 | 5.7 × 10−6 | 0.0014 | 15,767 | 48 | 39 | 4 | RNA polymerase II transcription factor activity, enhancer binding |
| GO:0043566 | 6.1 × 10−6 | 0.0015 | 15,767 | 48 | 151 | 6 | structure-specific DNA binding |
| GO:0032403 | 8.1 × 10−6 | 0.0019 | 15,767 | 48 | 243 | 7 | protein complex binding |
| 24 h Post-TBI | |||||||
| (1) Biological Processes | |||||||
| GO:0048522 | 4.0 × 10−10 | 4.8 × 10−7 | 14,791 | 46 | 1984 | 24 | positive regulation of cellular process |
| GO:0048518 | 4.5 × 10−10 | 5.4 × 10−7 | 14,791 | 46 | 2188 | 25 | positive regulation of biological process |
| GO:0009987 | 9.8 × 10−9 | 1.1 × 10−5 | 14,791 | 46 | 9216 | 45 | cellular process |
| GO:0050794 | 1.3 × 10−8 | 1.5 × 10−5 | 14,791 | 46 | 6896 | 40 | regulation of cellular process |
| GO:0031325 | 1.4 × 10−8 | 1.7 × 10−5 | 14,791 | 46 | 965 | 16 | positive regulation of cellular metabolic process |
| GO:0051716 | 1.9 × 10−8 | 2.3 × 10−5 | 14,791 | 46 | 847 | 15 | cellular response to stimulus |
| GO:0048523 | 2.1 × 10−8 | 2.5 × 10−5 | 14,791 | 46 | 1815 | 21 | negative regulation of cellular process |
| GO:0009893 | 3.0 × 10−8 | 3.5 × 10−5 | 14,791 | 46 | 1015 | 16 | positive regulation of metabolic process |
| GO:0044260 | 4.7 × 10−8 | 5.6 × 10−5 | 14,791 | 46 | 3428 | 28 | cellular macromolecule metabolic process |
| GO:0051128 | 4.9 × 10−8 | 5.9 × 10−5 | 14,791 | 46 | 529 | 12 | regulation of cellular component organization |
| (2) Cellular Components | |||||||
| GO:0044424 | 5.6 × 10−9 | 1.0 × 10−6 | 16,768 | 45 | 11,001 | 45 | intracellular part |
| GO:0005829 | 1.1 × 10−8 | 2.1 × 10−6 | 16,768 | 45 | 1269 | 17 | cytosol |
| GO:0005737 | 1.8 × 10−7 | 3.4 × 10−5 | 16,768 | 45 | 4549 | 29 | cytoplasm |
| GO:0043234 | 4.1 × 10−7 | 7.9 × 10−5 | 16,768 | 45 | 2748 | 22 | protein complex |
| GO:0005634 | 4.2 × 10−7 | 8.0 × 10−5 | 16,768 | 45 | 5037 | 30 | nucleus |
| GO:0032991 | 5.5 × 10−7 | 1.0 × 10−4 | 16,768 | 45 | 3312 | 24 | macromolecular complex |
| GO:0044428 | 4.7 × 10−6 | 9.1 × 10−4 | 16,768 | 45 | 1932 | 17 | nuclear part |
| GO:0000307 | 6.4 × 10−6 | 0.0012 | 16,768 | 45 | 14 | 3 | cyclin-dependent protein kinase holoenzyme complex |
| GO:0070435 | 7.0 × 10−6 | 0.0013 | 16,768 | 45 | 2 | 2 | Shc-EGFR complex (Src homology 2 domain containing transforming protein-epidermal growth factor receptor complex) |
| GO:0043229 | 2.6 × 10−5 | 0.0051 | 16,768 | 45 | 8759 | 37 | intracellular organelle |
| (3) Molecular Functions | |||||||
| GO:0005515 | 6.4 × 10−10 | 1.7 × 10−7 | 15,767 | 46 | 8097 | 43 | protein binding |
| GO:0005057 | 4.0 × 10−7 | 1.1 × 10−4 | 15,767 | 46 | 162 | 7 | receptor signaling protein activity |
| GO:0043560 | 5.0 × 10−6 | 0.0013 | 15,767 | 46 | 12 | 3 | insulin receptor substrate binding |
| GO:0019899 | 5.4 × 10−6 | 0.0015 | 15,767 | 46 | 584 | 10 | enzyme binding |
| GO:0032403 | 6.0 × 10−6 | 0.0016 | 15,767 | 46 | 243 | 7 | protein complex binding |
| GO:0004710 | 8.3 × 10−6 | 0.0022 | 15,767 | 46 | 2 | 2 | MAP/ERK kinase kinase activity (mitogen-activated protein kinases/extracellular signal-regulated kinases activity) |
| GO:0045309 | 1.0 × 10−5 | 0.0028 | 15,767 | 46 | 15 | 3 | protein phosphorylated amino acid binding |
| GO:0019900 | 2.4 × 10−5 | 0.0067 | 15,767 | 46 | 201 | 6 | kinase binding |
| GO:0005488 | 3.0 × 10−5 | 0.0083 | 15,767 | 46 | 12,581 | 46 | binding |
| GO:0004702 | 3.1 × 10−5 | 0.0086 | 15,767 | 46 | 62 | 4 | receptor signaling protein serine/threonine kinase activity |
| 72 h Post-TBI | |||||||
| (1) Biological Processes | |||||||
| GO:0048522 | 1.60 × 10−17 | 2.50 × 10−14 | 1.48 × 104 | 56 | 1984 | 35 | positive regulation of cellular process |
| GO:0048518 | 3.80 × 10−17 | 5.60 × 10−14 | 1.48 × 104 | 56 | 2188 | 36 | positive regulation of biological process |
| GO:0031325 | 1.60 × 10−12 | 2.40 × 10−09 | 1.48 × 104 | 56 | 965 | 22 | positive regulation of cellular metabolic process |
| GO:0009893 | 4.50 × 10−12 | 6.70 × 10−09 | 1.48 × 104 | 56 | 1015 | 22 | positive regulation of metabolic process |
| GO:0010604 | 9.20 × 10−12 | 1.30 × 10−08 | 1.48 × 104 | 56 | 939 | 21 | positive regulation of macromolecule metabolic process |
| GO:0032502 | 9.30 × 10−12 | 1.30 × 10−08 | 1.48 × 104 | 56 | 3032 | 35 | developmental process |
| GO:0048519 | 5.20 × 10−11 | 7.70 × 10−08 | 1.48 × 104 | 56 | 1983 | 28 | negative regulation of biological process |
| GO:0006950 | 1.00 × 10−10 | 1.50 × 10−07 | 1.48 × 104 | 56 | 1591 | 25 | response to stress |
| GO:0042981 | 1.00 × 10−10 | 1.50 × 10−07 | 1.48 × 104 | 56 | 842 | 19 | regulation of apoptosis |
| GO:0010033 | 1.20 × 10−10 | 1.80 × 10−07 | 1.48 × 104 | 56 | 850 | 19 | response to organic substance |
| (2) Cellular Components | |||||||
| GO:0005737 | 9.2 × 10−11 | 1.9 × 10−8 | 16,768 | 57 | 4549 | 39 | cytoplasm |
| GO:0043234 | 1.8 × 10−9 | 4.0 × 10−7 | 16,768 | 57 | 2748 | 29 | protein complex |
| GO:0005829 | 2.6 × 10−9 | 5.6 × 10−7 | 16,768 | 57 | 1269 | 20 | cytosol |
| GO:0032991 | 6.7 × 10−9 | 1.4 × 10−6 | 16,768 | 57 | 3312 | 31 | macromolecular complex |
| GO:0044428 | 2.4 × 10−8 | 5.2 × 10−6 | 16,768 | 57 | 1932 | 23 | nuclear part |
| GO:0044424 | 1.6 × 10−7 | 3.5 × 10−5 | 16,768 | 57 | 11,001 | 54 | intracellular part |
| GO:0044446 | 2.0 × 10−7 | 4.4 × 10−5 | 16,768 | 57 | 5015 | 36 | intracellular organelle part |
| GO:0044422 | 2.9 × 10−7 | 6.3 × 10−5 | 16,768 | 57 | 5082 | 36 | organelle part |
| GO:0005634 | 9.0 × 10−7 | 1.9 × 10−4 | 16,768 | 57 | 5037 | 35 | nucleus |
| GO:0070435 | 1.1 × 10−5 | 0.0024 | 16,768 | 57 | 2 | 2 | Shc-EGFR complex |
| (3) Molecular Functions | |||||||
| GO:0005515 | 2.8 × 10−17 | 6.5 × 10−15 | 15,767 | 57 | 8097 | 57 | protein binding |
| GO:0042802 | 8.7 × 10−8 | 1.9 × 10−5 | 15,767 | 57 | 677 | 14 | identical protein binding |
| GO:0019899 | 1.1 × 10−7 | 2.6 × 10−5 | 15,767 | 57 | 584 | 13 | enzyme binding |
| GO:0032403 | 2.4 × 10−6 | 5.5 × 10−4 | 15,767 | 57 | 243 | 8 | protein complex binding |
| GO:0005488 | 2.5 × 10−6 | 5.7 × 10−4 | 15,767 | 57 | 12,581 | 57 | binding |
| GO:0019900 | 7.5 × 10−6 | 0.0017 | 15,767 | 57 | 201 | 7 | kinase binding |
| GO:0016566 | 1.0 × 10−5 | 0.0023 | 15,767 | 57 | 38 | 4 | specific transcriptional repressor activity |
| GO:0019901 | 3.2 × 10−5 | 0.0072 | 15,767 | 57 | 169 | 6 | protein kinase binding |
| GO:0010843 | 4.9 × 10−5 | 0.0112 | 15,767 | 57 | 111 | 5 | promoter binding |
| GO:0044212 | 5.8 × 10−5 | 0.0132 | 15,767 | 57 | 115 | 5 | DNA regulatory region binding |
3. Experimental Section
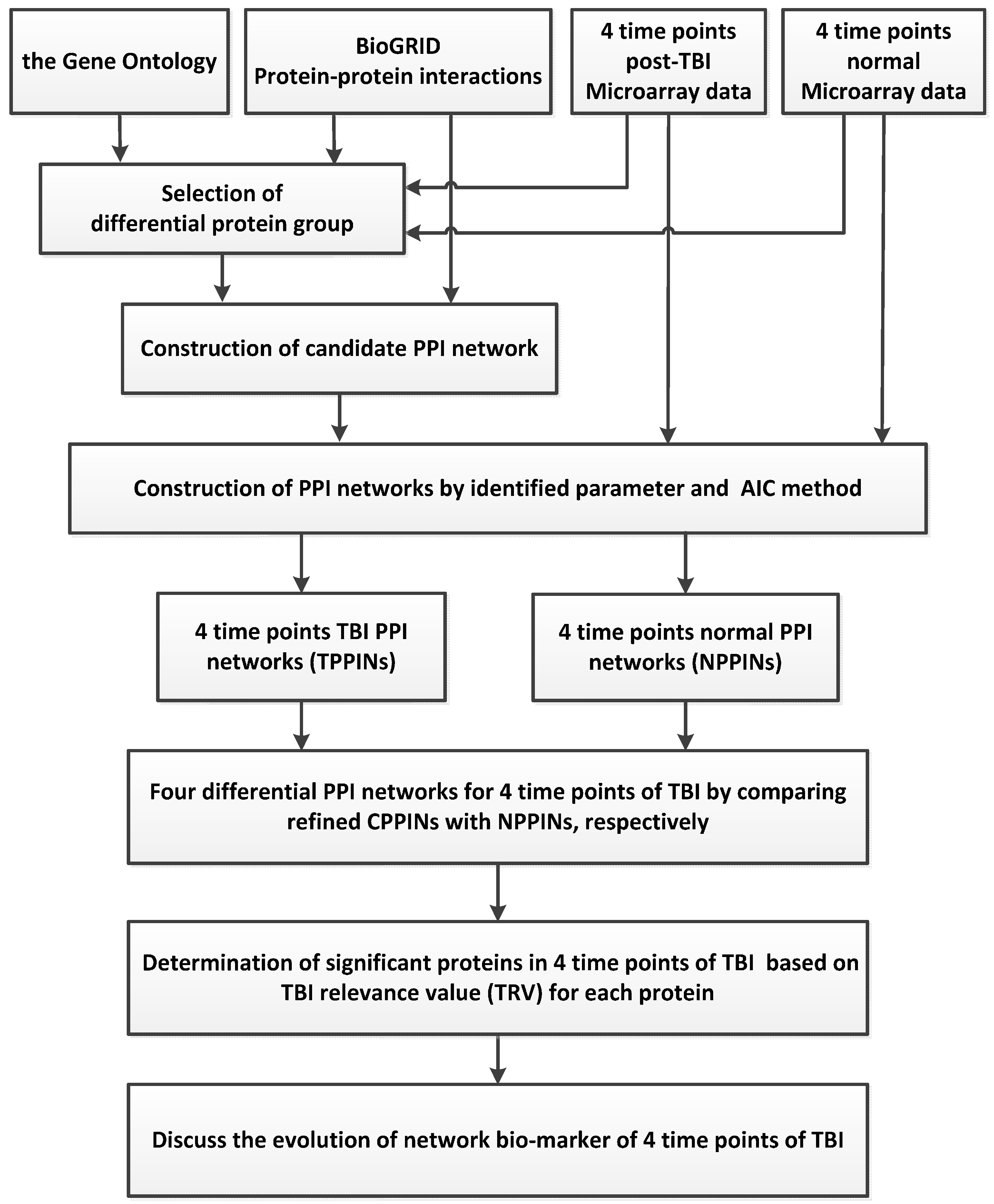
3.1. Overview of the Traumatic Brain Injury (TBI) Network Biomarkers Identification Process

3.2. Data Selection and Pre-Processing
3.3. Selection of the Differential Protein Groups and Identification of PPINs
3.4. Determination of Proteins with Top TBI Relevance Value (TRV)and Their Corresponding Network Structures
3.5. Pathway Analysis
3.6. Software and Databases Used in This Research
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Xiong, Y.; Mahmood, A.; Chopp, M. Animal models of traumatic brain injury. Nat. Rev. Neurosci. 2013, 14, 128–142. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Muneer, P.M.; Chandra, N.; Haorah, J. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol. Neurobiol. 2015, 51, 966–979. [Google Scholar] [CrossRef] [PubMed]
- Glass, T.F.; Fabian, M.J.; Schweitzer, J.B.; Weinberg, J.A.; Proctor, K.G. The impact of hypercarbia on the evolution of brain injury in a porcine model of traumatic brain injury and systemic hemorrhage. J. Neurotrauma 2001, 18, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Porth, C.; Porth, C. Essentials of Pathophysiology : Concepts of Altered Health States, 2nd ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007. [Google Scholar]
- Werner, C.; Engelhard, K. Pathophysiology of traumatic brain injury. Br. J. Anaesth. 2007, 99, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Prins, M.; Greco, T.; Alexander, D.; Giza, C.C. The pathophysiology of traumatic brain injury at a glance. Dis. Model. Mech. 2013, 6, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Bramlett, H.M.; Dietrich, W.D. Pathophysiology of cerebral ischemia and brain trauma: Similarities and differences. J. Cerebr. Blood Flow Metabol. 2004, 24, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, G.A. Neurological diseases in relation to the blood-brain barrier. J. Cerebr. Blood Flow Metabol. 2012, 32, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, K.; Smith, M. Disorders of sodium balance after brain injury. Cont. Educ. Anaesth. Crit. Care Pain 2008, 8, 129–133. [Google Scholar] [CrossRef]
- Hinzman, J.M.; Thomas, T.C.; Burmeister, J.J.; Quintero, J.E.; Huettl, P.; Pomerleau, F.; Gerhardt, G.A.; Lifshitz, J. Diffuse brain injury elevates tonic glutamate levels and potassium-evoked glutamate release in discrete brain regions at two days post-injury: An enzyme-based microelectrode array study. J. Neurotrauma 2010, 27, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Rodriguez, A.; Egea-Guerrero, J.J.; Murillo-Cabezas, F.; Carrillo-Vico, A. Oxidative stress in traumatic brain injury. Curr. Med. Chem. 2014, 21, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Kyritsis, N.; Kizil, C.; Zocher, S.; Kroehne, V.; Kaslin, J.; Freudenreich, D.; Iltzsche, A.; Brand, M. Acute Inflammation Initiates the Regenerative Response in the Adult Zebrafish Brain. Science 2012, 338, 1353–1356. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Chen, B.S. A network-based biomarker approach for molecular investigation and diagnosis of lung cancer. BMC Med. Genom. 2011, 4. [Google Scholar] [CrossRef] [PubMed]
- Kobeissy, F.H.; Sadasivan, S.; Oli, M.W.; Robinson, G.; Larner, S.F.; Zhang, Z.; Hayes, R.L.; Wang, K.K. Neuroproteomics and systems biology-based discovery of protein biomarkers for traumatic brain injury and clinical validation. Proteom. Clin. Appl. 2008, 2, 1467–1483. [Google Scholar] [CrossRef] [PubMed]
- Boutte, A.M.; Deng-Bryant, Y.; Johnson, D.; Tortella, F.C.; Dave, J.R.; Shear, D.A.; Schmid, K.E. serum glial fibrillary acidic protein predicts tissue glial fibrillary acidic protein break-down products and therapeutic efficacy after penetrating ballistic-like brain injury. J. Neurotrauma 2015. [Google Scholar] [CrossRef] [PubMed]
- Natale, J.E.; Ahmed, F.; Cernak, I.; Stoica, B.; Faden, A.I. Gene expression profile changes are commonly modulated across models and species after traumatic brain injury. J. Neurotrauma 2003, 20, 907–927. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.C.C.; Chen, K.Y.; Yo, Y.W.; Huang, S.W.; Shih, H.M.; Chiu, W.T.; Chiang, Y.H.; Shiau, C.Y. Different sham procedures for rats in traumatic brain injury experiments induce corresponding increases in levels of trauma markers. J. Surg. Res. 2013, 179, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.S.; Yang, S.K.; Lan, C.Y.; Chuang, Y.J. A systems biology approach to construct the gene regulatory network of systemic inflammation via microarray and databases mining. BMC Med. Genom. 2008, 1, 46. [Google Scholar] [CrossRef] [PubMed]
- White, T.E.; Ford, G.D.; Surles-Zeigler, M.C.; Gates, A.S.; Laplaca, M.C.; Ford, B.D. Gene expression patterns following unilateral traumatic brain injury reveals a local pro-inflammatory and remote anti-inflammatory response. BMC Genom. 2013, 14, 282. [Google Scholar] [CrossRef] [PubMed]
- Fulmer, T. Taking the inflammation out of epilepsy. Sci. Bus. X Chang. 2009, 2. [Google Scholar] [CrossRef]
- Zhang, Z.P. The Relations between the Expression of Bad, CDK2, STAT3 and Brain Fuctions after Traumatic Brain Injury in Rats. Master’s Thesis, Kunming Medical College, Kunming, China, 2010. Avalible online: http://www.dissertationtopic.net/doc/651281 (accessed on 22 May 2015). [Google Scholar]
- Lafuente, J.V.; Argandona, E.G.; Mitre, B. VEGFR-2 expression in brain injury: Its distribution related to brain-blood barrier markers. J. Neural Transm. 2006, 113, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.-H.; Wu, C.C.; Lai, H.-Y.; Jheng, B.-R.; Weng, H.-Y.; Chang, T.-H.; Chen, B.-S. Identification of network-based biomarker identification of cardioembolic stroke using a systems biology approach with time series data. BMC Syst. Biol. 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Gene: App (Amyloid β (A4) Precursor Protein) Rattus Norvegicus. Available online: http://rgd.mcw.edu/rgdweb/report/gene/main.html?id=2139 (accessed on 22 March 2015).
- Shimoyama, M.; De Pons, J.; Hayman, G.T.; Laulederkind, S.J.; Liu, W.; Nigam, R.; Petri, V.; Smith, J.R.; Tutaj, M.; Wang, S.J.; et al. The Rat Genome Database 2015: Genomic, phenotypic and environmental variations and disease. Nucleic Acids Res. 2015, 43, D743–D750. [Google Scholar] [CrossRef] [PubMed]
- Gene: NEDD8 (Neural Precursor Cell Expressed, Developmentally Down-Regulated 8) Homo Sapiens. Available online: http://rgd.mcw.edu/rgdweb/report/gene/main.html?id=735521 (accessed on 5 January 2015).
- Skliris, A.; Papadaki, O.; Kafasla, P.; Karakasiliotis, I.; Hazapis, O.; Reczko, M.; Grammenoudi, S.; Bauer, J.; Kontoyiannis, D. Neuroprotection requires the functions of the RNA-binding protein HuR. Cell Death Differ. 2015, 22, 703–718. [Google Scholar] [CrossRef] [PubMed]
- Ottens, A.K.; Bustamante, L.; Golden, E.C.; Yao, C.; Hayes, R.L.; Wang, K.K.; Tortella, F.C.; Dave, J.R. Neuroproteomics: A biochemical means to discriminate the extent and modality of brain injury. J. Neurotrauma 2010, 27, 1837–1852. [Google Scholar] [CrossRef] [PubMed]
- Di Giovanni, S.; Movsesyan, V.; Ahmed, F.; Cernak, I.; Schinelli, S.; Stoica, B.; Faden, A.I. Cell cycle inhibition provides neuroprotection and reduces glial proliferation and scar formation after traumatic brain injury. Proc. Natl. Acad. Sci. USA 2005, 102, 8333–8338. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Stoica, B.A.; Faden, A.I. Cell cycle activation and spinal cord injury. Neurother. J. Am. Soc. Exp. Neuro Ther. 2011, 8, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.H.; Chen, R.H.; Chen, B.S. Core and specific network markers of carcinogenesis from multiple cancer samples. J. Theor. Biol. 2014, 362, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.H.; Li, C.W.; Chen, B.S. Evolution of network biomarkers from early to late stage bladder cancer samples. Biomed. Res. Int. 2014, 2014, 159078. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.-H.; Wu, C.-C.; Lai, H.-Y.; Jheng, B.-R.; Weng, H.-Y.; Chang, T.-H.; Chen, B.-S. Identification of network-based biomarkers of cardioembolic stroke using a systems biology approach with time series data. BMC Genom. 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Stamova, B.; Jickling, G.C.; Ander, B.P.; Zhan, X.; Liu, D.; Turner, R.; Ho, C.; Khoury, J.C.; Bushnell, C.; Pancioli, A.; et al. Gene expression in peripheral immune cells following cardioembolic stroke is sexually dimorphic. PLoS ONE 2014, 9, e102550. [Google Scholar] [CrossRef] [PubMed]
- Chatr-aryamontri, A.; Breitkreutz, B.J.; Oughtred, R.; Boucher, L.; Heinicke, S.; Chen, D.C.; Stark, C.; Breitkreutz, A.; Kolas, N.; O’Donnell, L.; et al. The BioGRID interaction database: 2015 update. Nucleic Acids Res. 2015, 43, D470–D478. [Google Scholar] [CrossRef] [PubMed]
- Johansson, R. System Modeling and Identification. 1993. Avalible online: http://www.citeulike.org/group/8224/article/3853014 (accessed on 22 May 2015).
- Pagano, M.; Gauvreau, K. Principles of Biostatistics; Duxbury Press: Pacific Grove, CA, USA, 2000. [Google Scholar]
- Kanehisa, M. Molecular network analysis of diseases and drugs in KEGG. Methods Mol. Biol. 2013, 939, 263–275. [Google Scholar] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huang, Q.; Liu, Z.P.; Wang, Y.; Wu, L.Y.; Chen, L.; Zhang, X.S. NOA: A novel Network Ontology Analysis method. Nucleic Acids Res. 2011, 39, e87. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, J.; Hanspers, K.; Xu, D.; Chen, L.; Pico, A.R. NOA: A cytoscape plugin for network ontology analysis. Bioinformatics 2013, 29, 2066–2067. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.H.; Chen, B.S. Construction of a cancer-perturbed protein-protein interaction network for discovery of apoptosis drug targets. BMC Syst. Biol. 2008, 2. [Google Scholar] [CrossRef] [PubMed]
- MetaCore Advanced Training Manual. Available online: https://portal.genego.com/help/MetaCore_Advanced_Training_Manual_5.0.pdf (accessed on 21 May 2015).
- MetaCore Training Manual. Available online: https://portal.genego.com/help/MetaCore_bio_manual_5.0.pdf (accessed on 21 May 2015).
- Lopes, C.T.; Franz, M.; Kazi, F.; Donaldson, S.L.; Morris, Q.; Bader, G.D. Cytoscape Web: An interactive web-based network browser. Bioinformatics 2010, 26, 2347–2348. [Google Scholar] [CrossRef] [PubMed]
- GO Database. Available online: http://geneontology.org/page/go-database (accessed on 21 May 2015).
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, Y.-H.; Wu, C.-C.; Wu, J.C.-C.; Lai, H.-Y.; Chen, K.-Y.; Jheng, B.-R.; Chen, M.-C.; Chang, T.-H.; Chen, B.-S. Temporal Genetic Modifications after Controlled Cortical Impact—Understanding Traumatic Brain Injury through a Systematic Network Approach. Int. J. Mol. Sci. 2016, 17, 216. https://doi.org/10.3390/ijms17020216
Wong Y-H, Wu C-C, Wu JC-C, Lai H-Y, Chen K-Y, Jheng B-R, Chen M-C, Chang T-H, Chen B-S. Temporal Genetic Modifications after Controlled Cortical Impact—Understanding Traumatic Brain Injury through a Systematic Network Approach. International Journal of Molecular Sciences. 2016; 17(2):216. https://doi.org/10.3390/ijms17020216
Chicago/Turabian StyleWong, Yung-Hao, Chia-Chou Wu, John Chung-Che Wu, Hsien-Yong Lai, Kai-Yun Chen, Bo-Ren Jheng, Mien-Cheng Chen, Tzu-Hao Chang, and Bor-Sen Chen. 2016. "Temporal Genetic Modifications after Controlled Cortical Impact—Understanding Traumatic Brain Injury through a Systematic Network Approach" International Journal of Molecular Sciences 17, no. 2: 216. https://doi.org/10.3390/ijms17020216
APA StyleWong, Y.-H., Wu, C.-C., Wu, J. C.-C., Lai, H.-Y., Chen, K.-Y., Jheng, B.-R., Chen, M.-C., Chang, T.-H., & Chen, B.-S. (2016). Temporal Genetic Modifications after Controlled Cortical Impact—Understanding Traumatic Brain Injury through a Systematic Network Approach. International Journal of Molecular Sciences, 17(2), 216. https://doi.org/10.3390/ijms17020216