Sanger Sequencing for BRCA1 c.68_69del, BRCA1 c.5266dup and BRCA2 c.5946del Mutation Screen on Pap Smear Cytology Samples

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
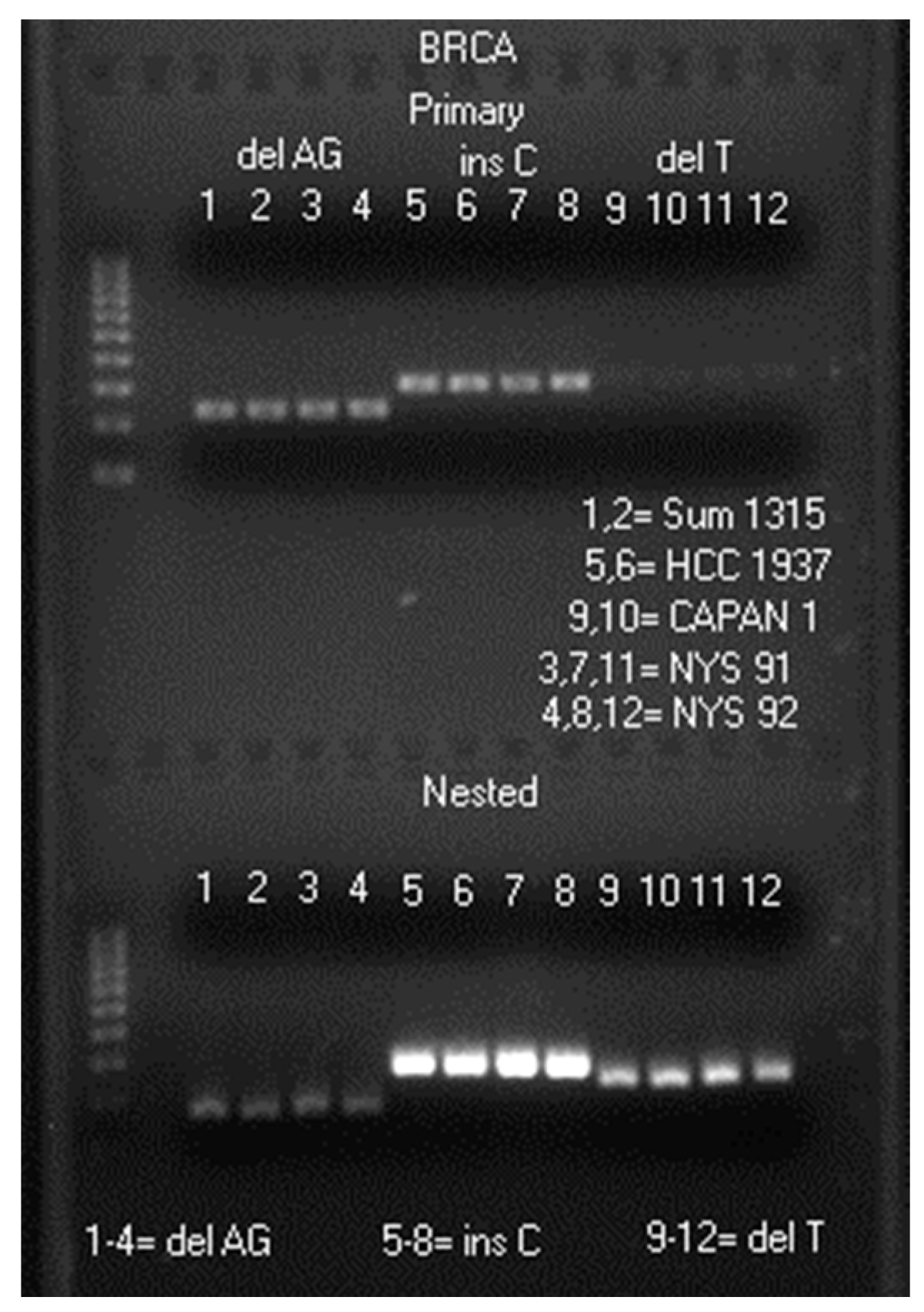
2.1. LoTemp® Nested PCR Amplicons as Sequencing Templates

2.2. Routine HPV Genotyping by Partial Sanger Sequencing of the L1 Gene

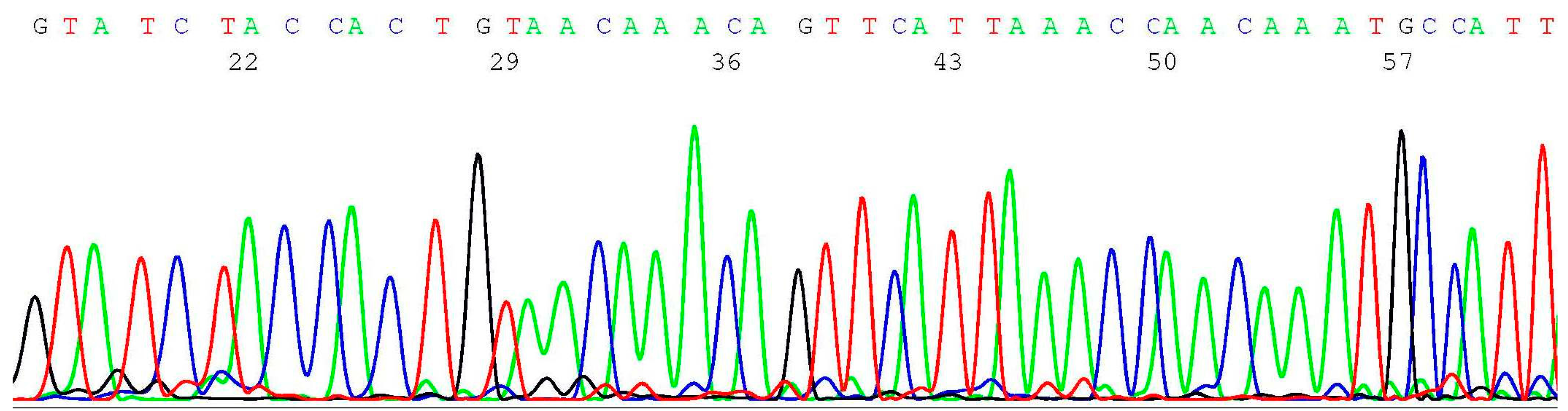
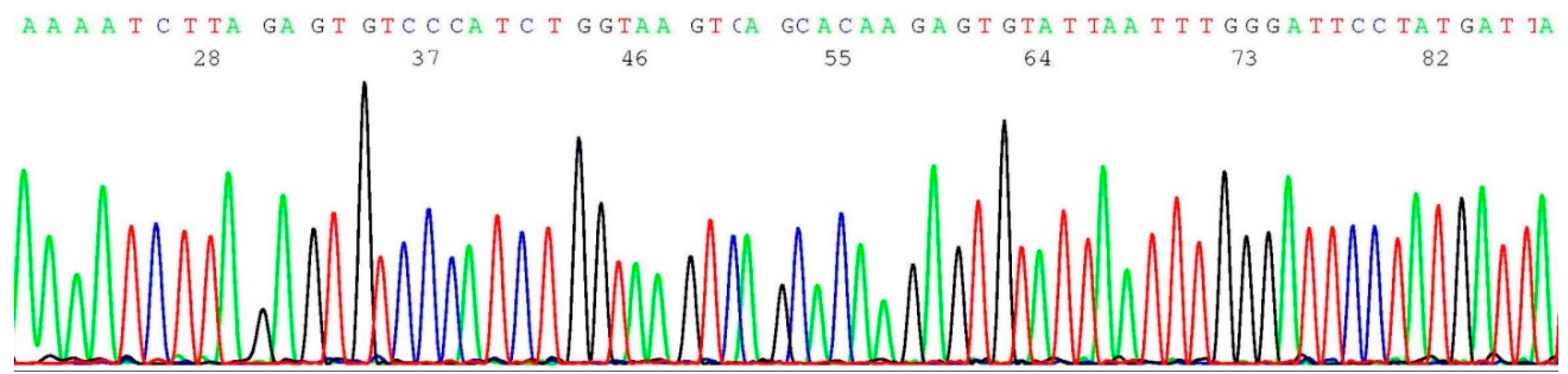
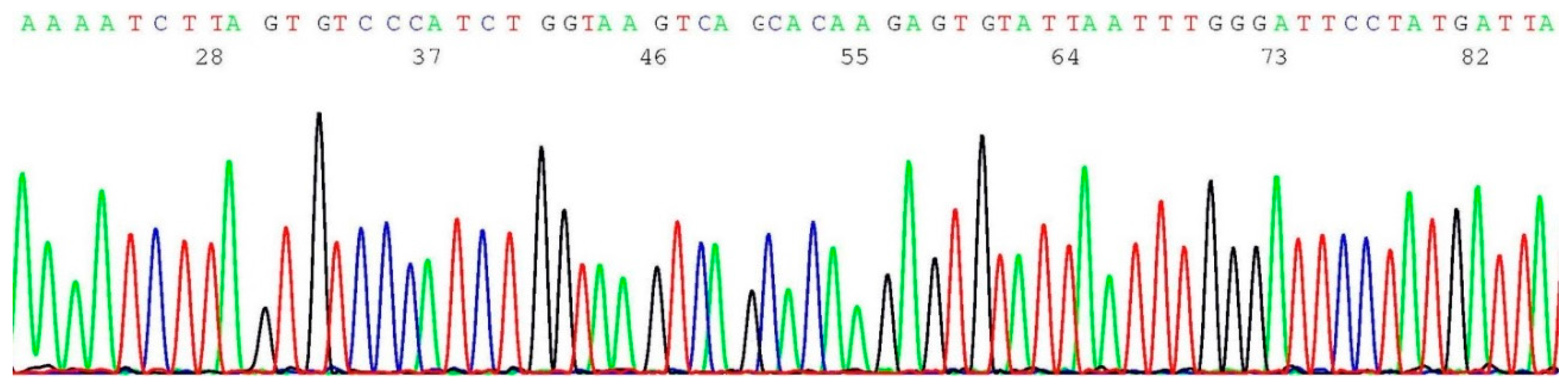
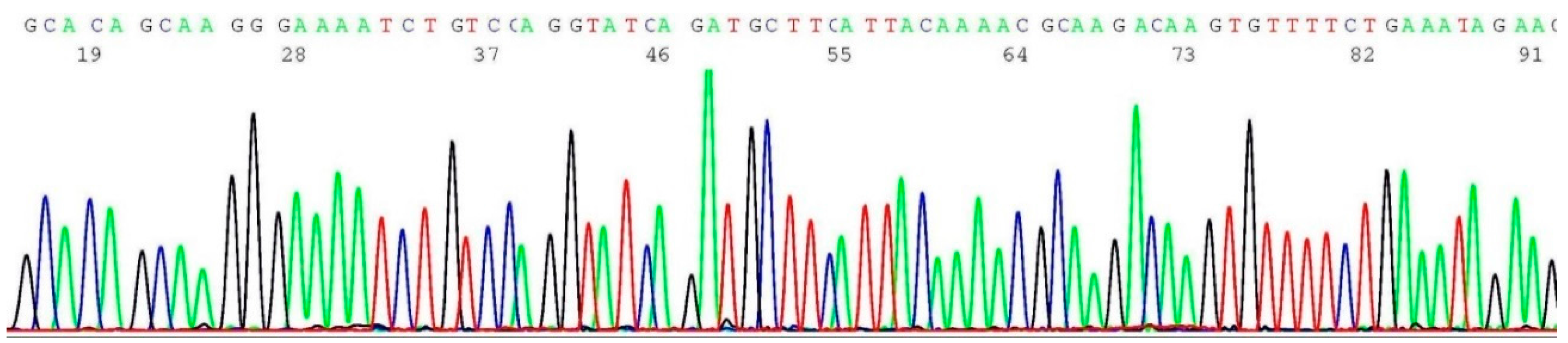
2.3. Documentation of Wild-Type Sequence without BRCA1 c.68_69del Mutation

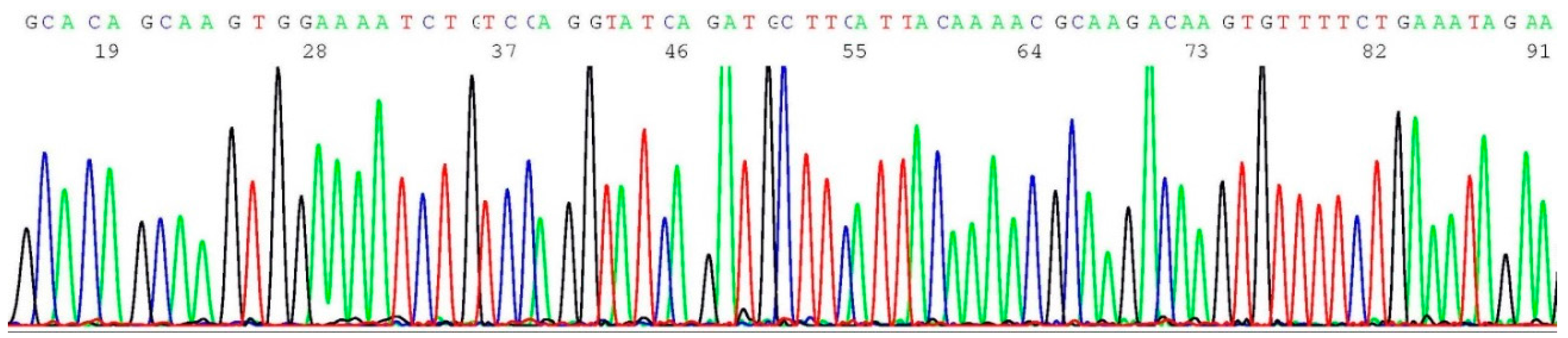
2.4. Validation of BRCA1 c.68_69del Mutation by Sanger Sequencing

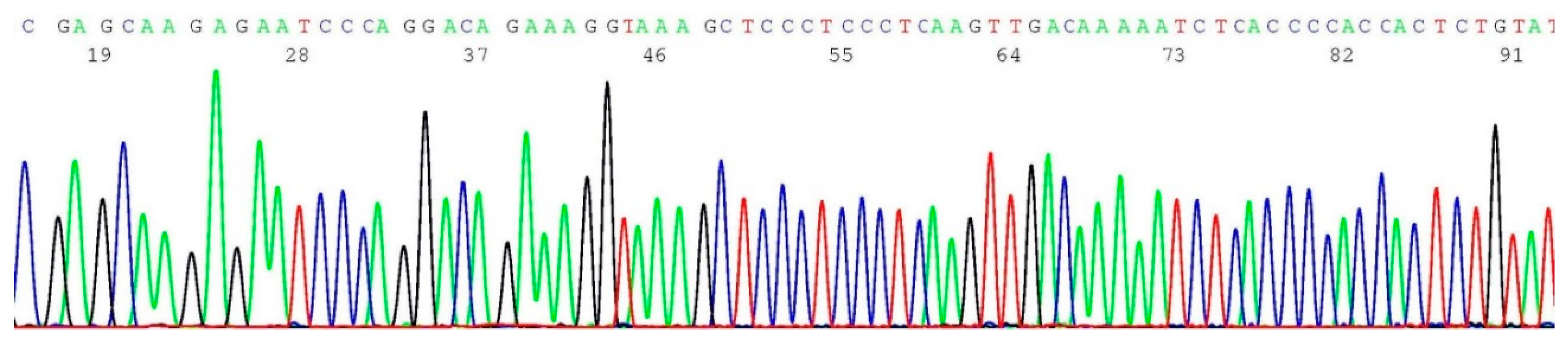
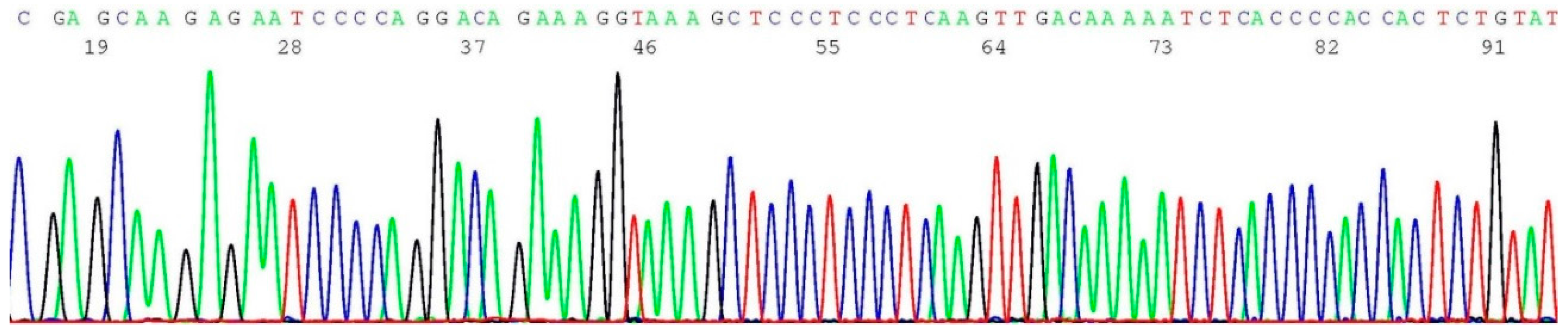
2.5. Documentation of Wild-Type Sequence without BRCA1 c.5266dup Mutation

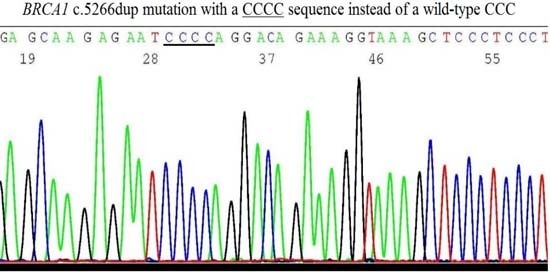
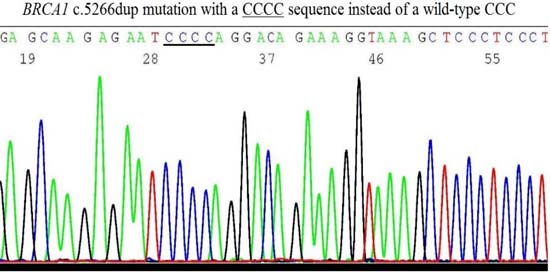
2.6. Validation of BRCA1 c.5266dup Mutation by Sanger Sequencing

2.7. Documentation of Wild-Type Sequence without BRCA2 c.5946del Mutation

2.8. Validation of BRCA2 c.5946del Mutation by Sanger Sequencing

2.9. Challenges of Translating Science into Clinical Practice
3. Experimental Section
3.1. Sources of Materials
3.2. Cell Digestion
3.3. Preparation of Heminested PCR Amplicons for DNA Sequencing
3.4. DNA Sequencing
3.5. Cross Contamination Control
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Breastcancer.org. US Breast Cancer Statistics. Available online: http://www.breastcancer.org/symptoms/understand_bc/statistics (accessed on 5 February 2016).
- Cancer.Net. Ovarian Cancer: Statistics. Available online: http://www.cancer.net/cancer-types/ovarian-cancer/statistics (accessed on 5 February 2016).
- Domchek, S.M.; Friebel, T.M.; Singer, C.F.; Evans, D.G.; Lynch, H.T.; Isaacs, C.; Garber, J.E.; Neuhausen, S.L.; Matloff, E.; Eeles, R.; et al. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA 2010, 304, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Schenberg, T.; Mitchell, G. Prophylactic bilateral salpingectomy as a prevention strategy in women at high-risk of ovarian cancer: A mini-review. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, L.C.; Sellers, T.A.; Schaid, D.J.; Frank, T.S.; Soderberg, C.L.; Sitta, D.L.; Frost, M.H.; Grant, C.S.; Donohue, J.H.; Woods, J.E.; et al. Efficacy of bilateral prophylactic mastectomy in BRCA1 and BRCA2 gene mutation carriers. J. Natl. Cancer Inst. 2001, 93, 1633–1637. [Google Scholar] [CrossRef] [PubMed]
- Domchek, S.; Weber, B.L. Genetic variants of uncertain significance: Flies in the ointment. J. Clin. Oncol. 2008, 26, 16–17. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.; Battelli, C.; Allen, B.; Kaldate, R.; Bhatnagar, S.; Bowles, K.; Timms, K.; Garber, J.E.; Herold, C.; Ellisen, L.; et al. Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer 2015, 121, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Trujillano, D.; Weiss, M.E.; Schneider, J.; Köster, J.; Papachristos, E.B.; Saviouk, V.; Zakharkina, T.; Nahavandi, N.; Kovacevic, L.; Rolfs, A. Next-generation sequencing of the BRCA1 and BRCA2 genes for the genetic diagnostics of hereditary breast and/or ovarian cancer. J. Mol. Diagn. 2015, 17, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Judkins, T.; Leclair, B.; Bowles, K.; Gutin, N.; Trost, J.; McCulloch, J.; Bhatnagar, S.; Murray, A.; Craft, J.; Wardell, B.; et al. Development and analytical validation of a 25-gene next generation sequencing panel that includes the BRCA1 and BRCA2 genes to assess hereditary cancer risk. BMC Cancer 2015, 15. [Google Scholar] [CrossRef] [PubMed]
- D’Argenio, V.; Esposito, M.V.; Telese, A.; Precone, V.; Starnone, F.; Nunziato, M.; Cantiello, P.; Iorio, M.; Evangelista, E.; D’Aiuto, M.; et al. The molecular analysis of BRCA1 and BRCA2: Next-generation sequencing supersedes conventional approaches. Clin. Chim. Acta 2015, 446, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Long, E.F.; Ganz, P.A. Cost-effectiveness of Universal BRCA1/2 Screening: Evidence-Based Decision Making. JAMA Oncol. 2015, 1, 1217–1218. [Google Scholar] [CrossRef] [PubMed]
- King, M.C.; Levy-Lahad, E.; Lahad, A. Population-based screening for BRCA1 and BRCA2: 2014 Lasker Award. JAMA 2014, 312, 1091–1092. [Google Scholar] [CrossRef] [PubMed]
- Frank, T.S.; Deffenbaugh, A.M.; Reid, J.E.; Hulick, M.; Ward, B.E.; Lingenfelter, B.; Gumpper, K.L.; Scholl, T.; Tavtigian, S.V.; Pruss, D.R.; et al. Clinical characteristics of individuals with germline mutations in BRCA1 and BRCA2: Analysis of 10,000 individuals. J. Clin. Oncol. 2002, 20, 1480–1490. [Google Scholar] [CrossRef] [PubMed]
- Gabai-Kapara, E.; Lahad, A.; Kaufman, B.; Friedman, E.; Segev, S.; Renbaum, P.; Beeri, R.; Gal, M.; Grinshpun-Cohen, J.; Djemal, K.; et al. Population-based screening for breast and ovarian cancer risk due to BRCA1 and BRCA2. Proc. Natl. Acad. Sci. USA 2014, 111, 14205–14210. [Google Scholar] [CrossRef] [PubMed]
- McClain, M.R.; Nathanson, K.L.; Palomaki, G.E.; Haddow, J.E. An evaluation of BRCA1 and BRCA2 founder mutations penetrance estimates for breast cancer among Ashkenazi Jewish women. Genet. Med. 2005, 7, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Manchanda, R.; Loggenberg, K.; Sanderson, S.; Burnell, M.; Wardle, J.; Gessler, S.; Side, L.; Balogun, N.; Desai, R.; Kumar, A.; et al. Population testing for cancer predisposing BRCA1/BRCA2 mutations in the Ashkenazi-Jewish community: A randomized controlled trial. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Levy-Lahad, E.; Lahad, A.; King, M.C. Precision medicine meets public health: Population screening for BRCA1 and BRCA2. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- FitzGerald, M.G.; MacDonald, D.J.; Krainer, M.; Hoover, I.; O’Neil, E.; Unsal, H.; Silva-Arrieto, S.; Finkelstein, D.M.; Beer-Romero, P.; Englert, C.; et al. Germ-line BRCA1 mutations in Jewish and non-Jewish women with early-onset breast cancer. N. Engl. J. Med. 1996, 334, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Negură, L.; Artenie, V.; Carasevici, E.; Negură, A. Multiplex-PCR generates false positives in detection of the BRCA1 185delAG recurrent mutation. Anal. Stiintifice Univ. 2009, 10, 1–8. [Google Scholar]
- Vuttariello, E.; Borra, M.; Calise, C.; Mauriello, E.; Greggi, S.; Vecchione, A.; Biffali, E.; Chiappetta, G. A new rapid methodological strategy to assess BRCA mutational status. Mol. Biotechnol. 2013, 54, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Strom, S.P.; Lee, H.; Das, K.; Vilain, E.; Nelson, S.F.; Grody, W.W.; Deignan, J.L. Assessing the necessity of confirmatory testing for exome-sequencing results in a clinical molecular diagnostic laboratory. Genet. Med. 2014, 16, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Dacheva, D.; Dodova, R.; Popov, I.; Goranova, T.; Mitkova, A.; Mitev, V.; Kaneva, R. Validation of an NGS Approach for Diagnostic BRCA1/BRCA2 Mutation Testing. Mol. Diagn. Ther. 2015, 19, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Vigliotti, V.S.; Vigliotti, J.S.; Pappu, S. Routine human papillomavirus genotyping by DNA sequencing in community hospital laboratories. Infect. Agents Cancer 2007, 2. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Vigliotti, J.S.; Vigliotti, V.S.; Jones, W. From human papillomavirus (HPV) detection to cervical cancer prevention in clinical practice. Cancers 2014, 6, 2072–2099. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H. Guidelines for the use of molecular tests for the detection and genotyping of human papilloma virus from clinical specimens. Methods Mol. Biol. 2012, 903, 65–101. [Google Scholar] [PubMed]
- Hong, G.; Lee, S.H.; Ge, S.; Zhou, S. A novel low temperature PCR assured high-fidelity DNA amplification. Int. J. Mol. Sci. 2013, 14, 12853–12862. [Google Scholar]
- Lee, S.H.; Vigliotti, V.S.; Pappu, S. Human papillomavirus (HPV) infection among women in a representative rural and suburban population of the United States. Int. J. Gynaecol. Obstet. 2009, 105, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Schneider, Erasmus. New York State Department of Health Evaluation of the Human Papilloma Virus (HPV) Proficiency Test from October 2015.
- Tafe, L.J.; Datto, M.B.; Palomaki, G.E.; Lacbawan, F.L. CAP/ACMG Biochemical and Molecular Genetics Resource Committee. Molecular testing for the BRCA1 and BRCA2 Ashkenazi Jewish founder mutations: A report on the College of American Pathologists proficiency testing surveys. Genet. Med. 2015, 17, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.; Lee, M.K.; Casadei, S.; Thornton, A.M.; Stray, S.M.; Pennil, C.; Nord, A.S.; Mandell, J.B.; Swisher, E.M.; King, M.C. Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 12629–12633. [Google Scholar] [CrossRef] [PubMed]
- ACOG. Ob-Gyns Oppose Payer Restrictions Limiting Patient Access to Genetic Testing. Available online: https://www.acog.org/About-ACOG/News-Room/News-Releases/2015/Ob-Gyns-Oppose-Payer-Restrictions-Limiting-Patient-Access-to-Genetic-Testing (accessed on 5 February 2016).
- Ge, S.; Gong, B.; Cai, X.; Yang, X.; Gan, X.; Tong, X.; Li, H.; Zhu, M.; Yang, F.; Zhou, H.; et al. Prevent cervical cancer by screening with reliable human papillomavirus detection and genotyping. Cancer Med. 2012, 1, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Elstrodt, F.; Hollestelle, A.; Nagel, J.H.; Gorin, M.; Wasielewski, M.; van den Ouweland, A.; Merajver, S.D.; Ethier, S.P.; Schutte, M. BRCA1 mutation analysis of 41 human breast cancer cell lines reveals three new deleterious mutants. Cancer Res. 2006, 66, 41–45. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.H.; Zhou, S.; Zhou, T.; Hong, G. Sanger Sequencing for BRCA1 c.68_69del, BRCA1 c.5266dup and BRCA2 c.5946del Mutation Screen on Pap Smear Cytology Samples. Int. J. Mol. Sci. 2016, 17, 229. https://doi.org/10.3390/ijms17020229
Lee SH, Zhou S, Zhou T, Hong G. Sanger Sequencing for BRCA1 c.68_69del, BRCA1 c.5266dup and BRCA2 c.5946del Mutation Screen on Pap Smear Cytology Samples. International Journal of Molecular Sciences. 2016; 17(2):229. https://doi.org/10.3390/ijms17020229
Chicago/Turabian StyleLee, Sin Hang, Shaoxia Zhou, Tianjun Zhou, and Guofan Hong. 2016. "Sanger Sequencing for BRCA1 c.68_69del, BRCA1 c.5266dup and BRCA2 c.5946del Mutation Screen on Pap Smear Cytology Samples" International Journal of Molecular Sciences 17, no. 2: 229. https://doi.org/10.3390/ijms17020229
APA StyleLee, S. H., Zhou, S., Zhou, T., & Hong, G. (2016). Sanger Sequencing for BRCA1 c.68_69del, BRCA1 c.5266dup and BRCA2 c.5946del Mutation Screen on Pap Smear Cytology Samples. International Journal of Molecular Sciences, 17(2), 229. https://doi.org/10.3390/ijms17020229