
Diabetes Mellitus Induces Alzheimer’s Disease Pathology: Histopathological Evidence from Animal Models

{kind=link}
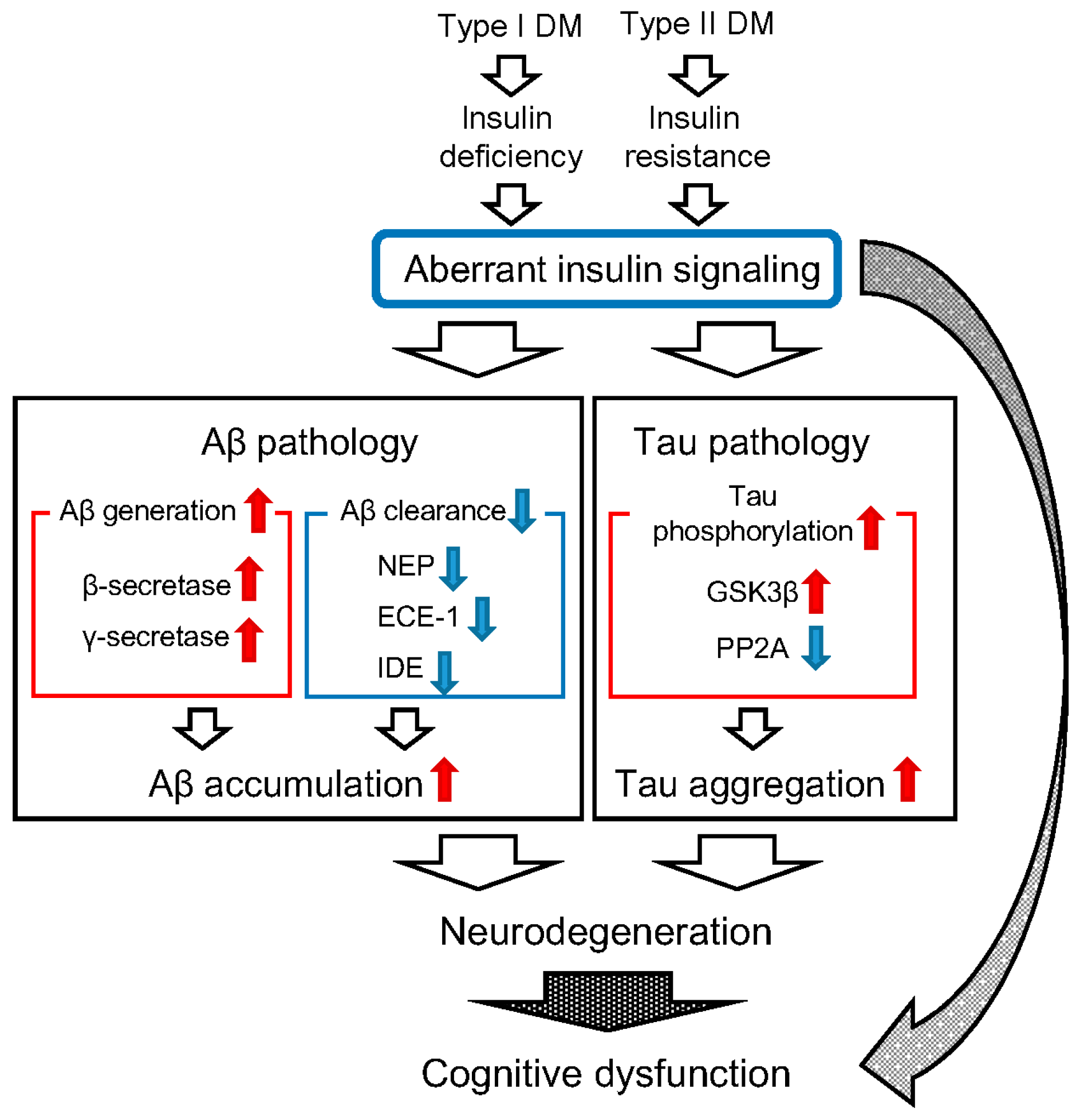
Abstract
:1. Introduction
2. Diabetes Mellitus (DM) and β-Amyloid Protein (Aβ) Pathology
3. DM and Tau Pathology
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Selkoe, D.J. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef]
- Mattson, M.P. Pathway towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, R.A. The molecular biology of senile plaques and neurofibrillary tangles in Alzheimer’s disease. Folia Neuropathol. 2009, 47, 289–299. [Google Scholar] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.; Vial, C.; Maccioni, R.B. A tau-like protein interacts with stress fibers and microtubules in human and rodent cultured cell lines. J. Cell Sci. 1993, 105, 51–60. [Google Scholar] [PubMed]
- Kosik, K.S.; Joachim, C.L.; Selkoe, D.J. Microtubule-associated protein tau is a major antigenic component of paired helical filaments in Alzheimer disease. PNAS 1986, 83, 4044–4048. [Google Scholar] [CrossRef] [PubMed]
- Maccioni, R.B.; Cambiazo, V. Role of microtubule-associated proteins in the control of microtubule assembly. Physiol. Rev. 1995, 75, 835–864. [Google Scholar] [PubMed]
- Mandelkow, E.M.; Biernat, J.; Drewes, G.; Gustke, N.; Trinczek, B.; Mandelkow, E. Tau domains, phosphorylation, and interactions with microtubules. Neurobiol. Aging 1995, 16, 355–363. [Google Scholar] [CrossRef]
- Kraegen, E.W.; Clark, P.W.; Jenkins, A.B.; Daley, E.A.; Chisholm, D.J.; Storlien, L.H. Development of muscle insulin resistance after liver insulin resistance in high-fat-fed rats. Diabetes 1991, 40, 1397–1403. [Google Scholar] [CrossRef] [PubMed]
- Lind, L.; Lithell, H. Hypertension, hyperlipidemia, insulin resistance and obesity: Parts of a metabolic syndrome. Blood Press. Suppl. 1992, 4, 49–54. [Google Scholar] [PubMed]
- Meigs, J.B. Invited commentary: Insulin resistance syndrome? Syndrome X? Multiple metabolic syndrom? A syndrome at all? Factor analysis reveals patterns in the fabric of correlated metabolic risk factors. Am. J. Epidemiol. 2000, 152, 908–911. [Google Scholar] [CrossRef] [PubMed]
- Leibson, C.L.; Rocca, W.A.; Hanson, V.A.; Cha, R.; Kokmen, E.; O’Brien, P.C.; Palumbo, P.J. Risk of dementia among persons with diabetes mellitus: A population-based cohort study. Am. J. Epidemiol. 1997, 145, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Arvanitakis, Z.; Wilson, R.S.; Bienias, J.L.; Evans, D.A.; Bennett, D.A. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch. Neurol. 2004, 61, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Frisardi, V.; Solfrizzi, V.; Seripa, D.; Capurso, C.; Santamato, A.; Sancarlo, D.; Vendemiale, G.; Pilotto, A.; Panza, F. Metabolic-cognitive syndrome: A cross-talk between metabolic syndrome and Alzheimer’s disease. Ageing Res. Rev. 2010, 9, 399–417. [Google Scholar] [CrossRef] [PubMed]
- Daviglus, M.L.; Plassman, B.L.; Pirzada, A.; Bell, C.C.; Bowen, P.E.; Burke, J.R.; Connolly, E.S., Jr.; Dunbar-Jacob, J.M.; Granieri, E.C.; McGarry, K.; et al. Risk factors and preventive interventions for Alzheimer disease: State of the science. Arch. Neurol. 2011, 68, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Crane, P.K.; Walker, R.; Hubbard, R.A.; Li, G.; Nathan, D.M.; Zheng, H.; Haneuse, S.; Craft, S.; Montine, T.J.; Kahn, S.E.; et al. Glucose levels and risk of dementia. N. Engl. J. Med. 2013, 369, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Ott, A.; Stolk, R.P.; van Harskamp, F.; Pols, H.A.P.; Hofman, A.; Breteler, M.M.B. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 1999, 53, 1937–1942. [Google Scholar] [CrossRef] [PubMed]
- Convit, A.; Wolf, O.T.; Tarshish, C.; de Leon, M.J. Reduced glucose tolerance is associated with poor memory performance and hippocampal atrophy among normal elderly. PNAS 2003, 100, 2019–2022. [Google Scholar] [CrossRef] [PubMed]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—Is this type 3 diabetes? J. Alzheimers Dis. 2005, 7, 63–80. [Google Scholar] [PubMed]
- Rönnemaa, E.; Zethelius, B.; Sundelöf, J.; Sundström, J.; Degerman-Gunnarsson, M.; Berne, C.; Lannfelt, L.; Kilander, L. Impaired insulin secretion increases the risk of Alzheimer disease. Neurology 2008, 71, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Frisardi, V.; Capurso, C.; Imbimbo, B.P.; Vendemiale, G.; Santamato, A.; D’Onofrio, G.; Seripa, D.; Sancarlo, D.; Pilotto, A.; et al. Metabolic syndrome and cognitive impairment: Current epidemiology and possible underlying mechanisms. J. Alzheimers Dis. 2010, 21, 691–724. [Google Scholar] [PubMed]
- Ohara, T.; Doi, Y.; Ninomiya, T.; Hirakawa, Y.; Hata, J.; Iwaki, T.; Kanba, S.; Kiyohara, Y. Glucose tolerance status and risk of dementia in the community: The Hisayama study. Neurology 2011, 77, 1126–1134. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Ferreira, S.T. Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes 2014, 63, 2262–2272. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, M.C.; Badonnel, K.; Meunier, N.; Tan, F.; Schlegel-Le Poupon, C.; Durieux, D.; Monnerie, R.; Baly, C.; Congar, P.; Salesse, R.; et al. Expression of insulin system in the olfactory epithelium: First approaches to its role and regulation. J. Neuroendocrinol. 2008, 20, 1176–1190. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, T.; Kagalwala, M.N.; Onuma, Y.; Ito, Y.; Warashina, M.; Terashima, K.; Sanosaka, T.; Nakashima, K.; Gage, F.H.; Asashima, M. Insulin biosynthesis in neuronal progenitors derived from adult hippocampus and the olfactory bulb. EMBO Mol. Med. 2011, 3, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Bingham, E.M.; Hopkins, D.; Smith, D.; Pernet, A.; Hallett, W.; Reed, L.; Marsden, P.K.; Amiel, S.A. The role of insulin in human brain glucose metabolism: An 18 fluoro-deoxyglucose positron emission tomography study. Diabetes 2002, 51, 3384–3390. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.Q.; Chen, H.; Quon, M.J.; Alkon, D.L. Insulin and the insulin receptor in experimental models of learning and memory. Eur. J. Pharmacol. 2004, 490, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.J.; Verkhratsky, A.; Fernyhough, P. Insulin enhances mitochondrial inner membrane potential and increases ATP levels through phosphoinositide 3-kinase in adult sensory neurons. Mol. Cell. Neurosci. 2005, 28, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.I.; Proença, T.; Oliveira, C.R.; Santos, M.S.; Rego, A.C. Insulin restores metabolic function in cultured cortical neurons subjected to oxidative stress. Diabetes 2006, 55, 2863–2870. [Google Scholar] [CrossRef] [PubMed]
- Salkovic-Petrisic, M.; Tribl, F.; Schmidt, M.; Hoyer, S.; Riederer, P. Alzheimer-like changes in protein kinase B and glycogen synthase kinase-3 in rat frontal cortex and hippocampus after damage to the insulin signalling pathway. J. Neurochem. 2006, 96, 1005–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, L.D.; Cross, D.J.; Minoshima, S.; Belongia, D.; Watson, G.S.; Craft, S. Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch. Neurol. 2010, 68, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.; Qin, W.; Pompl, P.N.; Xiang, Z.; Wang, J.; Zhao, Z.; Peng, Y.; Cambareri, G.; Rocher, A.; Mobbs, C.V.; et al. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J. 2004, 18, 902–904. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Duffy, K.B.; Ottinger, M.A.; Ray, B.; Bailey, J.A.; Holloway, H.W.; Tweedie, D.; Perry, T.; Mattson, M.P.; Kapogiannis, D.; et al. GLP-1 receptor stimulation reduces amyloid-β peptide accumulation and cytotoxicity in cellular and animal models of Alzheimer’s disease. J. Alzheimers Dis. 2010, 19, 1205–1219. [Google Scholar] [PubMed]
- Plaschke, K.; Kopitz, J.; Siegelin, M.; Schliebs, R.; Salkovic-Petrisic, M.; Riederer, P.; Hoyer, S. Insulin-resistant brain state after intracerebroventricular streptozotocin injection exacerbates Alzheimer-like changes in Tg2576 AβPP-overexpressing mice. J. Alzheimers Dis. 2010, 19, 691–704. [Google Scholar] [PubMed]
- Takeda, S.; Sato, N.; Uchio-Yamada, K.; Sawada, K.; Kunieda, T.; Takeuchi, D.; Kurinami, H.; Shinohara, M.; Rakugi, H.; Morishita, R. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Aβ deposition in an Alzheimer mouse model with diabetes. PNAS 2010, 107, 7036–7041. [Google Scholar] [CrossRef] [PubMed]
- Bitela, C.L.; Kasinathanb, C.; Kaswalab, R.H.; Klein, W.L.; Frederiksea, P.H. Amyloid-β and Tau Pathology of Alzheimer’s Disease Induced by Diabetes in a Rabbit Animal Model. J. Alzheimers Dis. 2012, 32, 291–305. [Google Scholar]
- Currais, A.; Prior, M.; Lo, D.; Jolivalt, C.; Schubert, D.; Maher, P. Diabetes exacerbates amyloid and neurovascular pathology in aging-accelerated mice. Aging Cell 2012, 11, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Maesako, M.; Uemura, K.; Kubota, M.; Kuzuya, A.; Sasaki, K.; Asada, M.; Watanabe, K.; Hayashida, N.; Ihara, M.; Ito, H.; et al. Environmental enrichment ameliorated high-fat diet-induced Aβ deposition and memory deficit in APP transgenic mice. Neurobiol. Aging 2012, 33, 1011.e11–1011.e23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, S.M.; Song, H.; Byun, J.; Park, K.S.; Jang, H.C.; Park, Y.J.; Mook-Jung, I. Accumulation of autophagosomes contributes to enhanced amyloidogenic APP processing under insulin-resistant conditions. Autophagy 2012, 8, 1842–1844. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Matsubara, T.; Sobue, K.; Tanida, M.; Kasahara, R.; Naruse, K.; Taniura, H.; Sato, T.; Suzuki, K. Brain insulin resistance accelerates Aβ fibrillogenesis by inducing GM1 ganglioside clustering in the presynaptic membranes. J. Neurochem. 2012, 121, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liang, Z.; Blanchard, J.; Dai, C.L.; Sun, S.; Lee, M.H.; Grundke-Iqbal, I.; Iqbal, K.; Liu, F.; Gong, C.X. A non-transgenic mouse model (icv-STZ mouse) of Alzheimer’s disease: Similarities to and differences from the transgenic model (3xTg-AD mouse). Mol. Neurobiol. 2013, 47, 711–725. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wu, Y.; Zhang, S.; Song, W. High glucose promotes Aβ production by inhibiting APP degradation. PLoS ONE 2013, 8, e69824. [Google Scholar] [CrossRef] [PubMed]
- Mehlaa, J.; Chauhanc, B.C.; Chauhana, N.B. Experimental Induction of Type 2 Diabetes in Aging-Accelerated Mice Triggered Alzheimer-Like Pathology and Memory Deficits. J. Alzheimers Dis. 2014, 39, 145–162. [Google Scholar]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloffs, R.; et al. β-Secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Sisodia, S.S.; St George-Hyslop, P.H. Gamma-Secretase, Notch, Aβ and Alzheimer’s disease: Where do the presenilins fit in? Nat. Rev. Neurosci. 2002, 3, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.M.; Lai, M.T.; Xu, M.; Huang, Q.; DiMuzio-Mower, J.; Sardana, M.K.; Shi, X.P.; Yin, K.C.; Shafer, J.A.; Gardell, S.J. Presenilin 1 is linked with γ-secretase activity in the detergent solubilized state. PNAS 2000, 97, 6138–6143. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Nishimura, M.; Arakawa, S.; Levitan, D.; Zhang, L.; Tandon, A.; Song, Y.Q.; Rogaeva, E.; Chen, F.; Kawarai, T.; et al. Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and βAPP processing. Nature 2000, 407, 48–54. [Google Scholar] [PubMed]
- Francis, R.; McGrath, G.; Zhang, J.; Ruddy, D.A.; Sym, M.; Apfeld, J.; Nicoll, M.; Maxwell, M.; Hai, B.; Ellis, M.C.; et al. Aph-1 and pen-2 are required for Notch pathway signaling, γ-secretase cleavage of βAPP, and presenilin protein accumulation. Dev. Cell 2000, 3, 85–97. [Google Scholar] [CrossRef]
- Goutte, C.; Tsunozaki, M.; Hale, V.A.; Priess, J.R. APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. PNAS 2002, 99, 775–779. [Google Scholar] [CrossRef] [PubMed]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J. Amyloid, the presenilins and Alzheimer’s disease. Trends Neurosci. 1997, 20, 154–159. [Google Scholar] [CrossRef]
- Borchelt, D.R.; Thinakaran, G.; Eckman, C.B.; Lee, M.K.; Davenport, F.; Ratovitsky, T.; Prada, C.M.; Kim, G.; Seekins, S.; Yager, D.; et al. Familial Alzheimer’s disease-linked presenilin 1 variants elevate Aβ1–42/1–40 ratio in vitro and in vivo. Neuron 1996, 17, 1005–1013. [Google Scholar] [CrossRef]
- Duff, K.; Eckman, C.B.; Zehr, C.; Yu, X.; Prada, C.M.; Perez-tur, J.; Hutton, M.; Buee, L.; Harigaya, Y.; Yager, D.; et al. Increased amyloid-β42(43) in brains of mice expressing mutant presenilin 1. Nature 1996, 383, 710–713. [Google Scholar] [CrossRef] [PubMed]
- Scheuner, D.; Eckman, C.B.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.D.; Hardy, J.; Hutton, M.; Kukull, W.; et al. Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 1996, 2, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Decker, H.; Lo, K.Y.; Unger, S.M.; Ferreira, S.T.; Silverman, M.A. Amyloid-β peptide oligomers disrupt axonal transport through an NMDA receptordependent mechanism that is mediated by glycogen synthase kinase 3β in primary cultured hippocampal neurons. J. Neurosci. 2010, 30, 9166–9171. [Google Scholar] [CrossRef] [PubMed]
- Vossel, K.A.; Zhang, K.; Brodbeck, J.; Daub, A.C.; Sharma, P.; Finkbeiner, S.; Cui, B.; Mucke, L. Tau reduction prevents Aβ-induced defects in axonal transport. Science 2010, 330, 198. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Scott, D.A.; Das, U.; Edland, S.D.; Radomski, K.; Koo, E.H.; Roy, S. Early and selective impairments in axonal transport kinetics of synaptic cargoes induced by soluble amyloid β-protein oligomers. Traffic 2012, 13, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Okamoto, K.; Hayashi, Y.; Sheng, M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 2004, 119, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.H.; Cai, Q. Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.H. Mitochondrial trafficking and anchoring in neurons: New insight and implications. J. Cell Biol. 2014, 204, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Luine, V.; Frankfurt, M. Interactions between estradiol, BDNF and dendritic spines in promoting memory. Neuroscience 2012, 239, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Kellner, Y.; Gödecke, N.; Dierkes, T.; Thieme, N.; Zagrebelsky, M.; Korte, M. The BDNF effects on dendritic spines of mature hippocampal neurons depend on neuronal activity. Front. Synaptic Neurosci. 2014, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Scharfman, H.E.; Chao, M.V. The entorhinal cortex and neurotrophin signaling in Alzheimer’s disease and other disorders. Cogn. Neurosci. 2013, 4, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Khatri, N.; Man, H.Y. Synaptic activity and bioenergy homeostasis: Implications in brain trauma and neurodegenerative diseases. Front. Neurol. 2013, 4, 199. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. PNAS 1998, 95, 6448–6453. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hong, S.; Shepardson, N.E.; Walsh, D.M.; Shankar, G.M.; Selkoe, D. Soluble oligomers of amyloid β protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 2009, 62, 788–801. [Google Scholar] [CrossRef] [PubMed]
- Lacor, P.N.; Buniel, M.C.; Furlow, P.W.; Clemente, A.S.; Velasco, P.T.; Wood, M.; Viola, K.L.; Klein, W.L. Aβ oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J. Neurosci. 2007, 27, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Bloodgood, B.L.; Townsend, M.; Walsh, D.M.; Selkoe, D.J.; Sabatini, B.L. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 2007, 27, 2866–2875. [Google Scholar] [CrossRef] [PubMed]
- Jolivalt, C.G.; Hurford, R.; Lee, C.A.; Dumaop, W.; Rockenstein, E.; Masliah, E. Type 1 diabetes exaggerates features of Alzheimer’s disease in APP transgenic mice. Exp. Neurol. 2010, 223, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Alldred, M.J.; Ginsberg, S.D.; Ohno, M. Mechanisms underlying insulin deficiency-induced acceleration of β-amyloidosis in a mouse model of Alzheimer’s disease. PLoS ONE 2012, 7, e32792. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, H.; Yang, J.; Liu, X.; Lu, S.; Wen, T.; Xie, L.; Wang, G. Increased amyloid β-peptide (1–40) level in brain of streptozotocin-induced diabetic rats. Neuroscience 2008, 153, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Q.; Yin, J.; Song, Y.F.; Zhang, L.; Ren, Y.X.; Wang, D.G.; Gao, L.P.; Jing, Y.H. Brain aging and AD-like pathology in streptozotocin-induced diabetic rats. J. Diabetes Res. 2014, 2014, 796840. [Google Scholar] [CrossRef] [PubMed]
- Plaschke, K.; Hoyer, S. Action of the diabetogenic drug streptozotocin on glycolytic and glycogenolytic metabolism in adult rat brain cortex and hippocampus. Int. J. Dev. Neurosci. 1993, 11, 477–483. [Google Scholar] [CrossRef]
- Duelli, R.; Schröck, H.; Kuschinsky, W.; Hoyer, S. Intracerebroventricular injection of streptozotocin induces discrete local changes in cerebral glucose utilization in rats. Int. J. Dev. Neurosci. 1994, 12, 737–743. [Google Scholar] [CrossRef]
- Hoyer, S.; Müller, D.; Plaschke, K. Desensitization of brain insulin receptor. Effect on glucose/energy and related metabolism. J. Neural Transm. Suppl. 1994, 44, 259–268. [Google Scholar] [PubMed]
- Lannert, H.; Hoyer, S. Intracerebroventricular administration of streptozotocin causes long-term diminutions in learning and memory abilities and in cerebral energy metabolism in adult rats. Behav. Neurosci. 1998, 112, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Jia, J.; Qin, W. Enhancement of β-amyloid oligomer accumulation after intracerebroventricular injection of streptozotocin, which involves central insulin signaling in a transgenic mouse model. Neuroreport 2014, 25, 1289–1295. [Google Scholar] [CrossRef] [PubMed]
- Iwata, N.; Tsubuki, S.; Takaki, Y.; Watanabe, K.; Sekiguchi, M.; Hosoki, E.; Kawashima-Morishima, M.; Lee, H.J.; Hama, E.; Sekine-Aizawa, Y.; et al. Identification of the major Aβ1–42-degrading catabolic pathway in brain parenchyma: Suppression leads to biochemical and pathological deposition. Nat. Med. 2000, 6, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Eckman, E.A.; Reed, D.K.; Eckman, C.B. Degradation of the Alzheimer’s amyloid β peptide by endothelin-converting enzyme. J. Biol. Chem. 2001, 276, 24540–24548. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.Q.; Walsh, D.M.; Ye, Z.; Vekrellis, K.; Zhang, J.; Podlisny, M.B.; Rosner, M.R.; Safavi, A.; Hersh, L.B.; Selkoe, D.J. Insulin-degrading enzyme regulates extracellular levels of amyloid β-protein by degradation. J. Biol. Chem. 1998, 273, 32730–32738. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, L.; Lu, S.; Wang, D.; Liu, X.; Xie, L.; Wang, G. Impaired amyloid β-degrading enzymes in brain of streptozotocin-induced diabetic rats. J. Endocrinol. Investig. 2011, 34, 26–31. [Google Scholar] [CrossRef]
- Vandal, M.; White, P.J.; Tremblay, C.; St-Amour, I.; Chevrier, G.; Emond, V.; Lefrançois, D.; Virgili, J.; Planel, E.; Giguere, Y.; et al. Insulin reverses the high-fat diet-induced increase in brain Aβ and improves memory in an animal model of Alzheimer disease. Diabetes 2014, 63, 4291–4301. [Google Scholar] [CrossRef] [PubMed]
- Mehla, J.; Chauhan, B.C.; Chauhan, N.B. Experimental induction of type 2 diabetes in aging-accelerated mice triggered Alzheimer-like pathology and memory deficits. J. Alzheimers Dis. 2014, 39, 145–162. [Google Scholar] [PubMed]
- Cao, D.; Lu, H.; Lewis, T.L.; Li, L. Intake of sucrose-sweetened water induces insulin resistance and exacerbates memory deficits and amyloidosis in a transgenic mouse model of Alzheimer disease. J. Biol. Chem. 2007, 282, 36275–36282. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.G.; Zhang, W.; Sima, A.A. Alzheimer-like changes in rat models of spontaneous diabetes. Diabetes 2007, 56, 1817–1824. [Google Scholar] [CrossRef] [PubMed]
- Nagai, N.; Ito, Y.; Tanino, T. Effect of high glucose levels on amyloid β production in retinas of spontaneous diabetes mellitus Otsuka Long-Evans Tokushima fatty rats. Biol. Pharm. Bull. 2015, 38, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.M.; Nixon, R.A. Enzymatically active lysosomal proteases are associated with amyloid deposits in Alzheimer brain. PNAS 1990, 87, 3861–3865. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.M.; Barnett, J.L.; Pieroni, C.; Nixon, R.A. Increased neuronal endocytosis and protease delivery to early endosomes in sporadic Alzheimer’s disease: Neuropathologic evidence for a mechanism of increased β-amyloidogenesis. J. Neurosci. 1997, 17, 6142–6151. [Google Scholar] [PubMed]
- Cataldo, A.M.; Petanceska, S.; Terio, N.B.; Peterhoff, C.M.; Durham, R.; Mercken, M.; Metha, P.D.; Buxbaum, J.; Haroutunian, V.; Nixon, R.A. Aβ localization in abnormal endosomes: Association with earliest Aβ elevations in AD a syndrome. Neurobiol. Aging 2004, 25, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. Endosome function and dysfunction in Alzheimer’s disease and other neurodegenerative diseases. Neurobiol. Aging 2005, 26, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. Autophagy, amyloidogenesis and Alzheimer disease. J. Cell Sci. 2007, 120, 4081–4091. [Google Scholar] [CrossRef] [PubMed]
- Kimura, N.; Inoue, M.; Okabayashi, S.; Ono, F.; Negishi, T. Dynein dysfunction induces endocytic pathology accompanied by an increase in Rab GTPases: A potential mechanism underlying age-dependent endocytic dysfunction. J. Biol. Chem. 2009, 284, 31291–31302. [Google Scholar] [CrossRef] [PubMed]
- Kimura, N.; Okabayashi, S.; Ono, F. Dynein dysfunction disrupts intracellular vesicle trafficking bidirectionally and perturbs synaptic vesicle docking via endocytic disturbances a potential mechanism underlying age-dependent impairment of cognitive function. Am. J. Pathol. 2012, 180, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Grbovic, O.M.; Mathews, P.M.; Jiang, Y.; Schmidt, S.D.; Dinakar, R.; Summers-Terio, N.B.; Ceresa, B.P.; Nixon, R.A.; Cataldo, A.M. Rab5-stimulated up-regulation of the endocytic pathway increases intracellular β-cleaved amyloid precursor protein carboxyl-terminal fragment levels and Aβ production. J. Biol. Chem. 2003, 278, 31261–31268. [Google Scholar] [CrossRef] [PubMed]
- Koh, Y.H.; von Arnim, C.A.; Hyman, B.T.; Tanzi, R.E.; Tesco, G. BACE is degraded via the lysosomal pathway. J. Biol. Chem. 2005, 280, 32499–32504. [Google Scholar] [CrossRef] [PubMed]
- Lefort, R.; Pozueta, J.; Shelanski, M. Cross-linking of cell surface amyloid precursor protein leads to increased β-amyloid peptide production in hippocampal neurons: Implications for Alzheimer’s disease. J. Neurosci. 2012, 32, 10674–10685. [Google Scholar] [CrossRef] [PubMed]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seshadri, S.; Fitzpatrick, A.L.; Ikram, M.A.; DeStefano, A.L.; Gudnason, V.; Boada, M.; Bis, J.C.; Smith, A.V.; Carassquillo, M.M.; Lambert, J.C.; et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 2010, 303, 1832–1840. [Google Scholar] [CrossRef] [PubMed]
- Vardarajan, B.N.; Bruesegem, S.Y.; Harbour, M.E.; St. George-Hyslop, P.; Seaman, M.N.; Farrer, L.A. Identification of Alzheimer disease associated variants in genes that regulate retromer function. Neurobiol. Aging 2012, 33, 2231. [Google Scholar] [CrossRef] [PubMed]
- Talwar, P.; Silla, Y.; Grover, S.; Gupta, M.; Agarwal, R.; Kushwaha, S.; Kukreti, R. Genomic convergence and network analysis approach to identify candidate genes in Alzheimer’s disease. BMC Genom. 2014, 15, 199. [Google Scholar] [CrossRef] [PubMed]
- Chouraki, V.; Seshadri, S. Genetics of Alzheimer’s disease. Adv. Genet. 2014, 87, 245–294. [Google Scholar] [PubMed]
- Okabayashi, S.; Shimozawa, N.; Yasutomi, Y.; Yanagisawa, K.; Kimura, N. Diabetes mellitus accelerates Aβ pathology in brain accompanied by enhanced GAβ generation in nonhuman primates. PLoS ONE 2015, 10, e0117362. [Google Scholar] [CrossRef] [PubMed]
- Lane, R.F.; Raines, S.M.; Steele, J.W.; Ehrlich, M.E.; Lah, J.A.; Small, S.A.; Tanzi, R.E.; Attie, A.D.; Gandy, S. Diabetes-associated SorCS1 regulates Alzheimer’s amyloid-β metabolism: Evidence for involvement of SorL1 and the retromer complex. J. Neurosci. 2010, 30, 13110–13115. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Alonso Adel, C.; Chen, S.; Chohan, M.O.; El-Akkad, E.; Gong, C.X.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta 2005, 1739, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Imahori, K.; Uchida, T. Physiology and pathology of tau protein kinases in relation to Alzheimer’s disease. J. Biochem. 1997, 121, 179–188. [Google Scholar] [PubMed]
- Ishiguro, K.; Omori, A.; Takamatsu, M.; Sato, K.; Arioka, M.; Uchida, T.; Imahori, K. Phosphorylation sites on tau by tau protein kinase I, a bovine derived kinase generating an epitope of paired helical filaments. Neurosci. Lett. 1992, 148, 202–206. [Google Scholar] [CrossRef]
- Michel, G.; Mercken, M.; Murayama, M.; Noguchi, K.; Ishiguro, K.; Imahori, H.; Takashima, A. Characterization of tau phosphorylation in glycogen synthase kinase-3β and cyclin dependent kinase-5 activator (p23) transfected cells. Biochim. Biophys. Acta 1998, 1380, 177–182. [Google Scholar] [CrossRef]
- Godeman, R.; Biernat, J.; Mandelkow, E.; Mandelkow, E.M. Phosphorylation of tau protein by recombinant GSK3β: Pronounced phosphorylation at select Ser/Thr-Pro motif but no phosphorylation at Ser267 in the repeat domain. FEBS Lett. 1999, 454, 157–164. [Google Scholar] [CrossRef]
- Hanger, D.P.; Hughes, K.; Woodgett, J.R.; Brion, J.P.; Anderton, B.H. Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: Generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 1992, 147, 58–62. [Google Scholar] [CrossRef]
- Ullrich, A.; Bell, J.R.; Chen, E.Y.; Herrera, R.; Petruzzelli, L.M.; Dull, T.J.; Gray, A.; Coussens, L.; Liao, Y.C.; Tsubokawa, M.; et al. Human insulin receptor and its relationship to the tyrosine kinase family of oncogenes. Nature 1985, 313, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.G., Jr.; Sun, X.J.; White, M.F. The IRS-1 signaling system. Trends Biochem. Sci. 1994, 19, 289–293. [Google Scholar] [CrossRef]
- O’Hare, T.; Pilch, P.F. Intrinsic kinase activity of the insulin receptor. Int. J. Biochem. 1990, 22, 315–324. [Google Scholar] [CrossRef]
- Ghasemi, R.; Haeri, A.; Dargahi, L.; Mohamed, Z.; Ahmadiani, A. Insulin in the brain: Sources, localization and functions. Mol. Neurobiol. 2013, 47, 145–171. [Google Scholar] [CrossRef] [PubMed]
- Adamo, M.L.; Shemer, J.; Roberts, C.T., Jr.; LeRoith, D. Insulin and insulin-like growth factor-I induced phosphorylation in neurally derived cells. Ann. N. Y. Acad. Sci. 1993, 692, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Abbott, M.A.; Wells, D.G.; Fallon, J.R. The insulin receptor tyrosine kinase substrate p58/53 and the insulin receptor are components of CNS synapses. J. Neurosci. 1999, 19, 7300–7308. [Google Scholar] [PubMed]
- Werther, G.A.; Hogg, A.; Oldfield, B.J.; McKinley, M.J.; Figdor, R.; Allen, A.M.; Mendelsohn, F.A. Localization and characterization of insulin receptors in rat brain and pituitary gland using in vitro autoradiography and computerized densitometry. Endocrinology 1987, 121, 1562–1570. [Google Scholar] [CrossRef] [PubMed]
- Dozza, B.; Smith, M.A.; Perry, G.; Tabaton, M.; Strocchi, P. Regulation of glycogen synthase kinase-3β by products of lipid peroxidation in human neuroblastoma cells. J. Neurochem. 2004, 89, 1224–1232. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.D.; Delerue, F.; Gladbach, A.; Götz, J.; Ittner, L.M. Experimental diabetes mellitus exacerbates tau pathology in a transgenic mouse model of Alzheimer’s disease. PLoS ONE 2009, 4, e7917. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.X.; Singh, T.J.; Grundke-Iqbal, I.; Iqbal, K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J. Neurochem. 1993, 61, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.X.; Shaikh, S.; Wang, J.Z.; Zaidi, T.; Grundke-Iqbal, I.; Iqbal, K. Phosphatase activity toward abnormally phosphorylated tau: Decrease in Alzheimer disease brain. J. Neurochem. 1995, 65, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Vogelsberg-Ragaglia, V.; Schuck, T.; Trojanowski, J.Q.; Lee, V.M. PP2A mRNA expression is quantitatively decreased in Alzheimer’s disease hippocampus. Exp. Neurol. 2001, 168, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Tanimukai, H.; Grundke-Iqbal, I.; Iqbal, K. Up-regulation of inhibitors of protein phosphatase-2A in Alzheimer’s disease. Am. J. Pathol. 2005, 166, 1761–1771. [Google Scholar] [CrossRef]
- Planel, E.; Tatebayashi, Y.; Miyasaka, T.; Liu, L.; Wang, L.; Herman, M.; Yu, W.H.; Luchsinger, J.A.; Wadzinski, B.; Duff, K.E.; et al. Insulin dysfunction induces in vivo tau hyperphosphorylation through distinct mechanisms. J. Neurosci. 2007, 27, 13635–13648. [Google Scholar] [CrossRef] [PubMed]
- Clodfelder-Miller, B.J.; Zmijewska, A.A.; Johnson, G.V.; Jope, R.S. Tau is hyperphosphorylated at multiple sites in mouse brain in vivo after streptozotocin-induced insulin deficiency. Diabetes 2006, 55, 3320–3325. [Google Scholar] [CrossRef] [PubMed]
- Papon, M.A.; El Khoury, N.B.; Marcouiller, F.; Julien, C.; Morin, F.; Bretteville, A.; Petry, F.R.; Gaudreau, S.; Amrani, A.; Mathews, P.M.; et al. Deregulation of protein phosphatase 2A and hyperphosphorylation of τ protein following onset of diabetes in NOD mice. Diabetes 2013, 62, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhao, Y.; Dai, C.L.; Liang, Z.; Run, X.; Iqbal, K.; Liu, F.; Gong, C.X. Intranasal insulin restores insulin signaling, increases synaptic proteins, and reduces Aβ level and microglia activation in the brains of 3xTg-AD mice. Exp. Neurol. 2014, 261, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Pedrós, I.; Petrov, D.; Allgaier, M.; Sureda, F.; Barroso, E.; Beas-Zarate, C.; Auladell, C.; Pallàs, M.; Vázquez-Carrera, M.; Casadesús, G.; et al. Early alterations in energy metabolism in the hippocampus of APPswe/PS1dE9 mouse model of Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R.; et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: A pilot clinical trial. Arch. Neurol. 2012, 69, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Claxton, A.; Baker, L.D.; Hanson, A.; Trittschuh, E.H.; Cholerton, B.; Morgan, A.; Callaghan, M.; Arbuckle, M.; Behl, C.; Craft, S. Longacting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or earlystage Alzheimer’s disease dementia. J. Alzheimers Dis. 2015, 44, 897–906. [Google Scholar] [PubMed]

© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kimura, N. Diabetes Mellitus Induces Alzheimer’s Disease Pathology: Histopathological Evidence from Animal Models. Int. J. Mol. Sci. 2016, 17, 503. https://doi.org/10.3390/ijms17040503
Kimura N. Diabetes Mellitus Induces Alzheimer’s Disease Pathology: Histopathological Evidence from Animal Models. International Journal of Molecular Sciences. 2016; 17(4):503. https://doi.org/10.3390/ijms17040503
Chicago/Turabian StyleKimura, Nobuyuki. 2016. "Diabetes Mellitus Induces Alzheimer’s Disease Pathology: Histopathological Evidence from Animal Models" International Journal of Molecular Sciences 17, no. 4: 503. https://doi.org/10.3390/ijms17040503
APA StyleKimura, N. (2016). Diabetes Mellitus Induces Alzheimer’s Disease Pathology: Histopathological Evidence from Animal Models. International Journal of Molecular Sciences, 17(4), 503. https://doi.org/10.3390/ijms17040503