
A Tumor-Specific Neo-Antigen Caused by a Frameshift Mutation in BAP1 Is a Potential Personalized Biomarker in Malignant Peritoneal Mesothelioma


Abstract
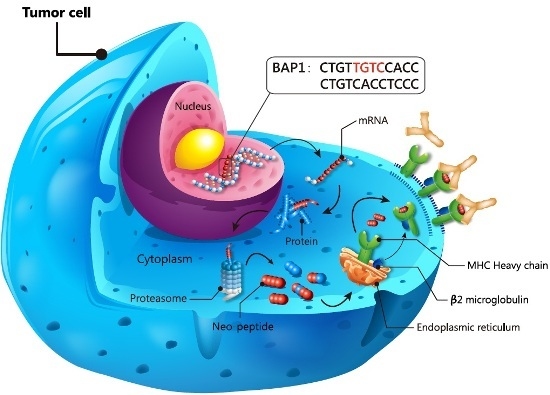
:
1. Introduction
2. Results
2.1. Next-Generation Sequencing (NGS) Data Analysis
2.2. Somatic Mutation Filtration and Validation
2.3. Prediction of Bap1 Neo-Peptide’s Protein Function and Major Histocompatibility Complex (MHC) Presentation
2.4. Western Blotting and Immunohistochemistry (IHC) Results
3. Discussion
4. Materials and Methods
4.1. Case Sample
4.2. Deep Targeted Sequencing
4.3. NGS Data Processing and Prediction of MHC Binding
4.4. Somatic Mutation Validation by PCR
4.5. Neo-Peptide Synthesis and Polyclonal Antibody Production
4.6. Western Blot Study and Immunohistochemistry (IHC)
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| MPM | Malignant peritoneal mesothelioma |
| NGS | Next generation sequencing |
| MHC | Major histocompatibility complex |
| HLA | Human leukocyte antigen |
References
- Bridda, A.; Padoan, I.; Mencarelli, R.; Frego, M. Peritoneal mesothelioma: A review. MedGenMed. 2007, 9, 32. [Google Scholar] [PubMed]
- Sharma, H.; Bell, I.; Schofield, J.; Bird, G. Primary peritoneal mesothelioma: Case series and literature review. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Remon, J.; Lianes, P.; Martinez, S.; Velasco, M.; Querol, R.; Zanui, M. Malignant mesothelioma: New insights into a rare disease. Cancer Treat. Rev. 2013, 39, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Sheffield, B.S.; Tinker, A.V.; Shen, Y.; Hwang, H.; Li-Chang, H.H.; Pleasance, E.; Ch’ng, C.; Lum, A.; Lorette, J.; McConnell, Y.J.; et al. Personalized oncogenomics: clinical experience with malignant peritoneal mesothelioma using whole genome sequencing. PLoS ONE 2015, 10, e0119689. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.Q.; Jhanwar, S.C.; Klein, W.M.; Bell, D.W.; Lee, W.C.; Altomare, D.A.; Nobori, T.; Olopade, O.I.; Buckler, A.J.; Testa, J.R. p16 alternations and deletion mapping of 9p21-p22 in malignant mesothelioma. Cancer Res. 1994, 54, 5547–5551. [Google Scholar] [PubMed]
- Andujar, P.; Pairon, J.C.; Renier, A.; Descatha, A.; Hysi, I.; Abd-Alsamad, I.; Billon-Galland, M.A.; Blons, H.; Clin, B.; Danel, C.; et al. Differential mutation profiles and similar intronic TP53 polymorphisms in asbestos-related lung cancer and pleural mesothelioma. Mutagenesis 2013, 28, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Bott, M.; Brevet, M.; Taylor, B.S.; Shimizu, S.; Ito, T.; Wang, L.; Creaney, J.; Lake, R.A.; Zakowski, M.F.; Reva, B.; et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat. Genet. 2011, 43, 668–672. [Google Scholar] [CrossRef] [PubMed]
- Rajasagi, M.; Shukla, S.A.; Fritsch, E.F.; Keskin, D.B.; DeLuca, D.; Carmona, E.; Zhang, W.; Sougnez, C.; Cibulskis, K.; Sidney, J.; et al. Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood 2014, 124, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; El-Gamil, M.; Dudley, M.E.; Li, Y.F.; Rosenberg, S.A.; Robbins, P.F. T cells associated with tumor regression recognize frameshifted products of the CDKN2A tumor suppressor gene locus and a mutated HLA class I gene product. J. Immunol. 2004, 172, 6057–6064. [Google Scholar] [CrossRef] [PubMed]
- Hacohen, N.; Fritsch, E.F.; Carter, T.A.; Lander, E.S.; Wu, C.J. Getting personal with neoantigen-based therapeutic cancer vaccines. Cancer Immunol. Res. 2013, 1, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Aithal, M.G.; Rajeswari, N. Role of Notch signaling pathway in cancer and its association with DNA methylation. J. Genet. 2014, 92, 667–675. [Google Scholar] [CrossRef]
- De Assis, L.V.; Locatelli, J.; Isoldi, M.C. The role of key genes and pathways involved in the tumorigenesis of Malignant Mesothelioma. Biochim. Biophys. Acta 2014, 1845, 232–247. [Google Scholar] [CrossRef] [PubMed]
- Zanella, C.L.; Posada, J.; Tritton, T.R.; Mossman, B.T. Asbestos causes stimulation of the extracellular signal-regulated kinase 1 mitogen-activated protein kinase cascade after phosphorylation of the epidermal growth factor receptor. Cancer Res. 1996, 56, 5334–5338. [Google Scholar] [PubMed]
- Tatton-Brown, K.; Rahman, N. The NSD1 and EZH2 overgrowth genes, similarities and differences. Am. J. Med. Genet. C Semin. Med. Genet. 2013, 163C, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Morishita, M.; di Luccio, E. Cancers and NSD family of histone lysine methyltransferases. Biochim. Biophys. Acta 2011, 1816, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Lo Iacono, M.; Monica, V.; Righi, L.; Grosso, F.; Libener, R.; Vatrano, S.; Bironzo, P.; Novello, S.; Musmeci, L.; Volante, M.; et al. Targeted next-generation sequencing of cancer genes in advanced stage malignant pleural mesothelioma: A retrospective study. J. Thorac. Oncol. 2015, 10, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Alakus, H.; Yost, S.E.; Woo, B.; French, R.; Lin, G.Y.; Jepsen, K.; Frazer, K.A.; Lowy, A.M.; Harismendy, O. BAP1 mutation is a frequent somatic event in peritoneal malignant mesothelioma. J. Transl. Med. 2015, 13, 122. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Yang, H.; Pass, H.I.; Krausz, T.; Testa, J.R.; Gaudino, G. BAP1 and cancer. Nat. Rev. Cancer 2013, 13, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Whitson, B.A.; Kratzke, R.A. Molecular pathways in malignant pleural mesothelioma. Cancer Lett. 2006, 239, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Van Cuijk, L.; Vermeulen, W.; Marteijn, J.A. Ubiquitin at work: The ubiquitous regulation of the damage recognition step of NER. Exp. Cell Res. 2014, 329, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Kulathu, Y.; Komander, D. Atypical ubiquitylation—The unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat. Rev. Mol. Cell Biol. 2012, 13, 508–523. [Google Scholar] [CrossRef] [PubMed]
- Husnjak, K.; Dikic, I. Ubiquitin-binding proteins: Decoders of ubiquitin-mediated cellular functions. Annu. Rev. Biochem. 2012, 81, 291–322. [Google Scholar] [CrossRef] [PubMed]
- Karasaki, T.; Nagayama, K.; Kawashima, M.; Hiyama, N.; Murayama, T.; Kuwano, H.; Nitadori, J.; Anraku, M.; Sato, M.; Miyai, M.; et al. Identification of Individual Cancer-Specific Somatic Mutations for Neoantigen-Based Immunotherapy of Lung Cancer. J. Thorac. Oncol. 2016, 11, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Gros, A.; Parkhurst, M.R.; Tran, E.; Pasetto, A.; Robbins, P.F.; Ilyas, S.; Prickett, T.D.; Gartner, J.J.; Crystal, J.S.; Roberts, I.M.; et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat. Med. 2016, 22, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.J.; Gartner, J.J.; Horovitz-Fried, M.; Shamalov, K.; Trebska-McGowan, K.; Bliskovsky, V.V.; Parkhurst, M.R.; Ankri, C.; Prickett, T.D.; Crystal, J.S.; et al. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J. Clin. Investig. 2015, 125, 3981–3991. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, S.; Muhitch, J.B. Unmasking targets of antitumor immunity via high-throughput antigen profiling. Curr. Opin. Biotechnol. 2016, 42, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Harbor, J.W.; Onken, M.D.; Roberson, E.D.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef] [PubMed]
- Pena-Llopis, S.; Vega-Rubin-de-Celis, S.; Liao, A.; Leng, N.; Pavia-Jimenez, A.; Wang, S.; Yamasaki, T.; Zhrebker, L.; Sivanand, S.; Spence, P.; et al. BAP1 loss defines a new class of renal cell carcinoma. Nat. Genet. 2012, 44, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Magnani, C.; Bianchi, C.; Chellini, E.; Consonni, D.; Fubini, B.; Gennaro, V.; Marinaccio, A.; Menegozzo, M.; Mirabelli, D.; Merler, E.; et al. III Italian Consensus Conference on Malignant Mesothelioma of the Pleura. Epidemiology, Public Health and Occupational Medicine related issues. Med. Lav. 2015, 106, 325–332. [Google Scholar] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Mutation | Compared to Peripheral Blood |
|---|---|
| Total Variants | |
| Base Substitution | 2897 |
| Small Indel | 218 |
| In the coding region, variant reads >3, 0% mutation frequency in blood | |
| Base Substitution | 88 |
| Small Indel | 9 |
| Mutation frequency >1% in tumor, non-repertoire in dbSNP and 1000Genome | |
| Base Substitution | 24 |
| Small Indel | 4 |
| Mutation frequency >5%, “causing disease” in SIFT and PolyPhen prediction | |
| Base Substitution | 8 |
| Small Indel | 2 |
| Validation by RT-PCR | |
| Base Substitution | 4 |
| Small Indel | 1 |
| Chromosome | Begin Position | End Position | Associated Gene | Reference | Alternative | Tumor Frequency | Mutation_Type | Validation by Sanger Sequencing |
|---|---|---|---|---|---|---|---|---|
| 1 | 120497774 | 120497774 | NOTCH2 | C | T | 17.63% | nonsynonymous | Yes |
| 1 | 144930847 | 144930847 | PDE4DIP | G | T | 8.82% | nonsynonymous | Yes |
| 1 | 186276044 | 186276046 | PRG4 | CCA | – | 17.65% | nonframeshift | No |
| 3 | 12626480 | 12626480 | RAF1 | T | C | 8.11% | nonsynonymous | No |
| 3 | 52437592 | 52437592 | BAP1 | – | GACA | 21.21% | frameshift | Yes |
| 5 | 160097517 | 160097517 | ATP10B | C | T | 12.28% | nonsynonymous | Yes |
| 5 | 176665298 | 176665298 | NSD1 | A | G | 5.08% | nonsynonymous | No |
| 5 | 176721814 | 176721814 | NSD1 | A | C | 15.89% | nonsynonymous | Yes |
| 14 | 95599707 | 95599707 | DICER1 | A | G | 5.77% | nonsynonymous | No |
| 22 | 28193936 | 28193936 | MN1 | C | A | 15.38% | nonsynonymous | No |
| HLA Typing | Peptide Sequence | Peptide Type | 1-log50k | nM | Rank | Banding Type |
|---|---|---|---|---|---|---|
| HLA-A*11:12 | ATCINGVNGFR | NOTCH2-703WT | 0.525 | 171.19 | 1.50 | Weak Binding |
| HLA-A*11:12 | CINGVNGFR | NOTCH2-703WT | 0.482 | 271.44 | 2.00 | Weak Binding |
| HLA-A*11:12 | ATCINGVNFR | NOTCH2-G703D | 0.5 | 224.39 | 2.00 | Weak Binding |
| HLA-A*11:12 | CINGVNFR | NOTCH2-G703D | 0.479 | 280.08 | 2.00 | Weak Binding |
| HLA-A*11:12 | KLELALSMIK | PDE4DIP_288WT | 0.476 | 290.35 | 2.00 | Weak Binding |
| HLA-A*11:12 | ELALSMIK | PDE4DIP_288WT | 0.446 | 402.87 | 3.00 | Weak Binding |
| HLA-A*11:12 | KELALSMIK | PDE4DIP_L288M | 0.528 | 165.47 | 1.50 | Weak Binding |
| HLA-A*11:12 | ELALSMIK | PDE4DIP_L288M | 0.446 | 402.87 | 3.00 | Weak Binding |
| HLA-B*35:42 | HKELALSM | PDE4DIP_L288M | 0.525 | 171.29 | 2.00 | Weak Binding |
| HLA-A*11:12 | ASLDGETNLK | ATP10B_210WT | 0.642 | 47.93 | 0.80 | Strong Binding |
| HLA-A*11:12 | SLDGETNLK | ATP10B_210WT | 0.523 | 174.12 | 1.50 | Weak Binding |
| HLA-A*11:12 | TASLDGETNLK | ATP10B_210WT | 0.451 | 380.57 | 3.00 | Weak Binding |
| HLA-A*11:12 | ASLDGTNLK | ATP10B_E210K | 0.638 | 50.5 | 0.80 | Strong Binding |
| HLA-A*11:12 | ATP10B_E210K | 0.546 | 135.45 | 1.50 | Strong Binding | |
| HLA-A*11:12 | SLDGKTNLK | ATP10B_E210K | 0.515 | 190.94 | 2.00 | Weak Binding |
| HLA-A*11:12 | TASLDGTNLK | ATP10B_E210K | 0.443 | 413.1 | 3.00 | Weak Binding |
| HLA-B*35:42 | MPVLESSSW | NSD1_2482WT | 0.755 | 14.22 | 0.30 | Strong Binding |
| HLA-B*35:42 | MPVLESSS | NSD1_2482WT | 0.497 | 232.2 | 2.00 | Weak Binding |
| HLA-B*35:42 | MPVLESSSW | NSD1_K2482T | 0.755 | 14.22 | 0.30 | Strong Binding |
| HLA-B*35:42 | NSD1_K2482T | 0.596 | 78.89 | 1.00 | Strong Binding | |
| HLA-B*35:42 | MPVLESSS | NSD1_K2482T | 0.497 | 232.2 | 2.00 | Weak Binding |
| HLA-B*35:42 | TPQADEMPVL | NSD1_K2482T | 0.451 | 379.75 | 3.00 | Weak Binding |
| HLA-B*35:42 | BAP1_V523fs | 0.736 | 17.46 | 0.40 | Strong Binding | |
| HLA-B*35:42 | LPHLQGAFW | BAP1_V523fs | 0.492 | 242.83 | 2.00 | Weak Binding |
| HLA-B*35:42 | SPVVHLPH | BAP1_V523fs | 0.465 | 326.57 | 3.00 | Weak Binding |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lai, J.; Zhou, Z.; Tang, X.-J.; Gao, Z.-B.; Zhou, J.; Chen, S.-Q. A Tumor-Specific Neo-Antigen Caused by a Frameshift Mutation in BAP1 Is a Potential Personalized Biomarker in Malignant Peritoneal Mesothelioma. Int. J. Mol. Sci. 2016, 17, 739. https://doi.org/10.3390/ijms17050739
Lai J, Zhou Z, Tang X-J, Gao Z-B, Zhou J, Chen S-Q. A Tumor-Specific Neo-Antigen Caused by a Frameshift Mutation in BAP1 Is a Potential Personalized Biomarker in Malignant Peritoneal Mesothelioma. International Journal of Molecular Sciences. 2016; 17(5):739. https://doi.org/10.3390/ijms17050739
Chicago/Turabian StyleLai, Jun, Zhan Zhou, Xiao-Jing Tang, Zhi-Bin Gao, Jie Zhou, and Shu-Qing Chen. 2016. "A Tumor-Specific Neo-Antigen Caused by a Frameshift Mutation in BAP1 Is a Potential Personalized Biomarker in Malignant Peritoneal Mesothelioma" International Journal of Molecular Sciences 17, no. 5: 739. https://doi.org/10.3390/ijms17050739
APA StyleLai, J., Zhou, Z., Tang, X. -J., Gao, Z. -B., Zhou, J., & Chen, S. -Q. (2016). A Tumor-Specific Neo-Antigen Caused by a Frameshift Mutation in BAP1 Is a Potential Personalized Biomarker in Malignant Peritoneal Mesothelioma. International Journal of Molecular Sciences, 17(5), 739. https://doi.org/10.3390/ijms17050739