1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is characterized by the presence of “desmoplasia”, which forms a barrier hindering the effective entry of chemotherapy drugs and promoting tumor growth. Leukocytes might account for approximately 50% of the total volume of solid tumors. Macrophages are one of the important cell subsets. In recent years, many studies have focused on the pathogenesis of PDAC inflammation and fibrosis, although the lack of oxygen is also a noteworthy feature of PDAC. To date, little research has been done on the relationship between hypoxia and macrophages.
Hypoxia is a common condition in the microenvironment of solid tumors [
1]. In a hypoxic environment, hypoxia can induce chemokines to attract monocytes/macrophages, such as vascular endothelial growth factor A (VEGF-A), around the surrounding area. Different tumor microenvironments can affect the infiltration of monocytes/macrophages [
2]. Macrophages in tumor tissues have their specific sources; tumor-derived chemokines and corresponding macrophage receptors play a critical role in the recruitment of monocytes/macrophages, such as C-C chemokine receptor type 2(CCR2)/ chemical chemokine 2 (CCL2), vascular endothelial growth factor receptor 1 VEGFR1/VEGF-A, L-selectin (CD62L)/CD62L ligands, and chemokine (C-X3-C motif) receptor 1 (CX3CR1)/ chemokine (C-X3-C motif) ligand 1 (CX3CL1). CCL2 is a potent cytokine in the recruitment of monocytes/macrophages to inflammation, tissue damage, and tumor locations; CCL2 is also known as the monocyte chemotactic protein-1 or small inducible cytokine 2. CCL2 is expressed in ovarian carcinoma, cervical squamous-cell carcinoma, melanoma, and other tumors; furthermore, CCL2 is proportional to the infiltration of monocytes/macrophages [
3,
4,
5].
Tumor stroma is a dynamic environment, which contains inflammatory cells, pancreatic stellate cells, endothelial cells, extracellular matrix, and different growth factors and cytokines; these cells interact with tumor cells to modify their behavior. In this study, we aimed (i) to analyze whether the expression of Hypoxia inducible factor (HIF)-1α/CCL2 and HIF-1α/CD68 has a correlation from the tissue; (ii) to investigate the regulation of HIF-1α on CCL2 was verified at the level of protein and nucleic acid in cells; and (iii) to investigate the recruitment of monocytes/macrophages and the activation of pancreatic stellate cells (PSCs) in vitro.
Little research has been done on PDAC hypoxia, inflammation, and fibrosis. The present study reports for the first time that HIF-1α promotes the PDAC secretion of CCL2 to recruit monocytes/macrophages, and to activate PSCs. This result will provide a new idea for inflammation and fibrosis formation.
3. Discussion
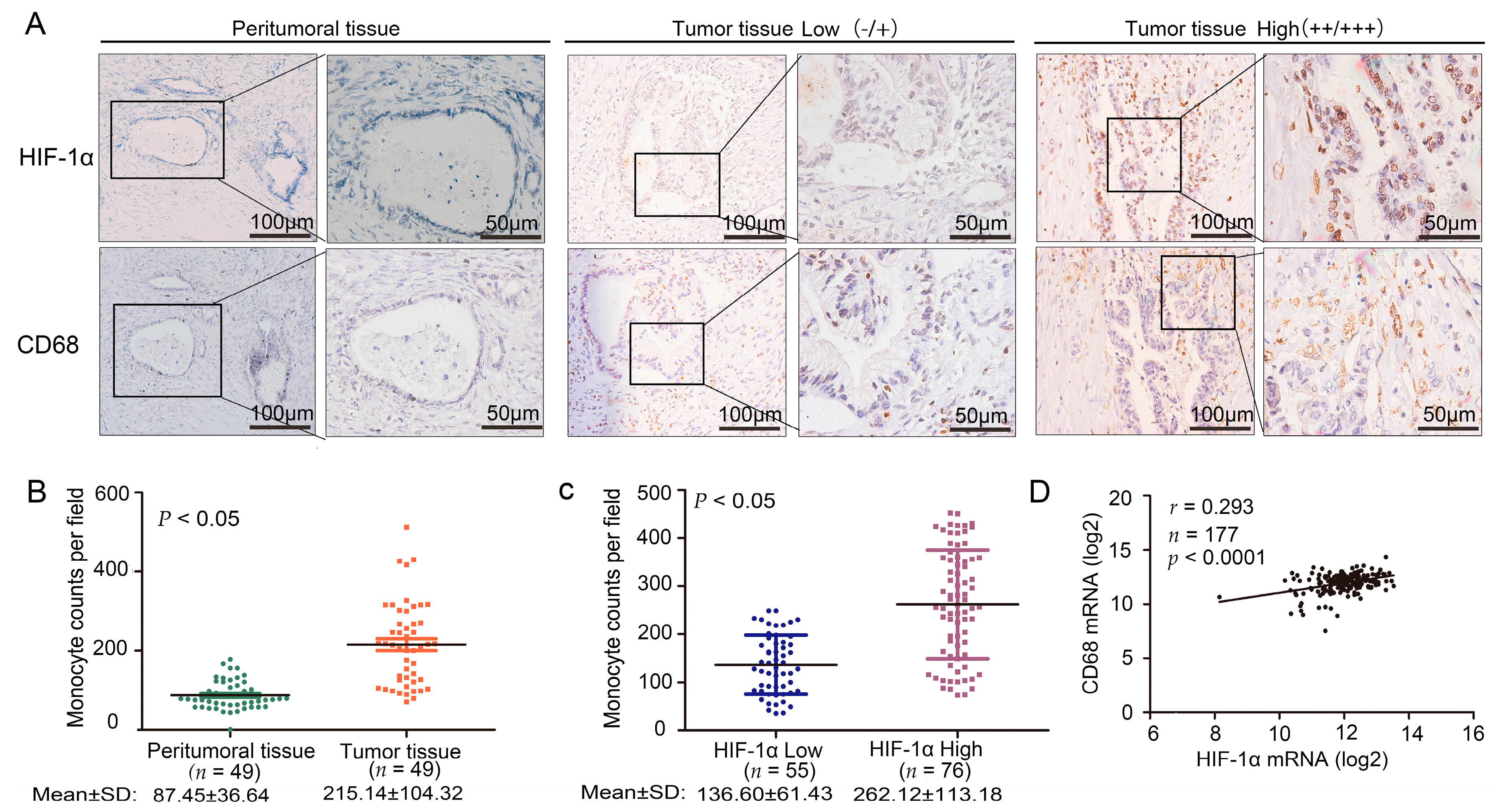
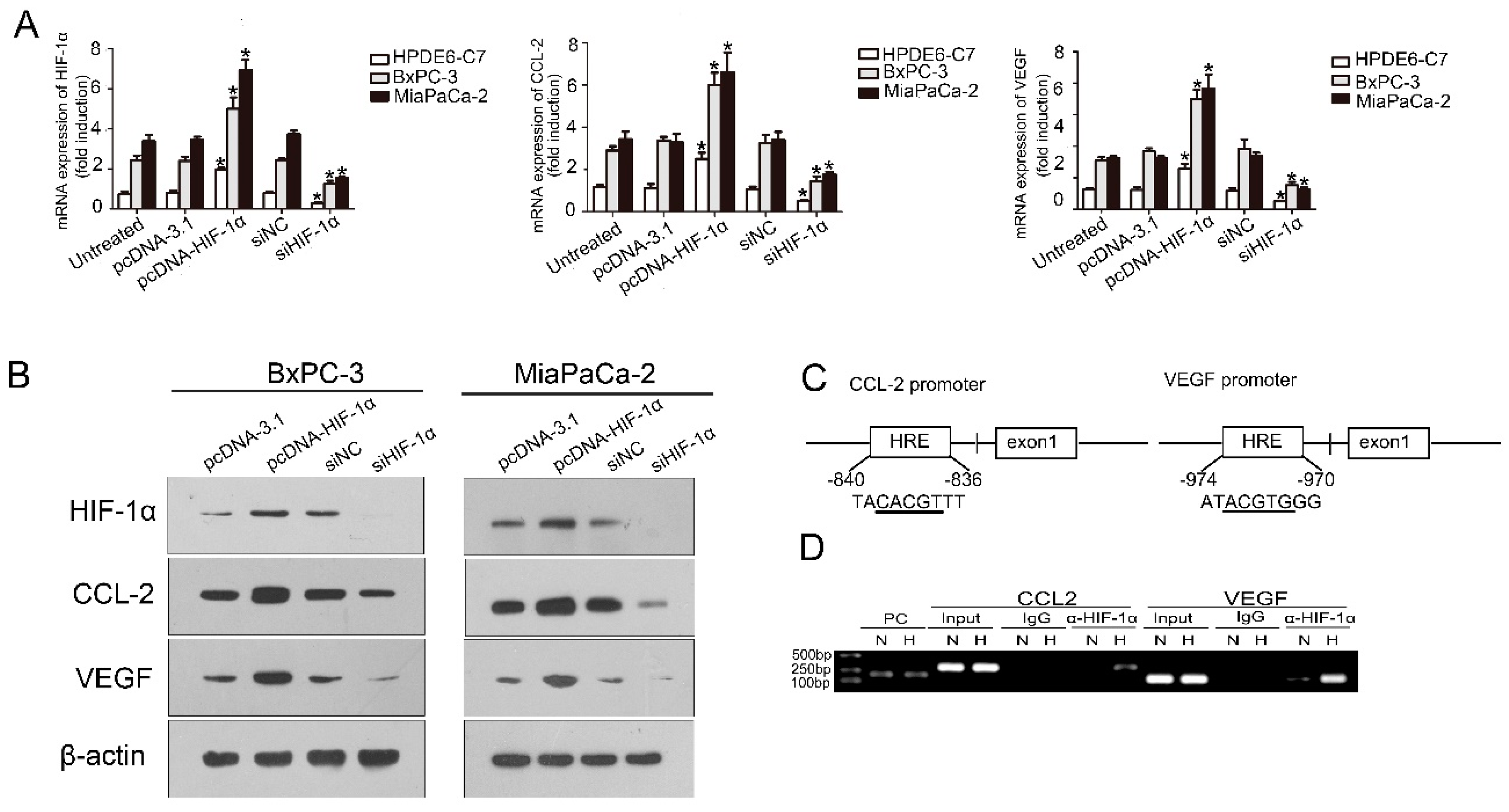
The expression level of HIF-1α in pancreatic cancer is linked to tumor progression, angiogenesis, invasion, and metastasis [
9,
10]. Macrophages originate mostly from the circulating mononuclear cells under the micro-environmental response. They gain different phenotypes [
10,
11]. Tumors in the hypoxic environment secrete numerous chemokines or other factors to promote the recruitment of monocytes/macrophages. More macrophages are present to infiltrate the hypoxia site of breast, prostate, and ovarian cancers [
12]. Macrophages in tumor metastasis, progression, angiogenesis, and other processes are also important regulators [
13,
14,
15]. The expression of CD68 and HIF-1α was studied in the samples through IHC staining. Samples showed a significant correlation between CD68 and HIF-1α (
t-test,
p < 0.05). PSCs and macrophages are two components playing an important role in the stroma. Hypoxia can directly increase the activity of PSCs to promote the formation of PDAC fibrosis. Interaction between PSCs and macrophages also promotes the fibrosis of pancreatic cancer. When quiescent PSCs and macrophages are co-cultured, astrocytes will be transformed into muscle fibroblasts [
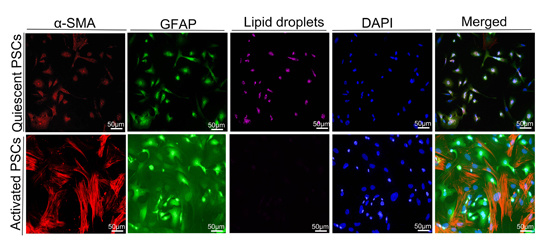
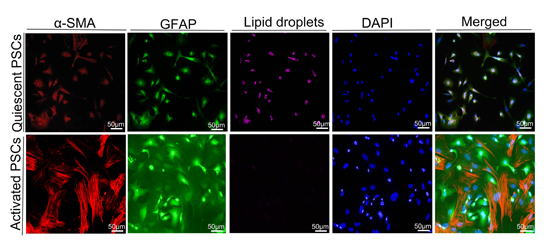
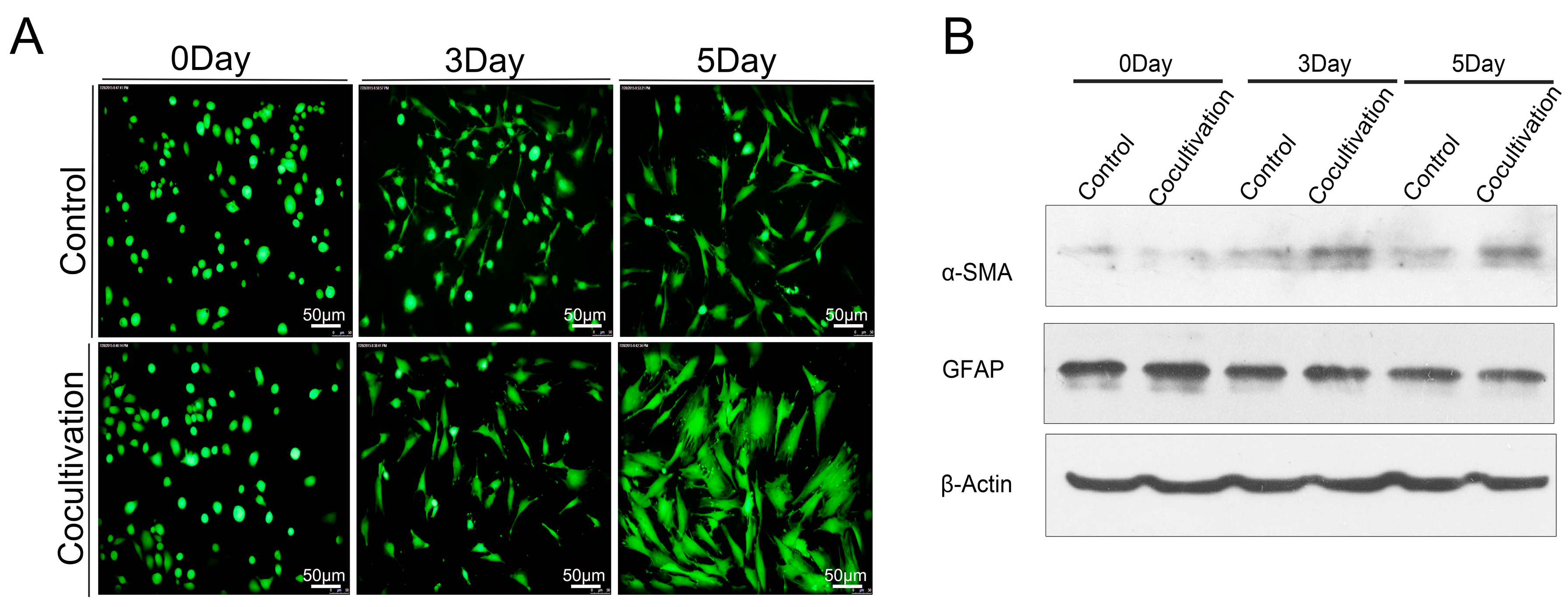
16]. Macrophages can produce several growth factors (transforming growth factor β1 [TGF-β1], fibroblast growth factor-2, TGF-α, and platelet-derived growth factor) and cytokines (tumor necrosis factor, interleukin-1 [IL-1], and IL-6) to activate PSCs. Activated PSCs show a myofibroblast-like phenotype. The principal changes in the PSCs include the loss of lipid droplets and the increase of α-SMA. Glial fibrillary acidic protein (GFAP) is a precise expression of intermediate filament cytoskeleton protein, which determines the structure and function of astrocytes. The expression of CD68 and collagen I was studied in the samples with high expression of HIF-1α through IHC staining. Samples with high HIF-1α expression showed a significant correlation between CD68 and collagen I (
t-test,
p < 0.05).
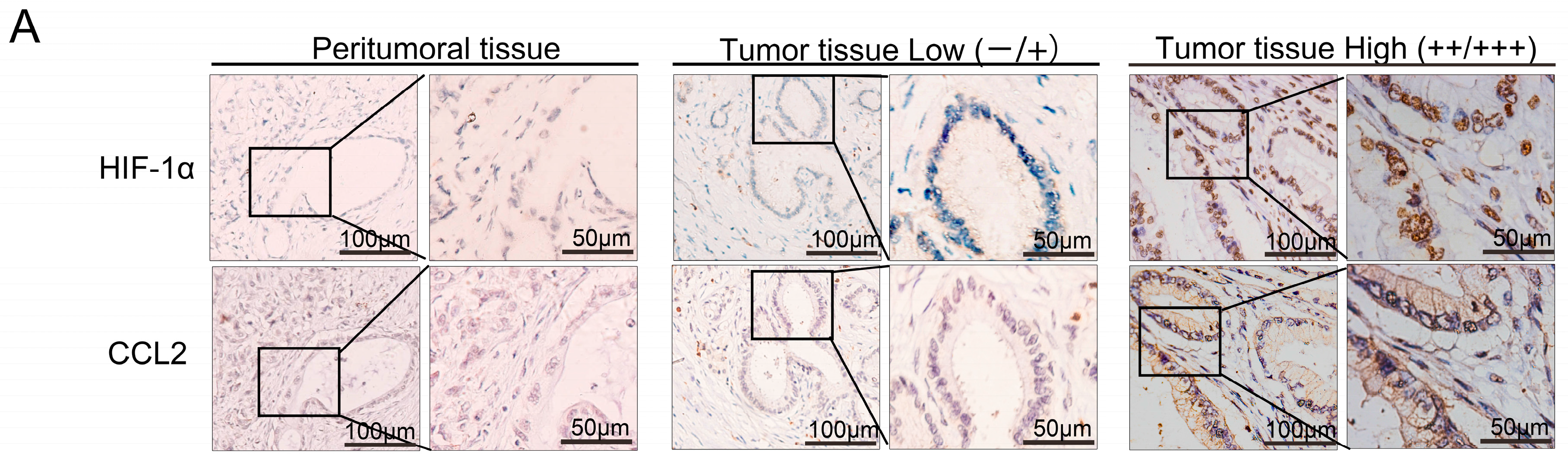
CCL2 acts as a classical chemokine that can effectively attract and activate monocytes/macrophages to local tissue damage, inflammation, tumor, and other sites. Endothelial cells, macrophages, smooth muscle cells, fibroblasts, and even tumor cells can produce CCL2 [
17]. CCL2 is highly expressed in the hypoxia region, and the expression level of CCL2 depends on the severity of hypoxia [
18]. The
CCL2 gene contains promoter sites associated with HIF-1α binding. Hence, the CCL2 expression can be improved by HIF-1α in a stable hypoxia environment [
19]. CCL2 expression also correlates closely with HIF-1α expression in gastric cancer [
20]. The expression of CCL2 and HIF-1α was studied in the samples through IHC staining; samples showed a significant correlation between CCL2 and HIF-1α (
r = 0.268,
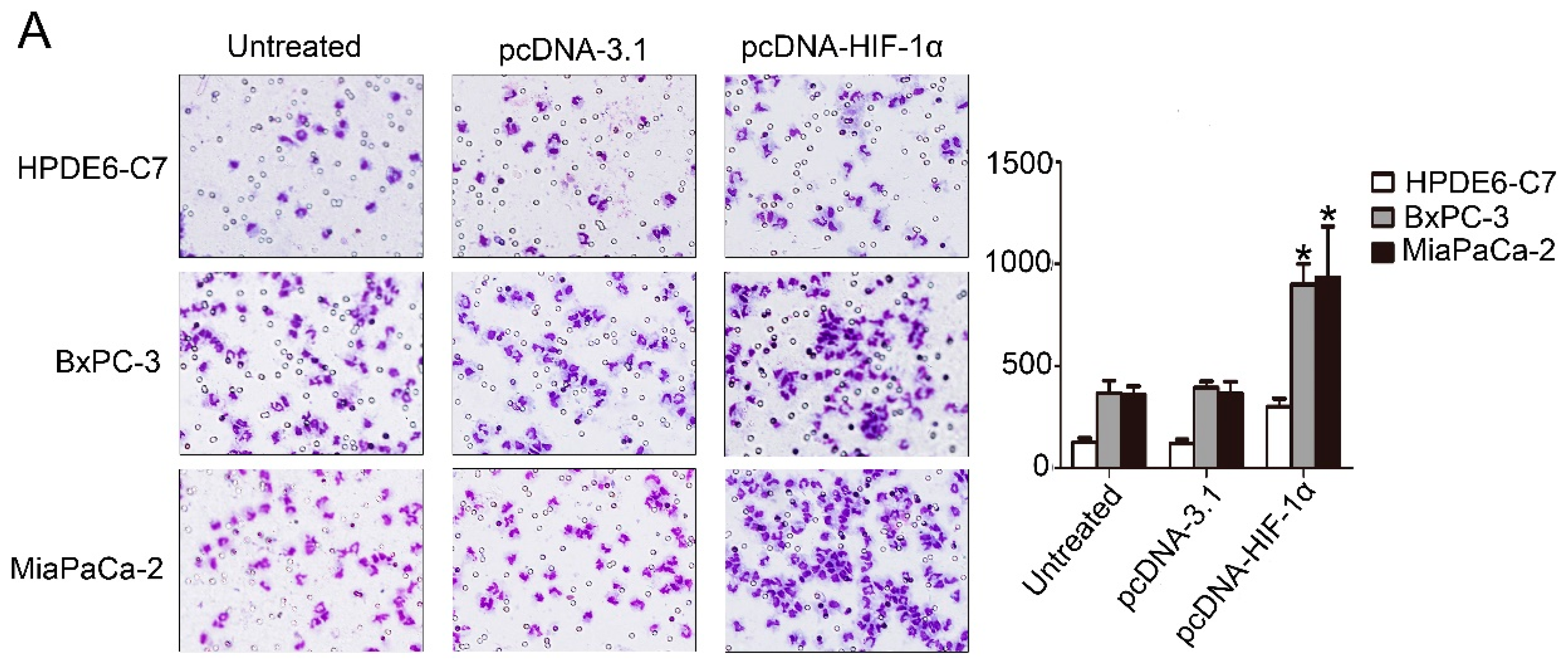
p < 0.05). Overall survival of PDAC patients with low CCL2 expression was significantly longer than that of patients with high CCL2 staining. Western blot analysis, qRT-PCR, chromatin immunoprecipitation assay, and TCGA confirmed that HIF-1α exerted a regulatory effect on CCL2 in PDAC. We utilized the Transwell assay to confirm that HIF-1α was beneficial for PDAC cells to recruit monocytes/macrophages by CCL2. We co-cultured macrophages and the newly isolated PSCs. The speed of activation in the co-cultured group was significantly faster than that in the control group.
Little research has been done on PDAC hypoxia and fibrosis. The present study reported for the first time that HIF-1α promoted the PDAC secretion of CCL2 to recruit monocytes/macrophages, as well as to form PDAC fibrosis. This result provides a novel idea for fibrosis formation. Overall, these results showed that the hypoxic microenvironment of pancreatic cancer is helpful for the recruitment of local monocytes/macrophages, and macrophages can accelerate the activation of PSCs in the stroma. Fibrosis and the lack of blood supply of the interstitial hinder the survival of cancer cells and affect the delivery of chemotherapy drugs. In pancreatic cancer, fibrosis and hypoxia can lead to tumor expansion. Substantial work is needed to explore the relationships among cancer cells, macrophages, and PSCs. The effect of HIF-1α on the inflammation and fibrosis of pancreatic cancer through regulating CCL2 has been shown in the present study. Hence, HIF-1α and CCL2 are potentially important targets. Since HIF-1α mainly exists in the nucleus, and is a transcription factor, blocking it poses a certain degree of difficulty, however, CCL2 is secreted, and has a relatively specific receptor of CCR2 and CCR4 [
21]. So in the hypoxia microenvironment of pancreatic cancer, blocking CCR2/CCR4 will likely have the potential to reduce the inflammation of pancreatic cancer tissue and fibrosis.
4. Materials and Methods
4.1. Immunohistochemistry
This experiment was approved by the Ethics Committee of Tianjin Medical University Cancer Hospital and Institute (Tianjin China) in 2 September 2014. The project identification code was E2014092. We collected 131 cases of PDAC from surgical specimens. IHC staining was done for HIF-1α, collagen I, CD68, and CCL2 in accordance with the manufacturers’ instructions. The staining intensity was classified as 0, 1, 2, and 3, corresponding to negative, low, medium, and high. The extent of staining was also assessed as 0, 1, 2, and 3, representing the values or range of 0%, 1%–25%, 26%–50%, and 51%–100%, respectively. The final score was the total of staining intensity and extent; each final score ranged from 0 to 9. Subsequently, the score was divided into four groups: 0, 1–2, 3–4, and 6–9, which represent the negative, low, medium, and high scores, respectively. Also, we used the symbols of −, +, ++ and+++ to form representatives of the four degrees of expression. We divided four levels of expression into two groups: low expression group (−/+) and high expression group (++/+++). All the scores were evaluated by two qualified pathological personnel.
4.2. Cell Culture and Hypoxic Treatment
Human PDAC cell lines (BxPC3) were purchased from the Chinese Academy of Sciences, Shanghai, China. MIAPaCa-2 cells were purchased from ATCC. The cells were grown at 37 °C in a humid atmosphere of 95% air and 5% CO2 and then cultured with RPMI-1640 or DMEM containing 10% fetal bovine serum (FBS). Hypoxia was produced in a sealing pot, with a debugger prepared in 94% N2, 5% CO2, and 1% O2 environment.
4.3. SiRNA Duplexes, Plasmid Constructs, and Transient Transfection
SiRNAs against HIF-1α and pcDNA-HIF-1α plasmids were used as previously described [
10]. For transfection, the PDAC cells were plated in six-well plates at a density of 5 × 10
5 cells/well. When the cells grew to 80%, about 50 nmol/L siRNAs or 4 µg of plasmids were transfected into the cells by using Lipofectamine-2000 (Invitrogen, Carlsbad, CA, USA) for 48 h.
4.4. Western-Blot Analysis
All cells were fully extracted using sodium dodecyl sulfate, sodium salt (SDS) cell lysis solution with protease inhibitor cocktail. Up to 20 µg of cleavage protein products were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and the objective protein was detected by Western blot analysis.
4.5. Real-Time Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
RNA was isolated from the transfected cells by using TRIzol reagent (Invitrogen). The reverse transcription polymerase chain reaction (qRT-PCR) system (TaKaRa Bio Group, Shiga, Japan) was utilized to obtain complementary DNA (cDNA). Each sample underwent experiments in triplicate, in which β-actin acted as loading control. The PCR primers are indicated in
supplementary Table S1.
4.6. Chromatin Immunoprecipitation Assay
A chromatin immunoprecipitation assay was performed using a commercial kit (Upstate Biotechnology, MILLIPORE, Darmstadt, Germany) according to the manufacturer’s instruction. MiaPaCa-2 was incubated under normoxia and hypoxia for 12 h and then added with 1% formaldehyde to cross link the DNA–protein complexes and lysis in SDS Lysis buffer. Lysate was required for shearing cross linked DNA to 200–1000 bp. Next, the cross linked protein was immunoprecipitated using anti-HIF-1 antibodies (Abcam, Cambridge, UK) at 4 °C overnight, the IgG was used as negative control. After washing, centrifugation, elution, and reverse, the DNA was invoked as a phenol/chloroform/isoamyl mixture and precipitated by alcohol. The DNA was used as a template for PCR of the CCL2 and VEGF binding sites (positive control). We found the promoter sequences of VEGF and CCL2 in University of California Santa Cruz (UCSC) Genome Bioinformatics Site and searched the binding sites of HIF-1α in their promoter region; then, we designed the primers with binding sites in Oligo7; finally, we used BLAST to sequence alignment. Forward and reverse primers were as follows: 5′-GGAGAATAGCTGCCATAACCA-3′, 5′-TTAAATAGCCTGCTCAAGGTC-3′ (CCL2), 5′CCTTTGGGTTTTGCCAGACTCC-3′, and 5′-TCCCTCTGACAATGTGCCATC-3′ (VEGF).
4.7. Isolation and Culture of Monocytes/Macrophages
This experiment was approved by the Ethics Committee of Tianjin Medical University Cancer Hospital and Institute. We performed density gradient centrifugation. Fresh peripheral blood of pancreatic cancer patients and phosphate buffered saline (PBS) were mixed with the same volume and spread above the Ficoll (Solarbio, Beijing, China). Ficoll had the equivalent volume of peripheral blood. After centrifugation (25 °C, 400× g, 25 min), we collected the turbid cell layer in the interface and placed it into the new tube. This layer was washed twice with PBS. When a supernatant was ruled out, cells were cultured using 1640, which contained 10% FBS. After overnight incubation, the suspended cells were deleted.
4.8. Transwell Assay
We used the 8 µm aperture of the 24-hole plate in this detection. Before this experiment, 2 × 105 pancreatic cells were seeded onto a 6-hole plate; the next day, the cells were treated with corresponding transfection after 36 h, we collected supernatant samples of each type of cells separately and then we centrifuged them while avoiding mixed cells. These supernatant samples were injected into the low chamber, and then we placed 1 × 105 macrophage cells in the upper chamber, with the culture medium of DMEM or 1640. After 24 h of incubation, macrophages were recruited to the bottom of the filter. Subsequently, a three-step method was performed (Thermo Scientific, Waltham, MA, USA). The results of each group were viewed under the microscope; we randomly selected 10 representative regions to count from which we took the average. All experiments were replicated at least thrice.
4.9. Isolation of PSCs
This experiment was subject to approval by the Ethics Committee of Tianjin Medical University Cancer Hospital and Institute. PSCs were isolated from the primary pancreatic cancer paraneoplastic tissues. The tissues were immersed in 3 mL of D-hank’s solution and cut into small pieces. Afterward, the tissue pieces were placed into the 37 °C digestive solution (0.05% collagenase V, 0.02% pronase, 0.03% DNase I, D-Hank’s solution, 0.5% BSA, 5 mM CaCl2, and 25 mM HEPES). These tissues were digested for about 20 min until no obvious tissue masses were observed. Samples were filtered, centrifuged at 450× g for 7 min under 4 °C, and then precipitated using a heavy suspension (0.3% BSA in D-Hank’s solution). Approximately 9.5 mL of cell suspension and 8 mL of 28.7% Nycodenz were mixed to spread under 6 mL of 0.3% BSA + D-Hank’s solution. After centrifugation (4 °C, 1400× g, 20 min), we collected the turbid cell layer in the interface and placed it into the new tube. This layer was washed twice with D-hank’s solution. The liquid was centrifuged at 450× g and 4 °C for 7 min. When the supernatant was discarded, the cells were cultured in DMEM, which contained 20% FBS.
4.10. Immunofluorescence
Freshly isolated PSCs were cultured on the glasses at the appropriate time the medium was drained. The PSCs were washed twice with PBS, fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.1% Triton X-100 in PBS for 10 min, and then blocked for 1 h with 3% BSA in PBS. Subsequently, the cells were incubated with the antibodies α-smooth muscle actin (α-SMA) and GFAP, and DAPI was utilized to stain the nuclei. Finally, the cells were observed under a fluorescence microscope. For the lipid droplets of PSC staining, we used Nile red staining (Cell neutral lipid of Nile Red Staining Kit, GENMED, Shanghai, China). Before the start of the experiment, we took the Reagent A out of the kit, and then removed the PBS owned 2 mL buffer to the 2 mL centrifuge tube. We added 10 µL Reagent A, fully mixed it, and marked it as the working fluid. We placed it at 37 °C to avoid light incubation of cells for 10 min. At last, fluorescence cells were observed under the fluorescence microscope, and the excitation wavelength was 543 nm.
4.11. Statistical Analysis
Spearman’s rank correlation coefficient was utilized to test the association of ordinal variables. t-Test was used to compare the mean .The log-rank test was conducted to obtain a p value for Kaplan–Meier curves’ divergence. All probability values were two-sided. Analyses were carried out using SPSS 21.0 statistical analysis software (Armonk, NY, USA).


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







