
DNA Damage and Repair in Schizophrenia and Autism: Implications for Cancer Comorbidity and Beyond


{kind=link}
Abstract
:1. Schizophrenia and Autism Spectrum Disorder: Symptoms and Origins
2. Schizophrenia, Autism Spectrum Disorder and Cancer Risk
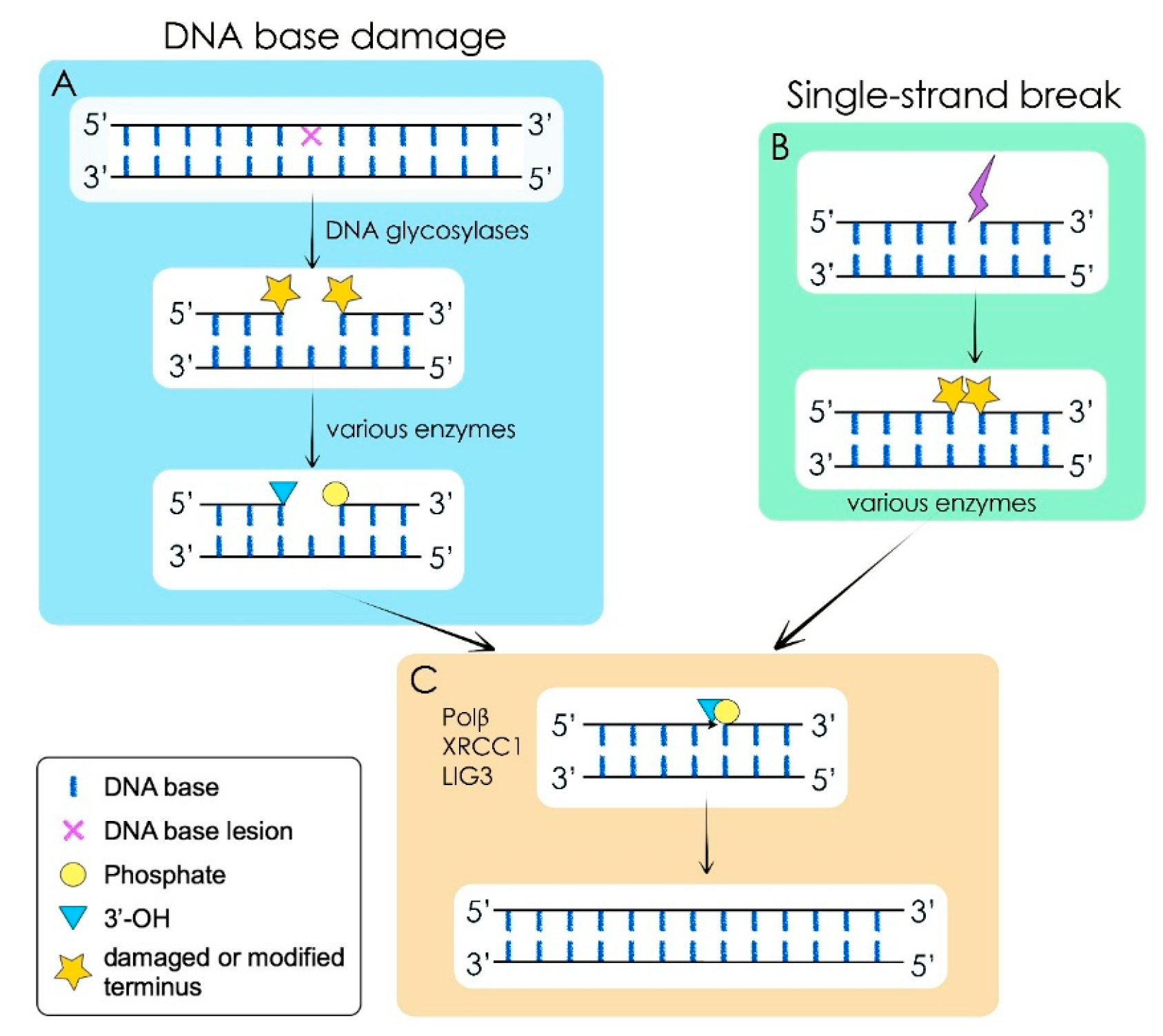
3. DNA Damage, DNA Repair and Cancer
4. Oxidative Stress, DNA Damage and DNA Repair in Schizophrenia and Autism Spectrum Disorder
4.1. Oxidative Stress, Mitochondrial Dysfunctions and DNA Damage
4.2. Dysregulated DNA Repair and DNA Damage
5. DNA Repair and Damage in Schizophrenia and Autism: A Role beyond Altered Cancer Risk?
6. Conclusions and Perspectives
Author Contributions
Conflicts of Interest
References
- Fombonne, E. Epidemiology of pervasive developmental disorders. Pediatr. Res. 2009, 65, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Tandon, R.; Nasrallah, H.A.; Keshavan, M.S. Schizophrenia, “just the facts” 4. Clinical features and conceptualization. Schizophr. Res. 2009, 110, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Rapin, I.; Tuchman, R.F. Autism: Definition, neurobiology, screening, diagnosis. Pediatr. Clin. N. Am. 2008, 55, 1129–1146. [Google Scholar] [CrossRef] [PubMed]
- Whiteford, H.A.; Degenhardt, L.; Rehm, J.; Baxter, A.J.; Ferrari, A.J.; Erskine, H.E.; Charlson, F.J.; Norman, R.E.; Flaxman, A.D.; Johns, N.; et al. Global burden of disease attributable to mental and substance use disorders: Findings from the Global Burden of Disease Study 2010. Lancet 2013, 382, 1575–1586. [Google Scholar] [CrossRef]
- Silberberg, D.; Anand, N.P.; Michels, K.; Kalaria, R.N. Brain and other nervous system disorders across the lifespan-Global challenges and opportunities. Nature 2015, 527, S151–S154. [Google Scholar] [CrossRef] [PubMed]
- Mehling, M.H.; Tassé, M.J. Severity of autism spectrum disorders: Current conceptualization, and transition to DSM-5. J. Autism. Dev. Disord. 2016, 46, 2000–2016. [Google Scholar] [CrossRef] [PubMed]
- Tarabeux, J.; Kebir, O.; Gauthier, J.; Hamdan, F.F.; Xiong, L.; Piton, A.; Spiegelman, D.; Henrion, É.; Millet, B.; Fathalli, F.; et al. Rare mutations in N-methyl-D-aspartate glutamate receptors in autism spectrum disorders and schizophrenia. Trans. Psychiatry 2011, 1, e55. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Penzes, P. Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders. Curr. Mol. Med. 2015, 15, 146–167. [Google Scholar] [CrossRef] [PubMed]
- Kruhøffer, M.; Jensen, J.L.; Laiho, P.; Dyrskjøt, L.; Salovaara, R.; Arango, D.; Birkenkamp-Demtroder, K.; Sørensen, F.B.; Christensen, L.L.; Buhl, L.; et al. Gene expression signatures for colorectal cancer microsatellite status and HNPCC. Br. J. Cancer 2005, 92, 2240–2248. [Google Scholar] [CrossRef] [PubMed]
- Uzunova, G.; Pallanti, S.; Hollander, E. Excitatory/inhibitory imbalance in autism spectrum disorders: Implications for interventions and therapeutics. World J. Biol. Psychiatry 2015, 17, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, D.R. Implications of normal brain development for the pathogenesis of schizophrenia. Arch. Gen. Psychiatry 1987, 44, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, J.L.; Giedd, J.N.; Gogtay, N. Neurodevelopmental model of schizophrenia: Update 2012. Mol. Psychiatry 2012, 17, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.; Yu, K.; Fung, G.; Leung, M.; Wong, C.; Li, Q.; Sham, P.; Chua, S.; McAlonan, G. Autistic disorders and schizophrenia: Related or remote? An anatomical likelihood estimation. PLoS ONE 2010, 5, e12233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ecker, C.; Bookheimer, S.Y.; Murphy, D.G.M. Neuroimaging in autism spectrum disorder: Brain structure and function across the lifespan. Lancet Neurol. 2015, 14, 1121–1134. [Google Scholar] [CrossRef]
- Selemon, L.D.; Zecevic, N. Schizophrenia: A tale of two critical periods for prefrontal cortical development. Trans. Psychiatry 2015, 5, e623. [Google Scholar] [CrossRef] [PubMed]
- Ziats, M.N.; Edmonson, C.; Rennert, O.M. The autistic brain in the context of normal neurodevelopment. Front. Neuroanat. 2015, 9, 115. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S. The environment and susceptibility to schizophrenia. Prog. Neurobiol. 2011, 93, 23–58. [Google Scholar] [CrossRef] [PubMed]
- Pinkham, A.E.; Hopfinger, J.B.; Pelphrey, K.A.; Piven, J.; Penn, D.L. Neural bases for impaired social cognition in schizophrenia and autism spectrum disorders. Schizophr. Res. 2008, 99, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Giusti-Rodríguez, P.; Sullivan, P.F. The genomics of schizophrenia: Update and implications. J. Clin. Investig. 2013, 123, 4557–4563. [Google Scholar] [CrossRef] [PubMed]
- Farrell, M.S.; Werge, T.; Sklar, P.; Owen, M.J.; Ophoff, R.A.; O’Donovan, M.C.; Corvin, A.; Cichon, S.; Sullivan, P.F. Evaluating historical candidate genes for schizophrenia. Mol. Psychiatry 2015, 20, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Leventhal, B.L. Genetic epidemiology and insights into interactive genetic and environmental effects in autism spectrum disorders. Biol. Psychiatry 2015, 77, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Mandy, W.; Lai, M.-C. Annual Research Review: The role of the environment in the developmental psychopathology of autism spectrum condition. J. Child Psychol. Psychiatry 2016, 57, 271–292. [Google Scholar] [CrossRef] [PubMed]
- Van Os, J.; Rutten, B.P.; Poulton, R. Gene-environment interactions in schizophrenia: Review of epidemiological findings and future directions. Schizophr. Bull. 2008, 34, 1066–1082. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Ripke, S.; Neale, B.M.; Faraone, S.V.; Purcell, S.M.; Perlis, R.H.; Mowry, B.J.; Thapar, A.; Goddard, M.E.; Witte, J.S.; et al. International Inflammatory Bowel Disease Genetics Consortium (IIBDGC). Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet. 2013, 45, 984–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, P.F.; Kendler, K.S.; Neale, M.C. Schizophrenia as a complex trait: Evidence from a meta-analysis of twin studies. Arch. Gen. Psychiatry 2003, 60, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Uher, R. Gene-environment interactions in common mental disorders: An update and strategy for a genome-wide search. Soc. Psychiatry Psychiatr. Epidemiol. 2014, 49, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Van Os, J.; Rutten, B.P.; Myin-Germeys, I.; Delespaul, P.; Viechtbauer, W.; van Zelst, C.; Bruggeman, R.; Reininghaus, U.; Morgan, C.; Murray, R.M.; et al. Identifying gene-environment interactions in schizophrenia: Contemporary challenges for integrated, large-scale investigations. Schizophr. Bull. 2014, 40, 729–736. [Google Scholar] [PubMed]
- Insel, T.R. Rethinking schizophrenia. Nature 2010, 468, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Tandon, R.; Nasrallah, H.A.; Keshavan, M.S. Schizophrenia, “just the facts” 5. Treatment and prevention. Past, present, and future. Schizophr. Res. 2010, 122, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Ruhela, R.K.; Prakash, A.; Medhi, B. An urgent need for experimental animal model of autism in drug development. Ann. Neurosci. 2015, 22, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Bauman, M.L. Medical comorbidities in autism: Challenges to diagnosis and treatment. Neurotherapeutics 2010, 7, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, B. Schizophrenia as a systemic disease. Schizophr. Bull. 2009, 35, 381–382. [Google Scholar] [CrossRef] [PubMed]
- Tabarés-Seisdedos, R.; Dumont, N.; Baudot, A.; Valderas, J.M.; Climent, J.; Valencia, A.; Crespo-Facorro, B.; Vieta, E.; Gómez-Beneyto, M.; Martinez, S.; et al. No paradox, no progress: Inverse cancer comorbidity in people with other complex diseases. Lancet Oncol. 2011, 12, 604–608. [Google Scholar] [CrossRef]
- Gejman, P.V.; Sanders, A.R.; Duan, J. The role of genetics in the etiology of schizophrenia. Psychiatr. Clin. N. Am. 2010, 33, 35–66. [Google Scholar] [CrossRef] [PubMed]
- Torrey, E.F. Are we overestimating the genetic contribution to schizophrenia? Schizophr. Bull. 1992, 18, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.; Susser, E. Commentary: What can epidemiology accomplish? Int. J. Epidemiol. 2006, 35, 587–590, discussion 593–596. [Google Scholar] [CrossRef] [PubMed]
- Patterson, P.H. Neuroscience. Maternal effects on schizophrenia risk. Science 2007, 318, 576–577. [Google Scholar] [CrossRef] [PubMed]
- Van Vugt, J.M.G.; Shulman, L.P. Prenatal Medicine; Taylor & Francis Group: New York, NY, USA, 2006. [Google Scholar]
- Hsiao, E.Y.; Patterson, P.H. Placental regulation of maternal-fetal interactions and brain development. Dev. Neurobiol. 2012, 72, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, R.; Wildgust, H.J.; Bushe, C.J. Cancer and schizophrenia: Is there a paradox? J. Psychopharmacol. (Oxf.) 2010, 24, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Bushe, C.J.; Bradley, A.J.; Wildgust, H.J.; Hodgson, R.E. Schizophrenia and breast cancer incidence: A systematic review of clinical studies. Schizophr. Res. 2009, 114, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, P.B. The incidence of cancer in schizophrenic patients. J. Epidemiol. Community Health 1989, 43, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, P.B. The occurrence of cancer in first admitted schizophrenic patients. Schizophr. Res. 1994, 12, 185–194. [Google Scholar] [CrossRef]
- Cohen, M.; Dembling, B.; Schorling, J. The association between schizophrenia and cancer: A population-based mortality study. Schizophr. Res. 2002, 57, 139–146. [Google Scholar] [CrossRef]
- Barak, Y.; Achiron, A.; Mandel, M.; Mirecki, I.; Aizenberg, D. Reduced cancer incidence among patients with schizophrenia. Cancer 2005, 104, 2817–2821. [Google Scholar] [CrossRef] [PubMed]
- Chou, F.H.-C.; Tsai, K.-Y.; Su, C.-Y.; Lee, C.-C. The incidence and relative risk factors for developing cancer among patients with schizophrenia: A nine-year follow-up study. Schizophr. Res. 2011, 129, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Sundquist, K.; Ning, Y.; Kendler, K.S.; Sundquist, J.; Chen, X. Incidence of cancer in patients with schizophrenia and their first-degree relatives: A population-based study in Sweden. Schizophr. Bull. 2013, 39, 527–536. [Google Scholar] [CrossRef]
- Lichtermann, D.; Ekelund, J.; Pukkala, E.; Tanskanen, A.; Lönnqvist, J. Incidence of cancer among persons with schizophrenia and their relatives. Arch. Gen. Psychiatry 2001, 58, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Ajdacic-Gross, V.; Tschopp, A.; Bopp, M.; Gutzwiller, F.; Rössler, W. Cancer comortality patterns in schizophrenia and psychotic disorders: A new methodological approach for unique databases. Int. J. Methods Psychiatr. Res. 2014, 23, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Catalá-López, F.; Suárez-Pinilla, M.; Suárez-Pinilla, P.; Valderas, J.M.; Gómez-Beneyto, M.; Martinez, S.; Balanzá-Martínez, V.; Climent, J.; Valencia, A.; McGrath, J.; et al. Inverse and direct cancer comorbidity in people with central nervous system disorders: A meta-analysis of cancer incidence in 577,013 participants of 50 observational studies. Psychother. Psychosom. 2014, 83, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.-M.; Chen, Y.-J.; Kuo, D.-J.; Jaiteh, L.E.S.; Wu, Y.-C.; Lo, T.-S.; Li, Y.-H. Cancer incidence in patients with schizophrenia or bipolar disorder: A nationwide population-based study in Taiwan, 1997–2009. Schizophr. Bull. 2013, 39, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Catts, V.S.; Catts, S.V.; O’Toole, B.I.; Frost, A.D.J. Cancer incidence in patients with schizophrenia and their first-degree relatives—A meta-analysis. Acta Psychiatr. Scand. 2008, 117, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Leucht, S.; Burkard, T.; Henderson, J.; Maj, M.; Sartorius, N. Physical illness and schizophrenia: A review of the literature. Acta Psychiatr. Scand. 2007, 116, 317–333. [Google Scholar]
- Howard, L.M.; Barley, E.A.; Davies, E.; Rigg, A.; Lempp, H.; Rose, D.; Taylor, D.; Thornicroft, G. Cancer diagnosis in people with severe mental illness: Practical and ethical issues. Lancet Oncol. 2010, 11, 797–804. [Google Scholar] [CrossRef]
- Fond, G.; Macgregor, A.; Attal, J.; Larue, A.; Brittner, M.; Ducasse, D.; Capdevielle, D. Antipsychotic drugs: Pro-cancer or anti-cancer? A systematic review. Med. Hypotheses 2012, 79, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Irwin, K.E.; Henderson, D.C.; Knight, H.P.; Pirl, W.F. Cancer care for individuals with schizophrenia. Cancer 2014, 120, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Crespi, B. Autism and cancer risk. Autism. Res. 2011, 4, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Kao, H.-T.; Buka, S.L.; Kelsey, K.T.; Gruber, D.F.; Porton, B. The correlation between rates of cancer and autism: An exploratory ecological investigation. PLoS ONE 2010, 5, e9372. [Google Scholar] [CrossRef] [PubMed]
- Crawley, J.N.; Heyer, W.-D.; LaSalle, J.M. Autism and cancer share risk genes, pathways, and drug targets. Trends Genet. 2016, 32, 139–146. [Google Scholar] [CrossRef] [PubMed]
- De Rubeis, S.; Buxbaum, J.D. Genetics and genomics of autism spectrum disorder: Embracing complexity. Hum. Mol. Genet. 2015, 24, R24–R31. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Takumi, T. Genomic and genetic aspects of autism spectrum disorder. Biochem. Biophys. Res. Commun. 2014, 452, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Vorstman, J.A.S.; Staal, W.G.; van Daalen, E.; van Engeland, H.; Hochstenbach, P.F.R.; Franke, L. Identification of novel autism candidate regions through analysis of reported cytogenetic abnormalities associated with autism. Mol. Psychiatry 2006, 11, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Van Loon, B.; Markkanen, E.; Hübscher, U. Oxygen as a friend and enemy: How to combat the mutational potential of 8-oxo-guanine. DNA Repair 2010, 9, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Horejsí, Z.; Koed, K.; Krämer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Beckman, K.B.; Ames, B.N. Oxidative decay of DNA. J. Biol. Chem. 1997, 272, 19633–19636. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Dianov, G.L.; Hübscher, U. Mammalian base excision repair: The forgotten archangel. Nucleic Acids Res. 2013, 2013, gkt076. [Google Scholar] [CrossRef] [PubMed]
- Lombard, D.B.; Chua, K.F.; Mostoslavsky, R.; Franco, S.; Gostissa, M.; Alt, F.W. DNA repair, genome stability, and aging. Cell 2005, 120, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [PubMed]
- Hegde, M.L.; Mantha, A.K.; Hazra, T.K.; Bhakat, K.K.; Mitra, S.; Szczesny, B. Oxidative genome damage and its repair: Implications in aging and neurodegenerative diseases. Mech. Ageing Dev. 2012, 133, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Canugovi, C.; Misiak, M.; Ferrarelli, L.K.; Croteau, D.L.; Bohr, V.A. The role of DNA repair in brain related disease pathology. DNA Repair 2013, 12, 578–587. [Google Scholar] [CrossRef] [PubMed]
- Bosshard, M.; Markkanen, E.; van Loon, B. Base excision repair in physiology and pathology of the central nervous system. Int. J. Mol. Sci. 2012, 13, 16172–16222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, A.L.; Schär, P. DNA glycosylases: In DNA repair and beyond. Chromosoma 2012, 121, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.S.; Murphy, D.L.; Sweasy, J.B. Base excision repair and cancer. Cancer Lett. 2012, 327, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Markkanen, E.; Dorn, J.; Hübscher, U. MUTYH DNA glycosylase: The rationale for removing undamaged bases from the DNA. Front. Genet. 2013, 4, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markkanen, E.; van Loon, B.; Ferrari, E.; Parsons, J.L.; Dianov, G.L.; Hübscher, U. Regulation of oxidative DNA damage repair by DNA polymerase λ and MutYH by cross-talk of phosphorylation and ubiquitination. Proc. Natl. Acad. Sci. USA 2012, 109, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Markkanen, E.; Hübscher, U.; van Loon, B. Regulation of oxidative DNA damage repair: The adenine: 8-oxo-guanine problem. Cell Cycle 2012, 11, 1070–1075. [Google Scholar] [CrossRef] [PubMed]
- Van Loon, B.; Hübscher, U. An 8-oxo-guanine repair pathway coordinated by MUTYH glycosylase and DNA polymerase λ. Proc. Natl. Acad. Sci. USA 2009, 106, 18201–18206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavin, M.F. Ataxia-telangiectasia: From a rare disorder to a paradigm for cell signalling and cancer. Nat. Rev. Mol. Cell Biol. 2008, 9, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Patterson, D. Molecular genetic analysis of down syndrome. Hum. Genet. 2009, 126, 195–214. [Google Scholar] [CrossRef] [PubMed]
- Rabin, K.R.; Whitlock, J.A. Malignancy in children with trisomy 21. Oncologist 2009, 14, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.K.; Jones, A.M. Evidence of toxicity, oxidative stress, and neuronal insult in autism. J. Toxicol. Environ. Health Part B 2006, 9, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Chauhan, V. Oxidative stress in autism. Pathophysiology 2006, 13, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Chauhan, V.; Brown, W.T.; Cohen, I. Oxidative stress in autism: Increased lipid peroxidation and reduced serum levels of ceruloplasmin and transferrin—The antioxidant proteins. Life Sci. 2004, 75, 2539–2549. [Google Scholar] [CrossRef] [PubMed]
- Clay, H.B.; Sillivan, S.; Konradi, C. Mitochondrial dysfunction and pathology in bipolar disorder and schizophrenia. Int. J. Dev. Neurosci. 2011, 29, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Pandya, C.D.; Howell, K.R.; Pillai, A. Antioxidants as potential therapeutics for neuropsychiatric disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 46, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Larsson, N.-G. Somatic mitochondrial DNA mutations in mammalian aging. Annu. Rev. Biochem. 2010, 79, 683–706. [Google Scholar] [CrossRef] [PubMed]
- Korotkova, E.I.; Misini, B.; Dorozhko, E.V.; Bukkel, M.V.; Plotnikov, E.V.; Linert, W. Study of OH radicals in human serum blood of healthy individuals and those with pathological schizophrenia. Int. J. Mol. Sci. 2011, 12, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Lagouge, M.; Larsson, N.-G. The role of mitochondrial DNA mutations and free radicals in disease and ageing. J. Int. Med. 2013, 273, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Herken, H.; Uz, E.; Ozyurt, H.; Söğüt, S.; Virit, O.; Akyol, O. Evidence that the activities of erythrocyte free radical scavenging enzymes and the products of lipid peroxidation are increased in different forms of schizophrenia. Mol. Psychiatry 2001, 6, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.; Moraes, C.T. Mechanisms linking mtDNA damage and aging. Free Radic. Biol. Med. 2015, 85, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Dietrich-Muszalska, A.; Olas, B. Modifications of blood platelet proteins of patients with schizophrenia. Platelets 2009, 20, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Sirota, P.; Gavrieli, R.; Wolach, B. Overproduction of neutrophil radical oxygen species correlates with negative symptoms in schizophrenic patients: Parallel studies on neutrophil chemotaxis, superoxide production and bactericidal activity. Psychiatry Res. 2003, 121, 123–132. [Google Scholar] [CrossRef]
- Grima, G.; Benz, B.; Parpura, V.; Cuénod, M.; Do, K.Q. Dopamine-induced oxidative stress in neurons with glutathione deficit: Implication for schizophrenia. Schizophr. Res. 2003, 62, 213–224. [Google Scholar] [CrossRef]
- Do, K.Q.; Trabesinger, A.H.; Kirsten-Krüger, M.; Lauer, C.J.; Dydak, U.; Hell, D.; Holsboer, F.; Boesiger, P.; Cuénod, M. Schizophrenia: Glutathione deficit in cerebrospinal fluid and prefrontal cortex in vivo. Eur. J. Neurosci. 2000, 12, 3721–3728. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.K.; Leonard, S.; Reddy, R.D. Increased nitric oxide radicals in postmortem brain from patients with schizophrenia. Schizophr. Bull. 2004, 30, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Andreazza, A.C.; Yeung, P.Y.; Isaacs-Trepanier, C.; Young, L.T. Oxidation and nitration in dopaminergic areas of the prefrontal cortex from patients with bipolar disorder and schizophrenia. J. Psychiatry Neurosci. 2014, 39, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, A.; Broedbaek, K.; Fink-Jensen, A.; Knorr, U.; Greisen Soendergaard, M.; Henriksen, T.; Weimann, A.; Jepsen, P.; Lykkesfeldt, J.; Poulsen, H.E.; et al. Increased systemic oxidatively generated DNA and RNA damage in schizophrenia. Psychiatry Res. 2013, 209, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Muraleedharan, A.; Menon, V.; Rajkumar, R.P.; Chand, P. Assessment of DNA damage and repair efficiency in drug naïve schizophrenia using comet assay. J. Psychiatr. Res. 2015, 68, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Mahadik, S.P.; Evans, D.; Lal, H. Oxidative stress and role of antioxidant and ω-3 essential fatty acid supplementation in schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2001, 25, 463–493. [Google Scholar] [CrossRef]
- Sertan Copoglu, U.; Virit, O.; Hanifi Kokacya, M.; Orkmez, M.; Bulbul, F.; Binnur Erbagci, A.; Semiz, M.; Alpak, G.; Unal, A.; Ari, M.; et al. Increased oxidative stress and oxidative DNA damage in non-remission schizophrenia patients. Psychiatry Res. 2015, 229, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Emiliani, F.E.; Sedlak, T.W.; Sawa, A. Oxidative stress and schizophrenia. Curr. Opin. Psychiatry 2014, 27, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, N.; Arnold, S.E. Evidence for oxidative DNA damage in the hippocampus of elderly patients with chronic schizophrenia. Am. J. Geriatr. Psychiatry 2004, 12, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Mamdani, F.; Rollins, B.; Morgan, L.; Sequeira, P.A.; Vawter, M.P. The somatic common deletion in mitochondrial DNA is decreased in schizophrenia. Schizophr. Res. 2014, 159, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Steullet, P.; Cabungcal, J.-H.; Kulak, A.; Kraftsik, R.; Chen, Y.; Dalton, T.P.; Cuénod, M.; Do, K.Q. Redox dysregulation affects the ventral but not dorsal hippocampus: Impairment of parvalbumin neurons, gamma oscillations, and related behaviors. J. Neurosci. 2010, 30, 2547–2558. [Google Scholar] [CrossRef] [PubMed]
- Cabungcal, J.-H.; Steullet, P.; Kraftsik, R.; Cuénod, M.; Do, K.Q. Early-life insults impair parvalbumin interneurons via oxidative stress: Reversal by N-acetylcysteine. Biol. Psychiatry 2013, 73, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Cabungcal, J.-H.; Counotte, D.S.; Lewis, E.M.; Tejeda, H.A.; Piantadosi, P.; Pollock, C.; Calhoon, G.G.; Sullivan, E.M.; Presgraves, E.; Kil, J.; et al. Juvenile antioxidant treatment prevents adult deficits in a developmental model of schizophrenia. Neuron 2014, 83, 1073–1084. [Google Scholar] [CrossRef] [PubMed]
- Cabungcal, J.-H.; Steullet, P.; Morishita, H.; Kraftsik, R.; Cuénod, M.; Hensch, T.K.; Do, K.Q. Perineuronal nets protect fast-spiking interneurons against oxidative stress. Proc. Natl. Acad Sci. USA 2013, 110, 9130–9135. [Google Scholar] [CrossRef] [PubMed]
- Trifunovic, A. Mitochondrial DNA and ageing. Biochim. Biophys. Acta 2006, 1757, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Shpyleva, S.; Ivanovsky, S.; de Conti, A.; Melnyk, S.; Tryndyak, V.; Beland, F.A.; James, S.J.; Pogribny, I.P. Cerebellar oxidative DNA damage and altered DNA methylation in the BTBR T+tf/J mouse model of autism and similarities with human post mortem cerebellum. PLoS ONE 2014, 9, e113712. [Google Scholar] [CrossRef] [PubMed]
- Sajdel-Sulkowska, E.M.; Xu, M.; Koibuchi, N. Increase in cerebellar neurotrophin-3 and oxidative stress markers in autism. Cerebellum 2009, 8, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Audhya, T.; Chauhan, V. Brain region-specific glutathione redox imbalance in autism. Neurochem. Res. 2012, 37, 1681–1689. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.; Melnyk, S.; Pavliv, O.; Bai, S.; Nick, T.G.; Frye, R.E.; James, S.J. Evidence of oxidative damage and inflammation associated with low glutathione redox status in the autism brain. Trans. Psychiatry 2012, 2, e134. [Google Scholar] [CrossRef] [PubMed]
- Melnyk, S.; Fuchs, G.J.; Schulz, E.; Lopez, M.; Kahler, S.G.; Fussell, J.J.; Bellando, J.; Pavliv, O.; Rose, S.; Seidel, L.; et al. Metabolic imbalance associated with methylation dysregulation and oxidative damage in children with autism. J. Autism. Dev. Disord. 2012, 42, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Napoli, E.; Wong, S.; Giulivi, C. Evidence of reactive oxygen species-mediated damage to mitochondrial DNA in children with typical autism. Mol. Autism. 2013, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Gutierrez Rios, P.; Kuo, S.-H.; Akman, H.O.; Rosoklija, G.; Tanji, K.; Dwork, A.; Schon, E.A.; Dimauro, S.; Goldman, J.; et al. Mitochondrial abnormalities in temporal lobe of autistic brain. Neurobiol. Dis. 2013, 54, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Saadat, M.; Pakyari, N.; Farrashbandi, H. Genetic polymorphism in the DNA repair gene XRCC1 and susceptibility to schizophrenia. Psychiatry Res. 2008, 157, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Derakhshandeh, S.; Saadat, I.; Farrashbandi, H.; Saadat, M. Association between genetic polymorphism of XRCC1 Arg194Trp and risk of schizophrenia. Psychiatry Res. 2009, 169, 186. [Google Scholar] [CrossRef] [PubMed]
- Odemis, S.; Tuzun, E.; Gulec, H.; Semiz, U.B.; Dasdemir, S.; Kucuk, M.; Yalcınkaya, N.; Bireller, E.S.; Cakmakoglu, B.; Küçükali, C.I. Association between polymorphisms of DNA repair genes and risk of schizophrenia. Genet. Test. Mol. Biomark. 2015, 20, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Rosales-Reynoso, M.A.; Barros-Núñez, P.; Peprah, E. DNA repair/replication transcripts are down regulated in patients with Fragile X Syndrome. BMC Res. Notes 2013, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Benes, F.M.; Lim, B.; Subburaju, S. Site-specific regulation of cell cycle and DNA repair in post-mitotic GABA cells in schizophrenic versus bipolars. Proc. Natl. Acad Sci. USA 2009, 106, 11731–11736. [Google Scholar] [CrossRef] [PubMed]
- Magin, G.K.; Robison, S.H.; Breslin, N.; Wyatt, R.J.; Alexander, R.C. DNA repair and mutant frequency in schizophrenia. Mutat. Res. 1991, 255, 241–246. [Google Scholar] [CrossRef]
- Psimadas, D.; Messini-Nikolaki, N.; Zafiropoulou, M.; Fortos, A.; Tsilimigaki, S.; Piperakis, S.M. DNA damage and repair efficiency in lymphocytes from schizophrenic patients. Cancer Lett. 2004, 204, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Young, J.; McKinney, S.B.; Ross, B.M.; Wahle, K.W.J.; Boyle, S.P. Biomarkers of oxidative stress in schizophrenic and control subjects. Prostaglandins Leukot. Essent. Fatty Acids 2007, 76, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Catts, V.S.; Catts, S.V.; Jablensky, A.; Chandler, D.; Weickert, C.S.; Lavin, M.F. Evidence of aberrant DNA damage response signalling but normal rates of DNA repair in dividing lymphoblasts from patients with schizophrenia. World J. Biol. Psychiatry 2012, 13, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Arrieta, I.; Núñez, T.; Martínez, B.; Pérez, A.; Télez, M.; Criado, B.; Gainza, I.; Lostao, C.M. Chromosomal fragility in a behavioral disorder. Behav. Genet. 2002, 32, 397–412. [Google Scholar] [CrossRef] [PubMed]
- Main, P.A.E.; Thomas, P.; Angley, M.T.; Young, R.; Esterman, A.; King, C.E.; Fenech, M.F. Lack of evidence for genomic instability in autistic children as measured by the cytokinesis-block micronucleus cytome assay. Autism. Res. 2015, 8, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Main, P.A.E.; Thomas, P.; Esterman, A.; Fenech, M.F. Necrosis is increased in lymphoblastoid cell lines from children with autism compared with their non-autistic siblings under conditions of oxidative and nitrosative stress. Mutagenesis 2013, 28, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Bjørge, M.D.; Hildrestrand, G.A.; Scheffler, K.; Suganthan, R.; Rolseth, V.; Kuśnierczyk, A.; Rowe, A.D.; Vågbø, C.B.; Vetlesen, S.; Eide, L.; et al. Synergistic actions of Ogg1 and mutyh DNA glycosylases modulate anxiety-like behavior in mice. Cell Rep. 2015, 13, 2671–2678. [Google Scholar] [CrossRef] [PubMed]
- Regnell, C.E.; Hildrestrand, G.A.; Sejersted, Y.; Medin, T.; Moldestad, O.; Rolseth, V.; Krokeide, S.Z.; Suganthan, R.; Luna, L.; Bjørås, M.; et al. Hippocampal adult neurogenesis is maintained by Neil3-dependent repair of oxidative DNA lesions in neural progenitor cells. Cell Rep. 2012, 2, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Arguello, P.A.; Gogos, J.A. Modeling madness in mice: One piece at a time. Neuron 2006, 52, 179–196. [Google Scholar] [CrossRef] [PubMed]
- Meyer, U.; Feldon, J.; Fatemi, S.H. In-vivo rodent models for the experimental investigation of prenatal immune activation effects in neurodevelopmental brain disorders. Neurosci. Biobehav. Rev. 2009, 33, 1061–1079. [Google Scholar] [CrossRef] [PubMed]
- Peleg-Raibstein, D.; Feldon, J.; Meyer, U. Behavioral animal models of antipsychotic drug actions. Handb. Exp. Pharmacol. 2012, 361–406. [Google Scholar]
- Lee, Y.; Katyal, S.; Li, Y.; El-Khamisy, S.F.; Russell, H.R.; Caldecott, K.W.; McKinnon, P.J. The genesis of cerebellar interneurons and the prevention of neural DNA damage require XRCC1. Nat. Neurosci. 2009, 12, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.L.; Tait, P.S.; Finch, D.; Dianova, I.I.; Allinson, S.L.; Dianov, G.L. CHIP-mediated degradation and DNA damage-dependent stabilization regulate base excision repair proteins. Mol. Cell 2008, 29, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Markkanen, E.; Fischer, R.; Ledentcova, M.; Kessler, B.M.; Dianov, G.L. Cells deficient in base-excision repair reveal cancer hallmarks originating from adjustments to genetic instability. Nucleic Acids Res. 2015, 43, 3667–3679. [Google Scholar] [CrossRef] [PubMed]
- Tebbs, R.S.; Flannery, M.L.; Meneses, J.J.; Hartmann, A.; Tucker, J.D.; Thompson, L.H.; Cleaver, J.E.; Pedersen, R.A. Requirement for the Xrcc1 DNA base excision repair gene during early mouse development. Dev. Biol. 1999, 208, 513–529. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.J. The hippocampus in schizophrenia: A review of the neuropathological evidence and its pathophysiological implications. Psychopharmacology 2004, 174, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Hampson, D.R.; Blatt, G.J. Autism spectrum disorders and neuropathology of the cerebellum. Front. Neurosci. 2015, 9, 420. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.A.; Hashimoto, T.; Volk, D.W. Cortical inhibitory neurons and schizophrenia. Nat. Rev. Neurosci. 2005, 6, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Blatt, G.J.; Fatemi, S.H. Alterations in GABAergic biomarkers in the autism brain: Research findings and clinical implications. Anat. Rec. 2011, 294, 1646–1652. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Burgos, G.; Fish, K.N.; Lewis, D.A. GABA neuron alterations, cortical circuit dysfunction and cognitive deficits in schizophrenia. Neural Plast. 2011, 2011, 723184. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.A.; Curley, A.A.; Glausier, J.R.; Volk, D.W. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 2012, 35, 57–67. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markkanen, E.; Meyer, U.; Dianov, G.L. DNA Damage and Repair in Schizophrenia and Autism: Implications for Cancer Comorbidity and Beyond. Int. J. Mol. Sci. 2016, 17, 856. https://doi.org/10.3390/ijms17060856
Markkanen E, Meyer U, Dianov GL. DNA Damage and Repair in Schizophrenia and Autism: Implications for Cancer Comorbidity and Beyond. International Journal of Molecular Sciences. 2016; 17(6):856. https://doi.org/10.3390/ijms17060856
Chicago/Turabian StyleMarkkanen, Enni, Urs Meyer, and Grigory L. Dianov. 2016. "DNA Damage and Repair in Schizophrenia and Autism: Implications for Cancer Comorbidity and Beyond" International Journal of Molecular Sciences 17, no. 6: 856. https://doi.org/10.3390/ijms17060856
APA StyleMarkkanen, E., Meyer, U., & Dianov, G. L. (2016). DNA Damage and Repair in Schizophrenia and Autism: Implications for Cancer Comorbidity and Beyond. International Journal of Molecular Sciences, 17(6), 856. https://doi.org/10.3390/ijms17060856