
Meta-Analysis of Differential Connectivity in Gene Co-Expression Networks in Multiple Sclerosis

 , , , and
, , , and
Abstract
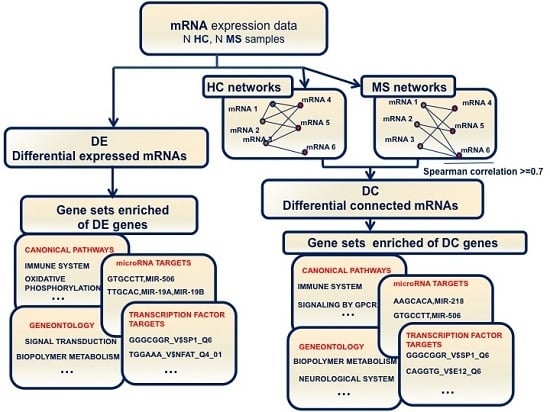
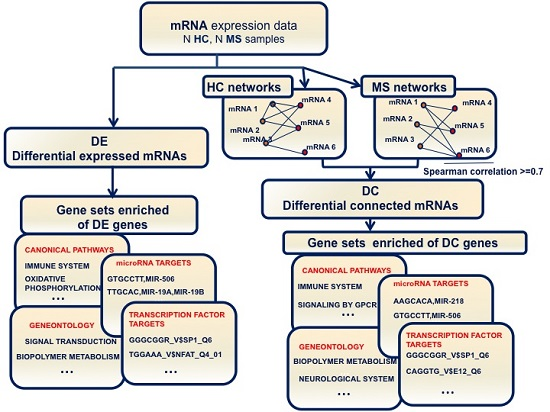
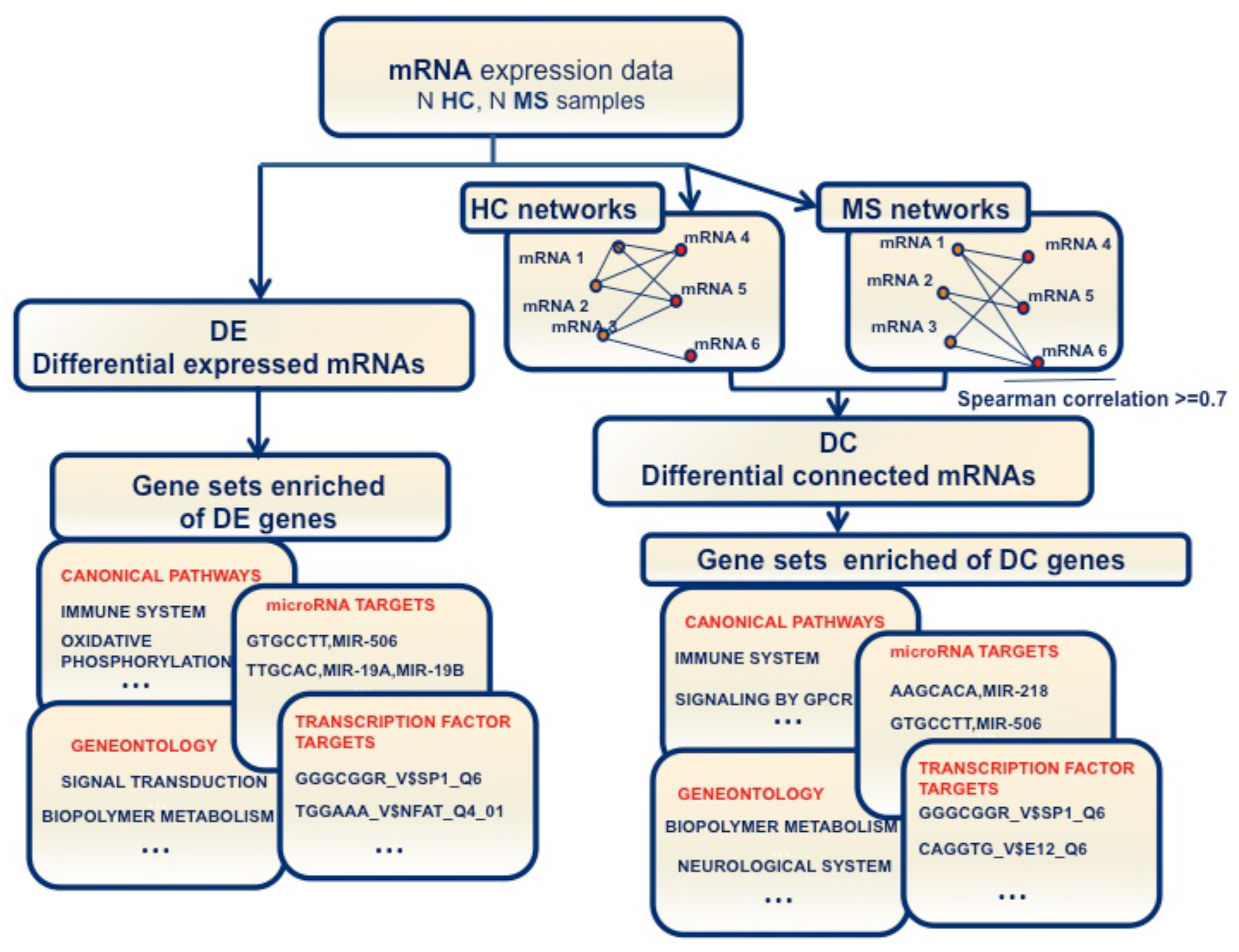
:
1. Introduction
2. Results
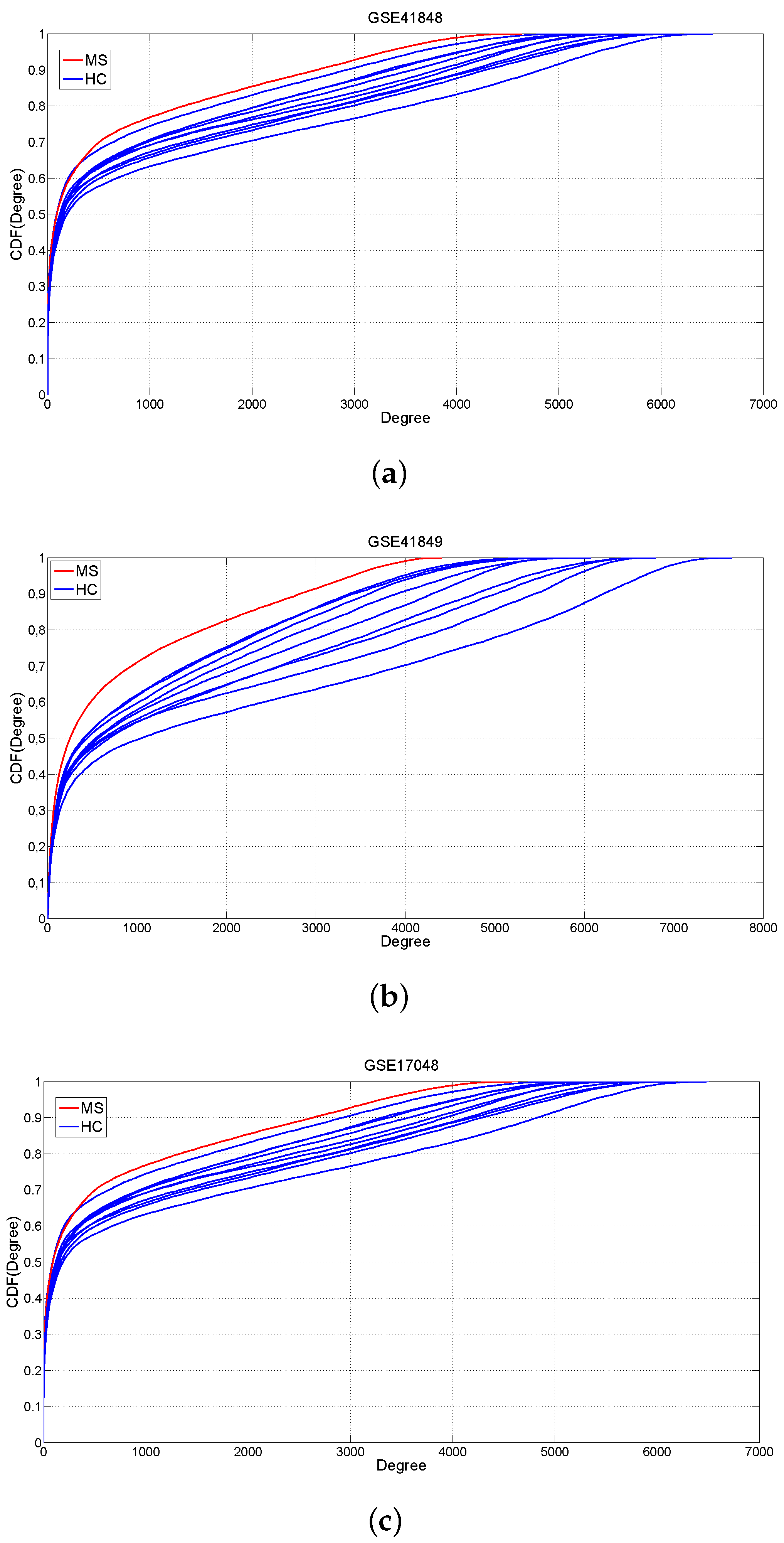
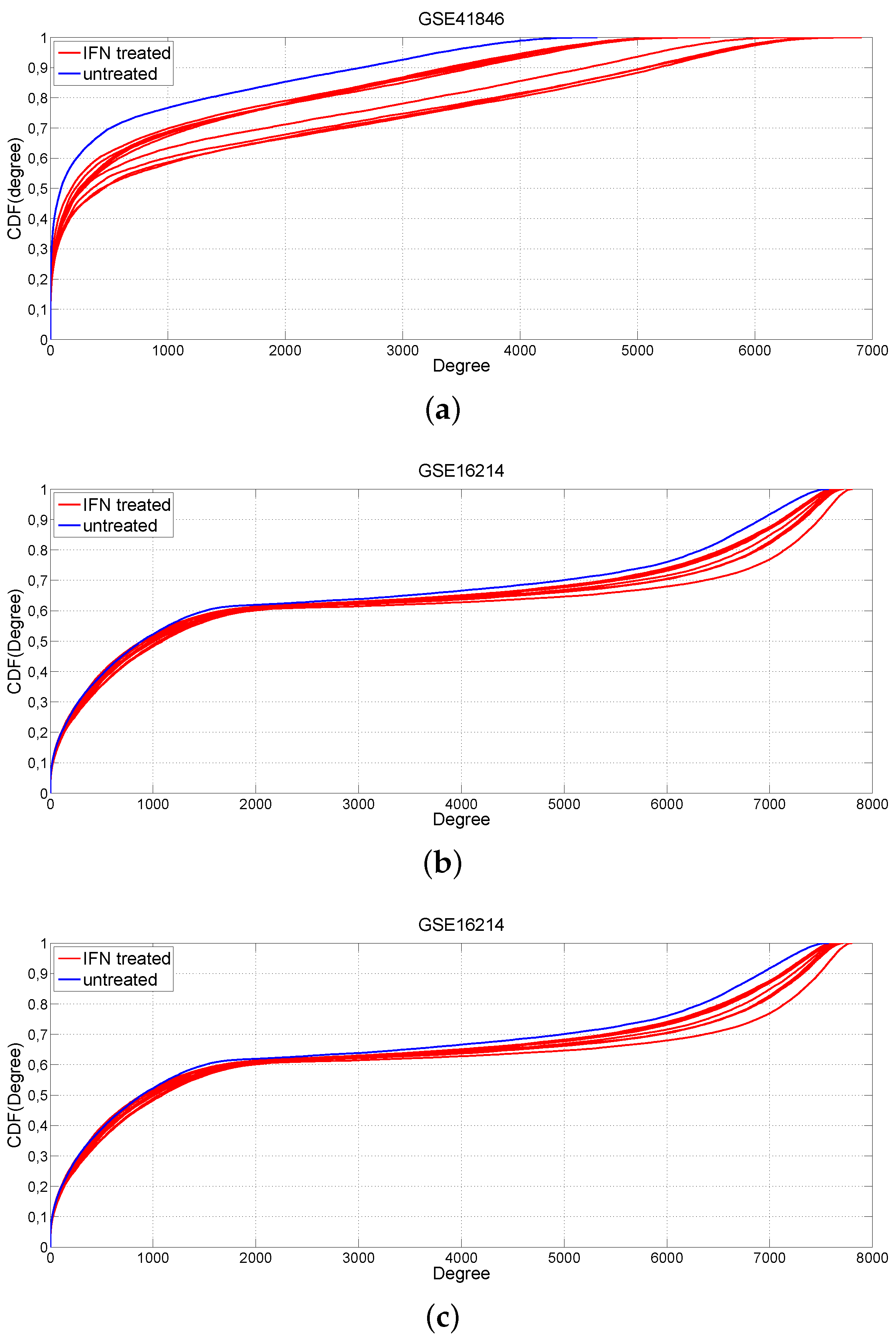
2.1. Changes of Connectivity in MS Networks
2.2. Differential Gene Expression and Connectivity
2.3. DE and DC Pathway Analysis
2.4. Analysis of Enriched Transcription Factor and MicroRNA Targets
3. Discussion
3.1. Comparison of MS vs. HCs
3.2. Comparison of IFN-β-Treated vs. Untreated MS Patients
3.3. Enrichment Pathway Analysis
3.4. Overrepresented 3’-UTR MicroRNA Binding Motifs
3.5. Overrepresented Transcription Factors Binding Sites
4. Materials and Methods
4.1. Data Collection
4.2. Data Analysis
4.2.1. Gene Differential Expression
4.2.2. Network Inference
4.2.3. Gene Differential Connectivity
4.2.4. Meta-Analysis of DC p-Values
4.2.5. Gene-Set Enrichment of SNPs by a Genome-Wide Association Study
4.2.6. Differential Expression and Connectivity Gene Set Enrichment Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| MS | Multiple Sclerosis |
| IFN | Interferon |
| HC | Healthy condition |
| DE | Differential expression |
| DC | Differentially connected |
| FDR | False discovery rate |
| DCA | Differential co-expression analysis |
| EAE | Transcription factor binding site |
| TFBS | Differential expression |
| CNS | Central nervous system |
| SNP | Single-nucleotide polymorphisms |
| GWAS | Genome-wide association study |
| HLA | Human leukocyte antigen |
References
- Ramagopalan, S.; Dobson, R.; Meyer, U.; Giovannoni, G. Multiple sclerosis: Risk factors, prodromes, and potential causal pathways. Lancet Neurol. 2010, 9, 727–739. [Google Scholar] [CrossRef]
- Sawcer, S.; Ban, M.; Maranian, M.; Yeo, T.W.; Compston, A.; Kirby, A.; Daly, M.J.; de Jager, P.L.; Walsh, E.; Lander, E.S.; et al. A high-density screen for linkage in multiple sclerosis. Am. J. Hum. Genet. 2005, 77, 454–467. [Google Scholar]
- International Multiple Sclerosis Genetics Consortium. Class II HLA interactions modulate genetic risk for multiple sclerosis. Nat. Genet. 2015, 47, 1107–1113. [Google Scholar]
- Patsopoulos, N.A.; Bayer Pharma, M.S.G.; Steering Committees of Studies Evaluating IFNbeta-1b and a CCR1-Antagonist; ANZgene Consortium; GeneMSA; IMGS Consortium; Esposito, F.; Reischl, J.; Lehr, S.; Bauer, D.; et al. Genome-wide meta-analysis identifies novel multiple sclerosis susceptibility loci. Ann. Neurol. 2011, 70, 897–912. [Google Scholar] [CrossRef] [PubMed]
- International Multiple Sclerosis Genetics Consortium; Wellcome Trust Case Control Consortium 2; Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; et al. Genetic risk and a primary role for cell-mediated immune mechanism in multiple sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Multiple Sclerosis Genetics Consortium; Beecham, A.H.; Patsopoulos, N.A.; Xifara, D.K.; Davis, M.F.; Kemppinen, A.; Cotsapas, C.; Shah, T.S.; Spencer, C.; Booth, D.; et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 2013, 45, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Pla, A.; Reverter, F.; Ruiz de Villa, M.; Comabella, M. Transcriptomics: mRNA and alternative splicing. J. Neuroimmunol. 2012, 248, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Fenoglio, C.; Ridolfi, E.; Cantoni, C.; de Riz, M.; Bonsi, R.; Serpente, M.; Villa, C.; Pietroboni, A.M.; Naismith, R.T.; Alvarez, E.; Parks, B.J.; et al. Decreased circulating miRNA levels in patients with primary progressive multiple sclerosis. Mult. Scler. 2013, 19, 1938–1942. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente, A. From ’differential expression’ to ’differential networking’—Identification of dysfunctional regulatory networks in diseases. Trends Genet. 2010, 26, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Xulvi-Brunet, R.; Li, H. Co-expression networks: Graph properties and topological comparisons. Bioinformatics 2010, 26, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Fairfax, B.; Humburg, P.; Makino, S.; Naranbhai, V.; Wong, D.; Lau, E.; Jostins, L.; Plant, K.; Andrews, R.; McGee, C.; et al. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science 2014, 343, 6175. [Google Scholar] [CrossRef] [PubMed]
- Anglani, R.; Creanza, T.; Liuzzi, V.; Piepoli, A.; Panza, A.; Andriulli, A.; Ancona, N. Loss of connectivity in cancer co-expression networks. PLoS ONE 2014, 9, e87075. [Google Scholar] [CrossRef] [PubMed]
- Taylor, I.W.; Linding, R.; Warde-Farley, D.; Liu, Y.; Pesquita, C.; Faria, D.; Bull, S.; Pawson, T.; Morris, Q.; Wrana, J.L. Dynamic modularity in protein interaction networks predicts breast cancer outcome. Nat. Biotechnol. 2009, 27, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Nayak, R.; Bernal, W.; Lee, J.; Kearns, M.; Cheung, V. Stress-induced changes in gene interactions in human cells. Nucleic Acids Res. 2014, 42, 1757–1771. [Google Scholar] [CrossRef] [PubMed]
- Rhinn, H.; Fujita, R.; Qiang, L.; Cheng, R.; Lee, J.; Abeliovich, A. Integrative genomics identifies APOE ϵ4 effectors in Alzheimer’s disease. Nature 2013, 500, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Nickles, D.; Chen, H.; Li, M.; Khankhanian, P.; Madireddy, L.; Caillier, S.; Santaniello, A.; Cree, B.; Pelletier, D.; Hauser, S.; et al. Blood RNA profiling in a large cohort of multiple sclerosis patients and healthy controls. Hum. Mol. Genet. 2013, 22, 4194–4205. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, K.; McKay, F.; Cox, M.; Riveros, C.; Armstrong, N.; Heard, R.N.; Vucic, S.; Williams, D.W.; Stankovich, J.; Brown, M.; et al. The multiple sclerosis whole blood mRNA transcriptome and genetic associations indicate dysregulation of specific T cell pathways in pathogenesis. Hum. Mol. Genet. 2010, 19, 2134–2143. [Google Scholar] [CrossRef] [PubMed]
- Ottoboni, O.; Keenan, B.; Tamayo, P.; Kuchroo, M.; Mesirov, J.; Buckle, G.; Khoury, S.; Hafler, D.; Weiner, H.; de Jager, P. An RNA profile identifies two subsets of multiple sclerosis patients differing in disease activity. Sci. Transl. Med. 2012, 4, 153ra131. [Google Scholar] [CrossRef] [PubMed]
- De Jager, P.; Jia, X.; Wang, J.; de Bakker, P.I.; Ottoboni, L.; Aggarwal, N.T.; Piccio, L.; Raychaudhuri, S.; Tran, D.; Aubin, C.; et al. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat. Genet. 2009, 41, 776–782. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- De Santis, G.; Ferracin, M.; Biondani, A.; Caniatti, L.; Tola, R.; Castellazzi, M.; Zagatti, B.; Battistini, L.; Borsellino, G.; Fainardi, E.; et al. Altered mirna expression in t regulatory cells in course of multiple sclerosis. J. Neuroimmunol. 2010, 226, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Gobin, S.; Montagne, L.; van Zutphen, M.; van der Valk, P.; van den Elsen, P.; de Groot, C. Upregulation of transcription factors controlling MHC expression in multiple sclerosis lesions. Glia 2001, 36, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Zhang, Y.; Katzaroff, A.; Edgar, B.; Buttitta, L. Hunting complex differential gene interaction patterns across molecular contexts. Nucleic Acids Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef] [PubMed]
- Bernardinelli, L.; Murgia, S.; Bitti, P.; Foco, L.; Ferrai, R.; Musu, L.; Prokopenko, I.; Pastorino, R.; Saddi, V.; Ticca, A.; et al. Association between the ACCN1 gene and multiple sclerosis in Central East Sardinia. PLoS ONE 2007, 2, e480. [Google Scholar] [CrossRef] [PubMed]
- Boiko, N.; Kucher, V.; Eaton, B.; Stockand, J. Inhibition of neuronal degenerin/epithelial Na+ channels by the multiple sclerosis drug 4-aminopyridine. J. Biol. Chem. 2013, 288, 9418–9427. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Hariri, D.; Caballero, B.; Zhang, S.; Bartlett, M.; Kaut, O.; Mount, D.W.; Wullner, U.; Sherman, S.J.; Falk, T. Comparative study of the neurotrophic effects elicited by VEGF-B and GDNF in preclinical in vivo models of Parkinson’s disease. Neuroscience 2014, 258, 385–400. [Google Scholar] [CrossRef] [PubMed]
- Fukada, Y.; Yasui, K.; Kitayama, M.; Doi, K.; Nakano, T.; Watanabe, Y.; Nakashima, K. Gene expression analysis of the murine model of amyotrophic lateral sclerosis: Studies of the Leu126delTT mutation in SOD1. Brain Res. 2007, 1160, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Majima, T.; Ogita, H.; Yamada, T.; Amano, H.; Togashi, H.; Sakisaka, T.; Tanaka–Okamoto, M.; Ishizaki, H.; Miyoshi, J.; Takai, Y. Involvement of afadin in the formation and remodeling of synapses in the hippocampus. Biochem. Biophys. Res. Commun. 2009, 385, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Vourc’h, P.; Martin, I.; Bonnet-Brilhault, F.; Marouillat, S.; Barthelemy, C.; Muh, P.J.; Andres, C. Mutation screening and association study of the UBE2H gene on chromosome 7q32 in autistic disorder. Psychiatr. Genet. 2003, 13, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Lesch, K.; Merker, S.; Reif, A.; Novak, M. Dances with black widow spiders: Dysregulation of glutamate signalling enters centre stage in ADHD. Eur. Neuropsychopharmacol. 2013, 23, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; O’Driscoll, C.; Bressler, J.; Kaufmann, W.; Rojas, C.; Slusher, B. Small molecule glutaminase inhibitors block glutamate release from stimulated microglia. Biochem. Biophys. Res. Commun. 2014, 443, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Azoitei, N.; Diepold, K.; Brunner, C.; Rouhi, A.; Genze, F.; Becher, A.; Kestler, H.; van Lint, J.; Chiosis, G.; Koren, J.; et al. HSP90 supports tumor growth and angiogenesis through PRKD2 protein stabilization. Cancer Res. 2014, 74, 7125–7136. [Google Scholar] [CrossRef] [PubMed]
- Fritschy, J.; Sidler, C.; Parpan, F.; Gassmann, M.; Kaupmann, K.; Bettler, B.; Benke, D. Independent maturation of the GABA(B) receptor subunits GABA(B1) and GABA(B2) during postnatal development in rodent brain. J. Comp. Neurol. 2004, 477, 235–252. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.G.; Wang, J.; Shan, Y.Z.; Yu, W.J.; Li, X.; Cong, C.H.; Wang, X. Identifying distinct candidate genes for early Parkinson’s disease by analysis of gene expression in whole blood. Neuro Endocrinol. Lett. 2014, 35, 398–404. [Google Scholar] [PubMed]
- Desplats, P.; Spencer, B.; Coffee, E.; Patel, P.; Michael, S.; Patrick, C.; Adame, A.; Rockenstein, E.; Masliah, E. Alpha-synuclein sequesters Dnmt1 from the nucleus: A novel mechanism for epigenetic alterations in Lewy body diseases. J. Biol. Chem. 2011, 286, 9031–9037. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Czech, T.; Felizardo, M.; Baumgartner, C.; Lubec, G. Aberrant expression of cytoskeleton proteins in hippocampus from patients with mesial temporal lobe epilepsy. Amino Acids 2006, 30, 477–493. [Google Scholar] [CrossRef] [PubMed]
- Gusareva, E.S.; Carrasquillo, M.M.; Bellenguez, C.; Cuyvers, E.; Colon, S.; Graff-Radford, N.R.; Petersen, R.C.; Dickson, D.W.; John, J.M.M.; Bessonov, K.; et al. Genome-wide association interaction analysis for Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2436–2443. [Google Scholar] [CrossRef] [PubMed]
- Lurbke, A.; Hagemeier, K.; Cui, Q.; Metz, I.; Bruck, W.; Antel, J.; Kuhlmann, T. Limited TCF7L2 expression in MS lesions. PLoS ONE 2013, 8, e72822. [Google Scholar] [CrossRef] [PubMed]
- Baranzini, S.; Wang, J.; Gibson, R.; Galwey, N.; Naegelin, Y.; Barkhof, F.; Radue, E.; Lindberg, R.L.; Uitdehaag, B.M.; Johnson, M.R.; et al. Genome-wide association analysis of susceptibility and clinical phenotype in multiple sclerosis. Hum. Mol. Genet. 2009, 18, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Zhang, K.; Gao, H. Paip1, an effective stimulator of translation initiation, is targeted by WWP2 for ubiquitination and degradation. Mol. Cell. Biol. 2014, 34, 4513–4522. [Google Scholar] [CrossRef] [PubMed]
- Martineau, Y.; Wanga, X.; Alaina, T.; Petroulakisa, E.; Shahbaziana, D.; Fabre, B.; Bousquet-Dubouch, M.P.; Monsarrat, B.; Pyronnet, S.; Sonenberg, N. Control of Paip1-eukayrotic translation initiation factor 3 interaction by amino acids through S6 kinase. Mol. Cell. Biol. 2014, 34, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Quadri, M.; Yang, X.; Cossu, G.; Olgiati, S.; Saddi, V.M.; Breedveld, G.J.; Ouyang, L.; Hu, J.; Xu, N.; Graafland, J.; et al. An exome study of Parkinson’s disease in Sardinia, a Mediterranean genetic isolate. Neurogenetics 2015, 16, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Shahmoradian, S.H.; Galaz-Montoya, J.G.; Schmid, M.F.; Cong, Y.; Ma, B.; Spiess, C.; Frydman, J.; Ludtke, S.J.; Chiu, W. TRiC’s tricks inhibit huntingtin aggregation. eLife 2013. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, L.L.; Wimmer, M.E.; Poplawski, S.G.; Tudor, J.C.; Kenworthy, C.A.; Liu, S.; Mizuno, K.; Garcia, B.A.; Zhang, N.R.; Giese, K.; et al. Memory acquisition and retrieval impact different epigenetic processes that regulate gene expression. BMC Genom. 2015, 16 (Suppl. 5), S5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allaire, N.; Bushnell, S.; Bienkowska, J.; Brock, G.; Carulli, J. Optimization of a high-throughput whole blood expression profiling methodology and its application to assess the pharmacodynamics of interferon (IFN) beta-1a or polyethylene glycol-conjugated IFN beta-1a in healthy clinical trial subjects. BMC Res. Notes 2013, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Palau, N.; Julia, A.; Ferrandiz, C.; Puig, L.; Fonseca, E.; Fernandez, E.; Lopez-Lasanta, M.; Tortosa, R.; Marsal, S. Genome-wide transcriptional analysis of T cell activation reveals differential gene expression associated with psoriasis. BMC Genom. 2013, 14, 285. [Google Scholar] [CrossRef] [PubMed]
- Hesse, D.; Krakauer, M.; Lund, H.; Sondergaard, H.; Limborg, S.; Soelberg Sorensen, P.; Sellebjerg, F. Disease protection and interleukin-10 induction by endogenous interferon-beta in multiple sclerosis? Eur. J. Neurol. 2011, 18, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Satoh, J.; Takitani, M.; Miyoshi, J.; Kino, Y. RNA-Seq data analysis identifies the comprehensive profile of in vivo interferon-beta-stimulated genes in multiple sclerosis. Clin. Exp. Neuroimmunol. 2015, 7, 39–51. [Google Scholar] [CrossRef]
- Thamilarasan, M.; Hecker, M.; Goertsches, R.; Paap, B.; Schroder, I.; Koczan, D.; Thiesen, H.; Zettl, U. Glatiramer acetate treatment effects on gene expression in monocytes of multiple sclerosis patients. Neuroinflammation 2013, 10, 126. [Google Scholar] [CrossRef] [PubMed]
- Veroni, C.; Marnetto, F.; Granieri, L.; Bertolotto, A.; Ballerini, C.; Repice, A.; Schirru, L.; Coghe, G.; Cocco, E.; Anastasiadou, E.; et al. Immune and Epstein-Barr virus gene expression in cerebrospinal fluid and peripheral blood mononuclear cells from patients with relapsing-remitting multiple sclerosis. Neuroinflammation 2015, 12, 132. [Google Scholar] [CrossRef] [PubMed]
- Henig, N.; Avidan, N.; Mandel, I.; Staun-Ram, E.; Ginzburg, E.; Paperna, T.; Pinter, R.; Miller, A. Interferon-beta induces distinct gene expression response patterns in human monocytes vs. T cells. PLoS ONE 2013, 8, e62366. [Google Scholar] [CrossRef] [PubMed]
- Cemana, S.; Saugstad, J. MicroRNAs: Meta-controllers of gene expression in synaptic activity emerge as genetic and diagnostic markers of human disease. Pharmacol. Ther. 2011, 130, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Hu, V.; Addington, A.; Hyman, A. Novel Autism subtype-dependent genetic variants are revealed by quantitative trait and subphenotype association analyses of published GWAS data. PLoS ONE 2011, 6, e19067. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Yang, J.; Chen, J.; Wu, Q.; Gong, W.; Zhang, J.; Shao, W.; Mu, J.; Yang, D.; Yang, Y.; et al. Differential co-expression and regulation analyses reveal different mechanisms underlying major depressive disorder and subsyndromal symptomatic depression. BMC Bioinform. 2015, 16, 112. [Google Scholar] [CrossRef] [PubMed]
- Derkow, K.; Bauer, J.; Hecker, M.; Paap, B.; Thamilarasan, M.; Koczan, D.; Schott, E.; Deuschle, K.; Bellmann-Strobl, J.; Paul, F.; et al. Multiple sclerosis: Modulation of Toll-Like Receptor (TLR) expression by interferon-beta includes upregulation of TLR7 in plasmacytoid dendritic cells. PLoS ONE 2013, 8, e70626. [Google Scholar] [CrossRef] [PubMed]
- Hecker, M.; Thamilarasan, M.; Koczan, D.; Schroder, I.; Flechtner, K.; Freiesleben, S.; Fullen, G.; Thiesen, H.; Zettl, U. MicroRNA expression changes during interferon-beta treatment in the peripheral blood of multiple sclerosis patients. Int. J. Mol. Sci. 2013, 14, 16087–16110. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, S.; Bustamante, M.; Porez-Miralles, F.; Rio, J.; Ruiz de Villa, M.; Vegas, E.; Nonell, L.; Deisenhammer, F.; Fissolo, N.; Nurtdinov, R.; et al. Search for specific biomarkers of IFN? Bioactivity in patients with multiple sclerosis. PLoS ONE 2011, 6, e23634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurevich, M.; Miron, G.; Falb, R.; Magalashvili, D.; Dolev, M.; Stern, Y.; Achiron, A. Transcriptional response to interferon beta-1a treatment in patients with secondary progressive multiple sclerosis. BMC Neurol. 2015, 15, 240. [Google Scholar] [CrossRef] [PubMed]
- Bornsen, L.; Christensen, J.; Ratzer, R.; Hedegaard, C.; Sondergaard, H.; Krakauer, M.; Hesse, D.; Nielsen, C.; Sorensen, P.; Sellebjerg, F. Endogenous interferon-β-inducible gene expression and interferon-β-treatment are associated with reduced T cell responses to myelin basic protein in multiple sclerosis. PLoS ONE 2015, 10, e0118830. [Google Scholar] [CrossRef] [PubMed]
- Van Baarsen, L.; Vosslamber, S.; Tijssen, M.; Baggen, J.; van der Voort, L.; Killestein, J.; van der Pouw Kraan, T.; Polman, C.; Verweij, C. Pharmacogenomics of Interferon-β Therapy in multiple sclerosis: Baseline IFN signature determines pharmacological differences between patients. PLoS ONE 2008, 3, e1927. [Google Scholar] [CrossRef] [PubMed]
- Bhan, V.; Hebb, A.; Holcik, M.; Korneluk, R.G.; Moore, C.; Robertson, G. Xaf-1, a Diagnostic Marker for Interferon Responsiveness in Multiple Sclerosis. WO Patent 2,007,101,341 A1, 13 September 2007. [Google Scholar]
- Hermanrud, C.; Ryner, M.; Engdahl, E.; Fogdell-Hahn, A. Anti-interferon beta antibody titers strongly correlate between two bioassays and in vivo biomarker expression, and indicates that a titer of 150 TRU/mL is a biologically functional cut-point. J. Interferon Cytokine Res. 2014, 34, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Samarajiwa, S.; Forster, S.; Auchettl, K.; Hertzog, P. INTERFEROME: The database of interferon regulated genes. Nucleic Acids Res. 2009, 37, D852–D857. [Google Scholar] [CrossRef] [PubMed]
- Adamczak, S.; de Rivero Vaccari, J.P.; Dale, G.; Brand, F.J.; Nonner, D.; Bullock, M.; Dahl, G.P.; Dietrich, W.D.; Keane, R.W. Pyroptotic neuronal cell death mediated by the AIM2 inflammasome. J. Cereb. Blood Flow Metab. 2014, 34, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, S.; Rio, J.; Urcelay, E.; Nurtdinov, R.; Bustamante, M.; Fernandez, O.; Oliver, B.; Zettl, U.; Brassat, D.; Killestein, J.; et al. NLRP3 inflammasome is associated with the response to IFN-β in patients with multiple sclerosis. Brain 2015, 138, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Naba, A.; Clauser, K.; Hoersch, S.; Liu, H.; Carr, S.; Hynes, R. The matrisome: In silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell. Proteom. 2012, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briggs, F.; Leung, L.; Barcellos, L. Annotation of functional variation within non-MHC MS susceptibility loci through bioinformatics analysis. Genes Immun. 2014, 15, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Arora, H.; Qureshi, R.; Park, W. miR-506 regulates epithelial mesenchymal transition in breast cancer cell lines. PLoS ONE 2013, 8, e64273. [Google Scholar] [CrossRef] [PubMed]
- McGuire, C.; Prinz, M.; Beyaert, R.; van Loo, G. Nuclear factor kappa B (NF-kB) in multiple sclerosis pathology. Trends Mol. Med. 2013, 19, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.; Cairns, M.; Gandhi, K.; Carroll, A.; Moscovis, S.; Stewart, G.; Broadley, S.; Scott, R.J.; Booth, D.R.; Lechner-Scott, J.; et al. MicroRNAs miR-17 and miR-20a inhibit T cell activation genes and are under-expressed in MS whole blood. PLoS ONE 2010, 5, e12132. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.; Leidinger, P.; Lange, J.; Borries, A.; Schroers, H.; Scheffler, M.; Lenhof, H.P.; Ruprecht, K.; Meese, E. Multiple sclerosis: MicroRNA expression profiles accurately differentiate patients with relapsing-remitting disease from healthy controls. PLoS ONE 2009, 4, e7440. [Google Scholar] [CrossRef] [PubMed]
- Noorbakhsh, F.; Ellestad, K.K.; Maingat, F.; Warren, K.G.; Han, M.H.; Steinman, L.; Baker, G.B.; Power, C. Impaired neurosteroid synthesis in multiple sclerosis. Brain 2011, 134, 2703–2721. [Google Scholar] [CrossRef] [PubMed]
- Thiebes, K.; Nam, H.; Cambronne, X.; Shen, R.; Glasgow, S.; Cho, H.; Kwon, J.; Goodman, R.; Lee, J.; Lee, S.; et al. miR-218 is essential to establish motor neuron fate as a downstream effector of Isl1-Lhx3. Nat. Commun. 2015, 6, 7718. [Google Scholar] [CrossRef] [PubMed]
- Ponomarev, E.; Veremeyko, T.; Barteneva, N.; Krichevsky, A.; Weiner, H. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-alpha-PU.1 pathway. Nat. Med. 2011, 17, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.; Chomyk, A.; Chang, A.; Ribaudo, M.V.; Deckard, S.A.; Doud, M.K.; Edberg, D.D.; Bai, B.; Li, M.; Baranzini, S.E.; et al. Hippocampal demyelination and memory dysfunction are associated with increased levels of the neuronal microRNA miR-124 and reduced AMPA receptors. Ann. Neurol. 2013, 73, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Pan, W.; Song, X.; Liu, Y.; Shao, X.; Tang, Y.; Liang, D.; He, D.; Wang, H.; Liu, W.; et al. The microRNA miR-23b suppresses IL-17-associated autoimmune inflammation by targeting TAB2, TAB3 and IKK-α. Nat. Med. 2012, 18, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.; Guerau-de Arellano, M.; Costinean, S.; Williams, J.; Bottoni, A.; Cox, G.; Satoskar, A.; Croce, C.; Racke, M.; Lovett-Racke, A.; et al. miR-29ab1-deficiency identifies a negative feedback loop controlling Th1 bias that is dysregulated in multiple sclerosis. J. Immunol. 2012, 189, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Guerau-de Arellano, M.; Smith, K.; Godlewski, J.; Liu, Y.; Winger, R.; Lawler, S.; Whitacre, C.; Racke, M.; Lovett-Racke, A. Micro-RNA dysregulation in multiple sclerosis favours pro-inflammatory T-cell-mediated autoimmunity. Brain 2011, 134, 3578–3589. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Carrasco, R. Crosstalk between microRNA-30a/b/c/d/e-5p and the canonical Wnt pathway: Implications for Multiple Myeloma therapy. Cancer Res. 2014, 74, 5351–5358. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Li, Z.; Zhang, G.; Guan, Y. Wnt signaling in remyelination in multiple sclerosis: Friend or foe? Mol. Neurobiol. 2014, 49, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Riveros, C.; Mellor, D.; Gandhi, K.; McKay, F.; Cox, M.; Berretta, R.; Vaezpour, S.; Inostroza-Ponta, M.; Broadley, S.; Heard, R.; et al. Transcription factor map as revealed by a genome-wide gene expression analysis of whole-blood mRNA transcriptome in multiple sclerosis. PLoS ONE 2010, 5, e14176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda, K.; Takaoka, A.; Taniguchi, T. Type I inteferon gene induction by the interferon regulatory factor family of transcription factors. Immunity 2006, 25, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.; Mony, J.; Lobner, M.; Khorooshi, R.; Owens, T. Interferon regulatory factor-7 modulates experimental autoimmune encephalomyelitis in mice. J. Neuroinflamm. 2011, 8, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menon, R.; di Dario, M.; Cordiglieri, C.; Musio, S.; la Mantia, L.; Milanese, C.; di Stefano, A.; Crabbio, M.; Franciotta, D.; Bergamaschi, R.; et al. Gender-based blood transcriptomes and interactomes in multiple sclerosis: Involvement of SP1 dependent gene transcription. J. Autoimmun. 2012, 38, J144–J155. [Google Scholar] [CrossRef] [PubMed]
- Kristjansdottir, G.; Sandling, J.K.; Bonetti, A.; Roos, I.M.; Milani, L.; Wang, C.; Gustafsdottir, S.M.; Sigurdsson, S.; Lundmark, A.; Tienari, P.J.; et al. Interferon regulatory factor 5 (IRF5) gene variants are associated with multiple sclerosis in three distinct populations. J. Med. Genet. 2008, 45, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Miskimins, W.K.; Miskimins, R. The Sp1 family of transcription factors is involved in p27Kip1-mediated activation of myelin basic protein gene expression. Mol. Cell. Biol. 2003, 23, 4035–4045. [Google Scholar] [CrossRef] [PubMed]
- Handel, A.; Sandve, G.; Disanto, G.; Berlanga-Taylor, A.; Gallone, G.; Hanwell, H.; Drabløs, F.; Gavin, G.; Ebers, G.; Ramagopalan, S. Vitamin D receptor ChIP-seq in primary CD4+ cells: Relationship to serum 25-hydroxyvitamin D levels and autoimmune disease. BMC Med. 2013, 11, 163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Fujiki, R.; Kitagawa, H.; Kato, S. 1alpha,25(OH)2D3-induced DNA methylation suppresses the human CYP27B1 gene. Mol. Cell. Endocrinol. 2007, 265–266, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.; Bernales, C.; Lee, J.; Sadovnick, A.; Traboulsee, A.; Vilarino-Guell, C. Analysis of CYP27B1 in multiple sclerosis. J. Neuroimmunol. 2014, 266, 64–66. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Haggarty, S.; Giacometti, E.; Dannenberg, J.H.; Joseph, N.; Gao, J.; Nieland, T.J.; Zhou, Y.; Wang, X.; Mazitschek, R.; et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 2009, 459, 55. [Google Scholar] [CrossRef] [PubMed]
- Kreple, C.; Lu, Y.; Taugher, R.; Schwager-Gutman, A.; Du, J.; Stump, M.; Wang, Y.; Ghobbeh, A.; Fan, R.; Cosme, C.V.; et al. Acid-sensing ion channels contribute to synaptic transmission and inhibit cocaine-evoked plasticity. Nat Neurosci. 2014, 17, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar]
- Altman, N.; Krzywinski, M. An RNA profile identifies two subsets of multiple sclerosis patients differing in disease activity. Nat. Methods 2015, 12, 899–900. [Google Scholar] [CrossRef] [PubMed]
- Segre, A.; Diagram Consortium; Magic Investigators; Groop, L.; Mootha, V.; Daly, M.J.; Altshuler, D. Common inherited variation in mitochondrial genes is not enriched for associations with type 2 diabetes or related glycemic traits. PLoS Genet. 2010, 6, e1001058. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | [17] | [18] | [19,20] | ||||||
|---|---|---|---|---|---|---|---|---|---|
| GEO datasets - SuperSeries | GSE41850 | GSE17048 | GSE16214 | ||||||
| Platform | Affymetrix Human Exon 1.0 ST Array | Illumina HumanHT-12 V3.0 expression beadchip | Affymetrix Human Genome U133 Plus 2.0 Array | ||||||
| Tissue | Whole blood | Whole blood | Peripheral blood mononuclear cells | ||||||
| Total genes studied | 17,551 | 13,162 | 19,520 | ||||||
| GEO datasets - SubSeries | Discovery sets | Replications sets | |||||||
| GSE41846 | GSE41848 | GSE41847 | GSE41849 | ||||||
| Comparisons | Untreated vs. IFN β-treated | MS vs. HC | Untreated vs IFN β-treated | MS vs. HC | MS vs. HC | Untreated vs. IFN β-treated | |||
| Condition of the analyzed arrays | MS | MS | HC | MS | MS | HC | MS | HC | MS |
| Number of the analyzed arrays | 98 | 37 | 79 | 63 | 15 | 46 | 36 | 45 | 176 |
| Untreated/IFN β-treated | 37/61 | 37/0 | - | 15/48 | 15/0 | - | 36/0 | - | 82/94 |
| Study | Genes | DE | DC | Reference |
|---|---|---|---|---|
| GSE41848 | 17,551 | 1520 | 741 | [17] |
| GSE41849 | 17,551 | 2164 | 449 | [17] |
| GSE17048 | 13,162 | 272 | 1223 | [18] |
| GSE41846 | 17,551 | 1358 | 759 | [17] |
| GSE41847 | 17,551 | 631 | 207 | [17] |
| GSE16214 | 19,520 | 1522 | 790 | [19,20] |
| MS vs. HC (in 2 of 3 datasets) | 7941 | 227 | 29 | [17,18] |
| MS vs. HC (in 3 datasets) | 7941 | 12 | 4 | [17,18] |
| IFN treated vs. untreated (in 2 of 3 datasets) | 14,794 | 563 | 60 | [17,19,20] |
| IFN treated vs. untreated (in 3 datasets) | 14,794 | 188 | 15 | [17,19,20] |
| Gene Symbol | Chr | Gene Name | Function | Reports in MS | Other Associations | References |
|---|---|---|---|---|---|---|
| with CNS Diseases | ||||||
| ACCN1 | 17 | acid sensing (proton gated) | ion transport, neuronal transmission, | yes | [26,27] | |
| ion channel 2 (ASICS2) | CNS development | |||||
| OR7A17 | 19 | olfactory receptor, family 7, | signal transducer activity | |||
| subfamily A, member 17 | ||||||
| FH | 1 | fumarate hydratase | protein binding, lyase activity | Parkinson’s disease | [28] | |
| GJA5 | 1 | gap junction protein, alpha 5, 40 kDa | cell communication, component | angiogenesis | ||
| of plasma membrane | ||||||
| PAIP1 | 5 | poly(A) binding protein interacting protein 1 | RNA and protein binding | FALS | [29] | |
| MLL4 | 12 | lysine (K)-specific methyltransferase 2D (KMT2D) | transcription regulatory DNA | developmental brain | [30] | |
| DNAJC2 | 7 | DnaJ (Hsp40) homolog, subfamily C, member 2 | ubiquitin and histone binding | |||
| KIF18A | 11 | kinesin family member 18A | microtubule binding | |||
| UBE2H | 7 | ubiquitin-conjugating enzyme E2H | ubiquitin-dependent protein catabolic processes | autism | [31] | |
| FLRT3 | 20 | fibronectin leucine-rich transmembrane protein 3 | cell adhesion, axonal guidance, | ADHD | [32] | |
| neuron projectiondevelopment | ||||||
| GLS | 2 | glutaminase | neurotransmitter secretion, synaptic transmission | yes | [33] | |
| LMBR1L | 12 | limb development membrane protein 1-like | endocytosis processes | |||
| KRTAP5-4 | 11 | keratin-associated protein 5-4 | component of keratin filament | |||
| PRKD2 | 19 | protein kinase D2 | adhesion, cell death, immunity | glioma | [34] | |
| GABRR2 | 6 | gamma-aminobutyric acid (GABA) A receptor, rho 2 | GABA-receptor activity, synapses component | epilepsy | [35] | |
| HIST2H2AB | 1 | histone cluster 2, H2ab | chromatin organization and silencing | |||
| PDE6D | 2 | phosphodiesterase 6D, cGMP-specific, rod, delta | visual perception, cell projection |
| Gene Symbol | Chr | Gene Name | Function | Reports in MS | Other Associations | Reference |
|---|---|---|---|---|---|---|
| with CNS Diseases | ||||||
| DLG1 | 3 | discs, large homolog 1 | axon guidance, regulation of | Parkinson’s disease | [36] | |
| myelination, immunological synapses | ||||||
| EYA3 | 1 | EYA transcriptional co-activator | histone dephosphorylation, DNA repair | |||
| and phosphatase 3 | ||||||
| PPA2 | 4 | pyrophosphatase (inorganic) 2 | mitochondrial matrix component | |||
| SPTA1 | 1 | spectrin, alpha, erythrocytic 1 | axon guidance, innate immune response | |||
| ELSPBP1 | 19 | epididymal sperm binding protein 1 | component of extracellular regions | |||
| PRKAR2A | 3 | protein kinase, cAMP-dependent, | innate immune response | Lewis body diseases | [37] | |
| regulatory, type II, alpha | ||||||
| TCP1 | 6 | t-complex 1 | myelin sheath component, | epilepsy | [38] | |
| DNA and protein binding | ||||||
| KHDRBS2 | 6 | KH domain containing, RNA binding, | protein heterodimerization activity | Alzheimer Disease | [39] | |
| signal transduction associated 2 | ||||||
| TRIM35 | 8 | tripartite motif containing 35 | apoptosis, immunity | |||
| SIKE1 | 1 | suppressor of IKBKE 1 | immunity, cell proliferation | |||
| HDAC2 | 6 | histone deacetylase 2 | transcriptional regulation, cell cycle | yes | [40] | |
| progression and developmental events | ||||||
| RNASE11 | 14 | ribonuclease, RNase A family, 11 | hydrolase activity |
| Gene Symbol | Chr | Gene Name | Function | Reports in MS | Other Associations | Reference |
|---|---|---|---|---|---|---|
| with CNS Diseases | ||||||
| SPATS2L | 2 | spermatogenesis associated, serine-rich 2-like | protein binding, poly(A) RNA binding | [47,48] | ||
| LAMP3 | 3 | lysosomal-associated membrane protein 3 | dendritic cell function, in adaptive immunity | yes | [49,50] | |
| ETV7 | 6 | Etsvariant 7 | transcriptional repressor activity, sequence-specific | yes | [51] | |
| DNA binding, protein binding | ||||||
| TRIM14 | 14 | tripartite motif containing 14 | protein binding, zinc ion binding | |||
| USP18 | 22 | ubiquitin-specific peptidase 18 | cysteine-type endopeptidase activity, | yes | [52] | |
| ubiquitin-specific protease activity, protein binding | ||||||
| OASL | 12 | 2’-5’-oligoadenylate synthetase-like | DNA, RNA and protein binding, | yes | [19] | |
| thyroid hormone receptor binding | ||||||
| MOV10 | 1 | Mov10 RISC complex RNA helicase | helicase activity, protein and ATP binding | synaptic plasticity | [53,54] | |
| OTOF | 2 | otoferlin | calcium ion binding, protein binding, | autism depressive disorder | [55,56] | |
| AP-2 adaptor complex binding | ||||||
| GBP1 | 1 | guanylate binding protein 1, | GTPase activity, protein binding | yes | [57] | |
| Interferon-Inducible | ||||||
| XAF1 | 17 | XIAP-associated factor 1 | protein binding, zinc ion binding | yes | [58] | |
| LY6E | 8 | lymphocyte antigen 6 complex, locus E | signal transduction | yes | [59] | |
| RSAD2 | 2 | radical S-adenosyl | catalytic activity, protein binding, | yes | [17] | |
| methionine domain containing 2 | metal ion binding | |||||
| ISG15 | 1 | ISG15 ubiquitin-like modifier | protein binding, protein tag | yes | [60] | |
| OAS3 | 12 | 2’-5’-oligoadenylate synthetase 3 | RNA binding, protein binding, | yes | [61] | |
| ATP binding, synthetase activity | ||||||
| DHX58 | 17 | DEXH (Asp-Glu-X-His) box polypeptide 58 | DNA, RNA and protein binding, helicase activity | yes | [62] |
| Reference | [17] | [17] | [18] | [19,20] | |||
|---|---|---|---|---|---|---|---|
| GEO datasets | GSE41846-GSE41848 | GSE41847-GSE41849 | GSE17048 | GSE16214 | |||
| Discovery sets | Replications sets | ||||||
| MS | HC | MS | HC | MS | HC | MS | |
| Number of the recruited subjects | 98 | 41 | 63 | 25 | 36 | 45 | 176 |
| Untreated/IFN β-treated | 37/61 | - | 15/48 | - | 36/0 | - | 82/94 |
| % Female | 70 | 75 | 65 | 55 | 81 | 64 | 82 |
| Clinical MS forms (CIS/RR/SP/PP in %) | 13/80/7/0 | - | 11/87/2/0 | - | 0/100/0/0 | - | 5/95/0/0 |
| Age at MS onset (mean; range) | - | - | - | - | 33 (19-56) | - | 36 (17-63) |
| Disease duration (mean/median; range/SD) | 8 (2-47) | - | 6 (2-27) | - | 15 (1-36) | - | 8 (0-43) |
| EDSS (mean/median; range/SD) | 2 (0-7) | - | 2 (0-6) | - | 2.7 (0-6.5) | - | - |
| Age (median/mean + range; mean + SD) | 45 (24-66) | 46 (26-66) | 46 (25-61) | 42 (27-61) | 48.5 (29-56) | 48.5 (23-77) | - |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Creanza, T.M.; Liguori, M.; Liuni, S.; Nuzziello, N.; Ancona, N. Meta-Analysis of Differential Connectivity in Gene Co-Expression Networks in Multiple Sclerosis. Int. J. Mol. Sci. 2016, 17, 936. https://doi.org/10.3390/ijms17060936
Creanza TM, Liguori M, Liuni S, Nuzziello N, Ancona N. Meta-Analysis of Differential Connectivity in Gene Co-Expression Networks in Multiple Sclerosis. International Journal of Molecular Sciences. 2016; 17(6):936. https://doi.org/10.3390/ijms17060936
Chicago/Turabian StyleCreanza, Teresa Maria, Maria Liguori, Sabino Liuni, Nicoletta Nuzziello, and Nicola Ancona. 2016. "Meta-Analysis of Differential Connectivity in Gene Co-Expression Networks in Multiple Sclerosis" International Journal of Molecular Sciences 17, no. 6: 936. https://doi.org/10.3390/ijms17060936
APA StyleCreanza, T. M., Liguori, M., Liuni, S., Nuzziello, N., & Ancona, N. (2016). Meta-Analysis of Differential Connectivity in Gene Co-Expression Networks in Multiple Sclerosis. International Journal of Molecular Sciences, 17(6), 936. https://doi.org/10.3390/ijms17060936