
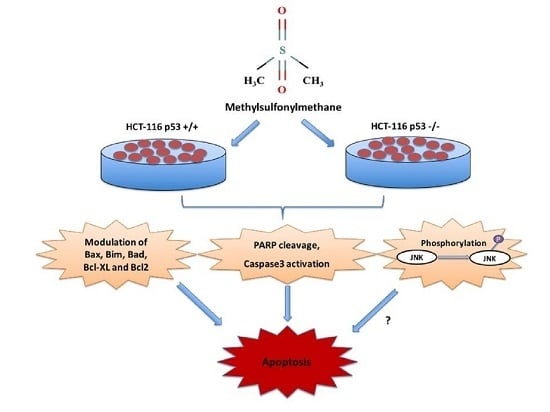
Methylsulfonylmethane Induces p53 Independent Apoptosis in HCT-116 Colon Cancer Cells

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Methylsulfonylmethane (MSM) Inhibited Proliferation of HCT-116 p53 +/+ and HCT-116 p53 −/− Colon Cancer Cells
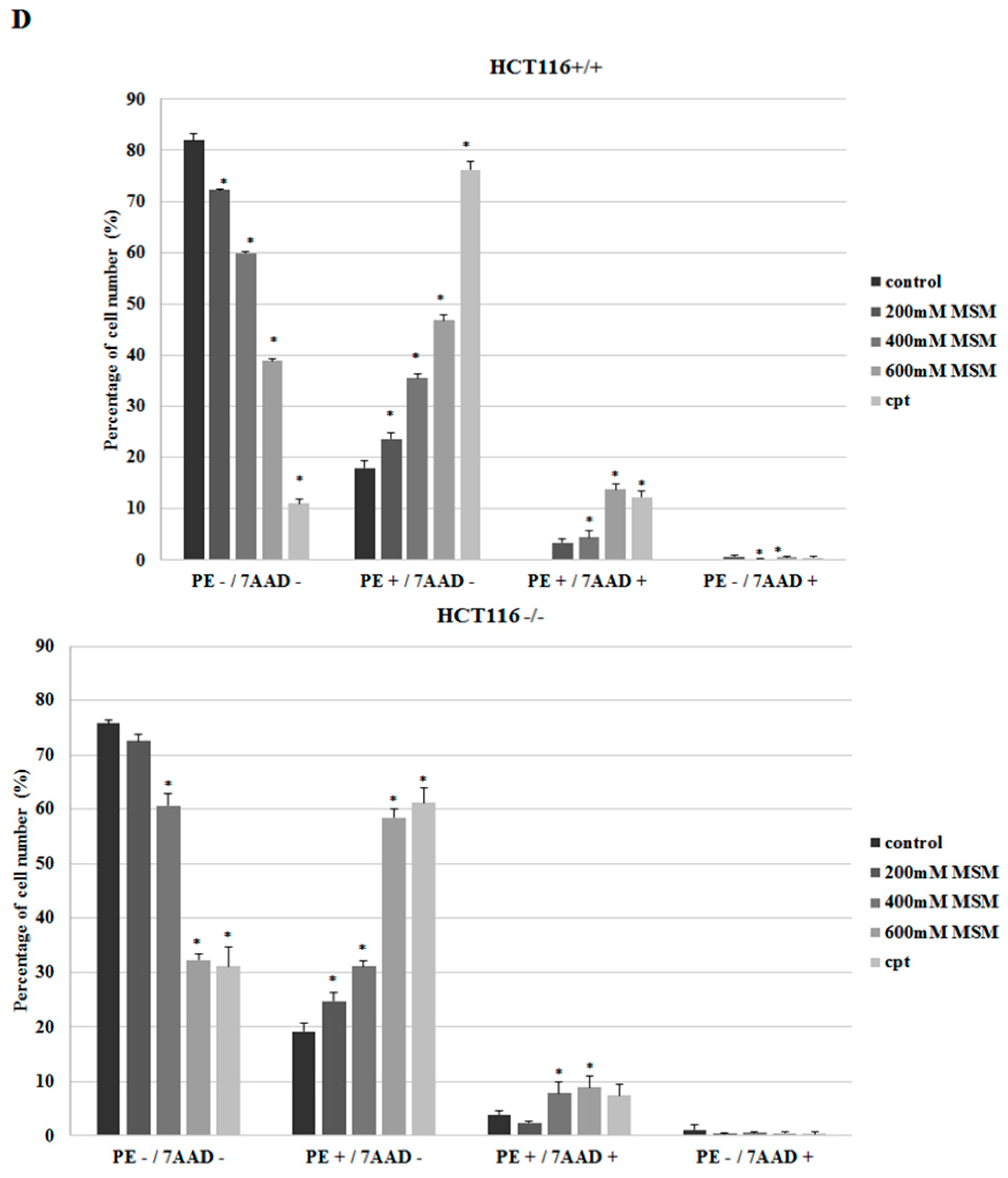
2.2. MSM Induced Apoptosis of HCT-116 p53 +/+ and HCT-116 p53 −/− Colon Cancer Cells
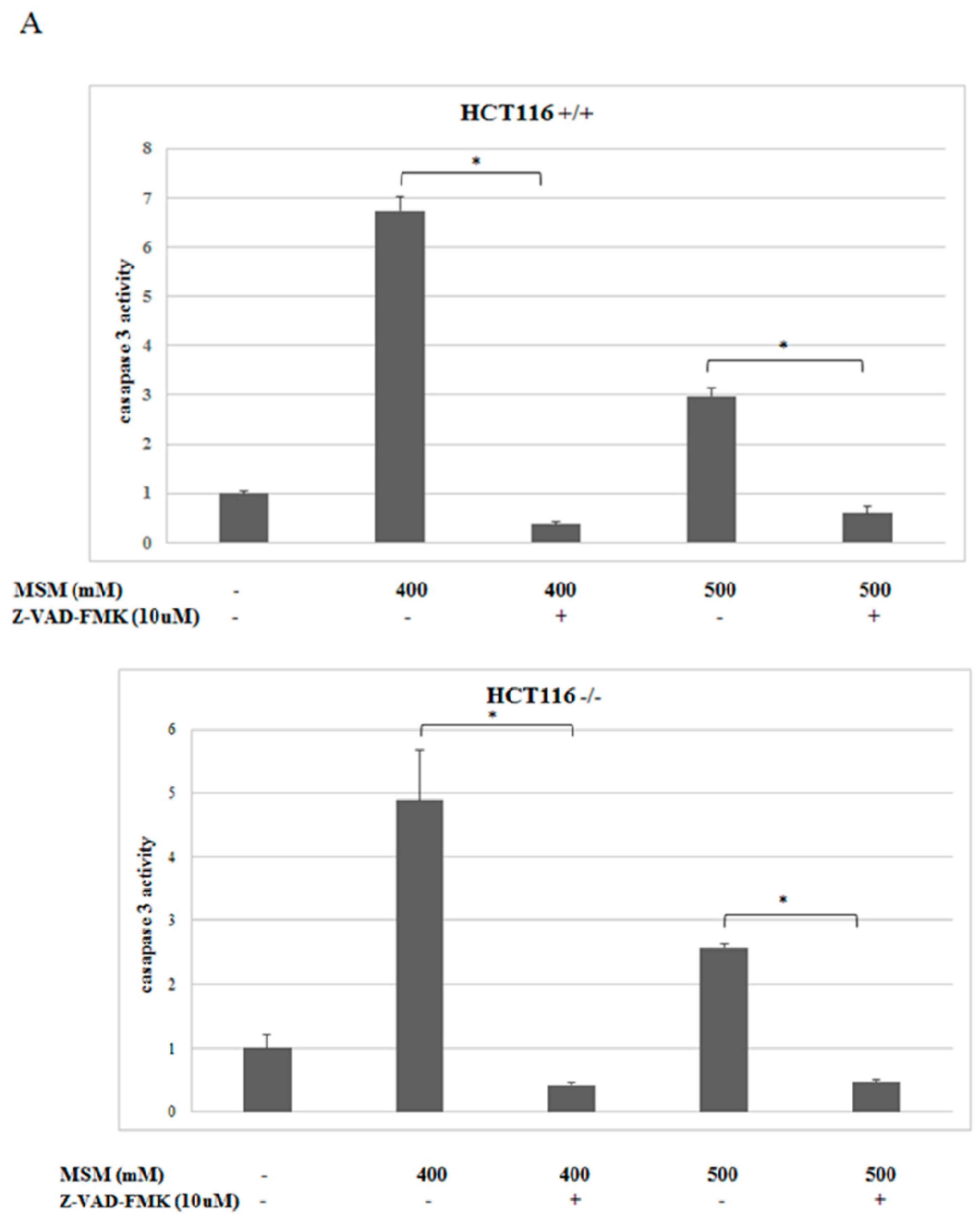
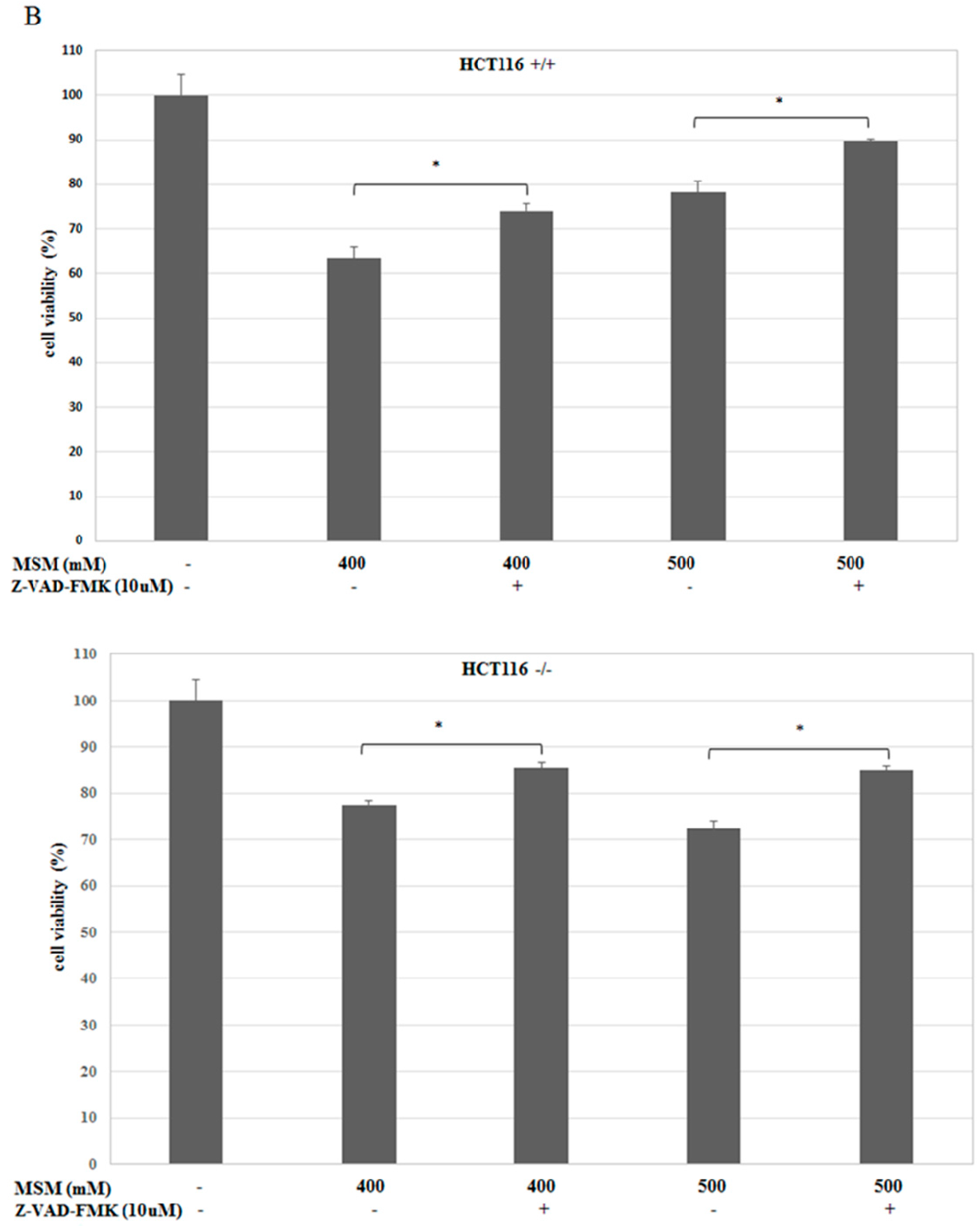
2.3. MSM-Increased Caspase-3 Activity in HCT-116 p53 +/+ and p53 −/− Colon Cancer Cells and Caspase-3 Inhibitor (Z-VAD-fmk) Rescued HCT-116 p53 +/+ and p53 −/− Colon Cancer Cells from MSM-Induced Cell Death
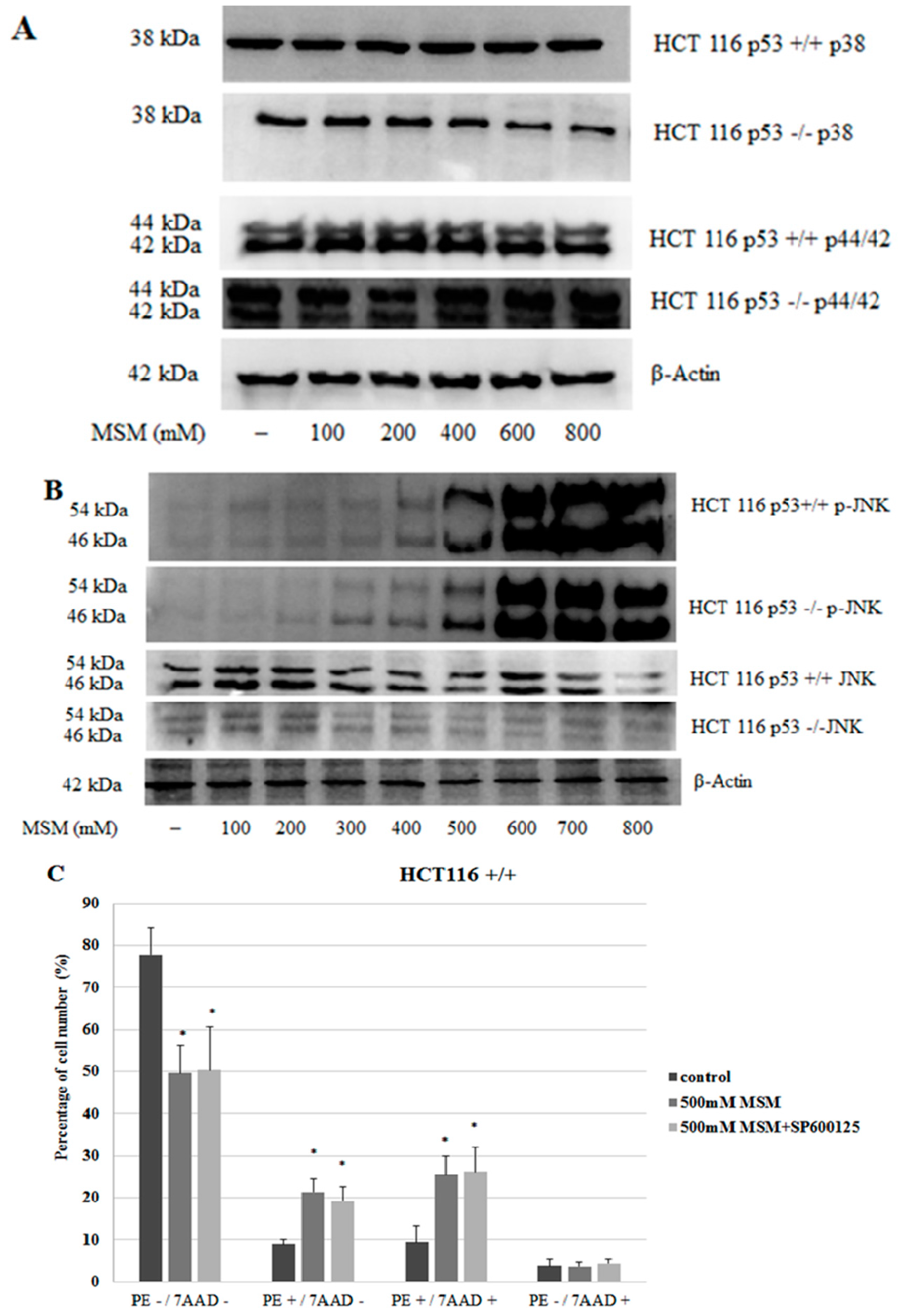
2.4. MSM Induced c-Jun N-Terminal Kinases (JNK) Phosphorylation in Both HCT-116 p53 +/+ and p53 −/− Colon Cancer Cells
2.5. MSM Treatment Modulated the Expressions of Various Genes and Proteins Involved in Apoptosis
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. Apoptosis Analysis by Flow Cytometry
4.5. Light and Fluorescence Microscopy
4.6. Caspase-3 Activity Assay
4.7. Western Blotting Analysis
4.8. RNA Extraction, cDNA Synthesis, and Quantitative Real-Time Polymerase Chain Reaction (qPCR)
4.9. Statistical Analysis
Author Contributions
Conflicts of Interest
References
- Rajamanickam, S.; Agarwal, R. Natural products and colon cancer: Current status and future prospects. Drug Dev. Res. 2008, 69, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Horvath, K.; Noker, P.E.; Somfai-Relle, S.; Glavits, R.; Financsek, I.; Schauss, A.G. Toxicity of methylsulfonylmethane in rats. Food Chem. Toxicol. 2002, 40, 1459–1462. [Google Scholar] [CrossRef]
- Magnuson, B.A.; Appleton, J.; Ames, G.B. Pharmacokinetics and Distribution of [35S]Methylsulfonylmethane following Oral Administration to Rats. J. Agric. Food Chem. 2007, 55, 1033–1038. [Google Scholar] [CrossRef] [PubMed]
- Ahn, H.; Kim, J.; Lee, M.J.; Kim, Y.J.; Cho, Y.W.; Lee, G.S. Methylsulfonylmethane inhibits NLRP3 inflammasome activation. Cytokine 2015, 71, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Caron, J.M.; Monteagudo, L.; Sanders, M.; Bannon, M.; Deckers, P.J. Methyl sulfone manifests anticancer activity in a metastatic murine breast cancer cell line and in human breast cancer tissue-part 2: Human breast cancer tissue. Chemotherapy 2013, 59, 24–34. [Google Scholar] [PubMed]
- Amirshahrokhi, K.; Bohlooli, S. Effect of methylsulfonylmethane on paraquat-induced acute lung and liver injury in mice. Inflammation 2013, 36, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Nipin, S.P.; Darvin, P.; Yoo, Y.B.; Joung, Y.H.; Kang, D.Y.; Kim, D.N.; Hwang, T.S.; Kim, S.Y.; Kim, W.S.; Lee, H.K.; et al. The combination of methylsulfonylmethane and tamoxifen inhibits the Jak2/STAT5b pathway and synergistically inhibits tumor growth and metastasis in ER-positive breast cancer xenografts. BMC Cancer 2015, 15, 474. [Google Scholar]
- Kim, J.H.; Shin, H.J.; Ha, H.L.; Park, Y.H.; Kwon, T.H.; Jung, M.R.; Moon, H.B.; Cho, E.S.; Son, H.Y.; Yu, D.Y. Methylsulfonylmethane suppresses hepatic tumor development through activation of apoptosis. World J. Hepatol. 2014, 6, 98–106. [Google Scholar] [PubMed]
- Joung, Y.H.; Na, Y.M.; Yoo, Y.B.; Darvin, P.; Sp, N.; Kang, D.Y.; Kim, S.Y.; Kim, H.S.; Choi, Y.H.; Lee, H.K.; et al. Combination of AG490, a Jak2 inhibitor, and methylsulfonylmethane synergistically suppresses bladder tumor growth via the Jak2/STAT3 pathway. Int. J. Oncol. 2014, 44, 883–895. [Google Scholar] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.G.; Choi, E.J. Apoptotic Signaling Pathways: Caspases and Stress-Activated Protein Kinases. J. Biochem. Mol. Biol. 2002, 35, 24–27. [Google Scholar] [CrossRef] [PubMed]
- Davids, M.S.; Letai, A. Targeting the B-cell lymphoma/leukemia 2 family in cancer. J. Clin. Oncol. 2012, 30, 3127–3135. [Google Scholar] [CrossRef] [PubMed]
- Yip, K.W.; Reed, J.C. Bcl-2 family proteins and cancer. Oncogene 2008, 27, 6398–6406. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, T.; Tanaka, S. Bim: Guardian of tissue homeostasis and critical regulator of the immune system, tumorigenesis and bone biology. Arch. Immunol. Ther. Exp. (Warsz) 2011, 59, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Gillings, A.S.; Balmanno, K.; Wiggins, C.M.; Johnson, M.; Cook, S.J. Apoptosis and autophagy: BIM as a mediator of tumour cell death in response to oncogene-targeted therapeutics. FEBS J. 2009, 276, 6050–6062. [Google Scholar] [CrossRef] [PubMed]
- Pflaum, J.; Schlosser, S.; Müller, M. p53 Family and Cellular Stress Responses in Cancer. Front. Oncol. 2014, 4, 285. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, L.A.; Manfredi, J.J. Another fork in the road-life or death decisions by the tumour suppressor p53. EMBO Rep. 2013, 14, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Pandit, B.; Gartel, A.L. Proteasome inhibitors induce p53-independent apoptosis in human cancer cells. Am. J. Pathol. 2011, 178, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yeung, J.H.; Hu, T.; Lee, W.Y.; Lu, L.; Zhang, L.; Shen, J.; Chan, R.L.; Wu, W.K.; Cho, C.H. Dihydrotanshinone induces p53-independent but ROS-dependent apoptosis in colon cancer cells. Life Sci. 2013, 93, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Mahyar-Roemer, M.; Kasten, A.; Mestres, P.; Roemer, K. Resveratrol induces colon tumor cell apoptosis independently of p53 and precede by epithelial differentiation, mitochondrial proliferation and membrane potential collapse. Int. J. Cancer 2001, 94, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Yu, J.; Zhang, L. PUMA, a potent killer with or without p53. Oncogene 2008, 27 (Suppl. S1), S71–S83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Kyriakis, J.M.; Banerjee, P.; Nikolakaki, E.; Dai, T.; Rubie, E.A.; Ahmad, M.F.; Avruch, J.; Woodgett, J.R. The stress-activated protein kinase subfamily of c-Jun kinases. Nature 1994, 369, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Lin, A. Activation of the JNK signaling pathway: Breaking the brake on apoptosis. Bioessays 2003, 25, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lin, A. Role of JNK activation in apoptosis: A double-edged sword. Cell Res. 2005, 15, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Graves, J.D.; Draves, K.E.; Craxton, A.; Saklatvala, J.; Krebs, E.G.; Clark, E.A. Involvement of stress-activated protein kinase and p38 mitogen-activated protein kinase in mIgM-induced apoptosis of human B lymphocytes. Proc. Natl. Acad. Sci. USA 1996, 93, 13814–13818. [Google Scholar] [CrossRef] [PubMed]
- Raingeaud, J.; Gupta, S.; Rogers, J.S.; Dickens, M.; Han, J.; Ulevitch, R.J.; Davis, R.J. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J. Biol. Chem. 1995, 270, 7420–7426. [Google Scholar] [PubMed]
- Xia, Z.; Dickens, M.; Raingeaud, J.; Davis, R.J.; Greenberg, M.E. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995, 270, 1326–1331. [Google Scholar] [CrossRef] [PubMed]
- Tournier, C.; Hess, P.; Yang, D.D.; Xu, J.; Turner, T.K.; Nimnual, A.; Bar-Sagi, D.; Jones, S.N.; Flavell, R.A.; Davis, R.J. Requirement of JNK for Stress-Induced Activation of the Cytochrome c-Mediated Death Pathway. Science 2000, 288, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Maundrell, K.; Antonsson, B.; Magnenat, E.; Camps, M.; Muda, M.; Chabert, C.; Gillieron, C.; Boschert, U.; Vial-Knecht, E.; Martinou, J.C.; et al. Bcl-2 undergoes phosphorylation by c-Jun N-terminal kinase/stress-activated protein kinases in the presence of the constitutively active GTP-binding protein Rac1. J. Biol. Chem. 1997, 272, 25238–25242. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Ichijo, H.; Korsmeyer, S.J. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G2/M. Mol. Cell. Biol. 1999, 19, 8469–8478. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef]
- Jafari, N.; Bohlooli, S.; Mohammadi, S.; Mazani, M. Cytotoxicity of methylsulfonylmethane on gastrointestinal (AGS, HepG2, and KEYSE-30) cancer cell lines. J. Gastrointest. Cancer 2012, 43, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Potapova, O.; Sergey, V.A.; Gorospe, M.; Dougherty, R.H.; Gaarde, W.A.; Boheler, K.R.; Holbrook, N.J. Targets of c-Jun NH2-terminal Kinase 2-mediated Tumor Growth Regulation Revealed by Serial Analysis of Gene Expression. Cancer Res. 2002, 62, 3257–3263. [Google Scholar] [PubMed]
- Potapova, O.; Gorospe, M.; Dougherty, R.H.; Dean, N.M.; Gaarde, W.A.; Holbrook, N.J. Inhibition of c-Jun N-terminal kinase 2 expression suppresses growth and induces apoptosis of human tumor cells in a p53-dependent manner. Mol. Cell. Biol. 2000, 20, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, Z.; Chen, S.; Jiao, Y.; Bai, C. ROS-mediated activation of JNK/p38 contributes partially to the pro-apoptotic effect of ajoene on cells of lung adenocarcinoma. Tumour Biol. 2016, 37, 3727–3738. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, S.Y.; Adler, V.; Buschmann, T.; Yin, Z.; Wu, X.; Jones, S.N.; Ronai, Z. JNK targets p53 ubiquitination and degradation in nonstressed cells. Genes Dev. 1998, 12, 2658–2663. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, T.; Potapova, O.; Bar-Shira, A.; Ivanov, V.N.; Fuchs, S.Y.; Henderson, S.; Fried, V.A.; Minamoto, T.; Alarcon-Vargas, D.; Pincus, M.R.; et al. Jun NH2-terminal kinase phosphorylation of p53 on Thr-81 is important for p53 stabilization and transcriptional activities in response to stress. J. Cancer Ther. 2012, 3, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Oleinik, N.V.; Krupenko, N.I.; Krupenko, S.A. Cooperation between JNK1 and JNK2 in activation of p53 apoptotic pathway. Oncogene 2007, 26, 7222–7230. [Google Scholar] [CrossRef] [PubMed]
- Karabay, A.Z.; Aktan, F.; Sunguroglu, A.; Buyukbingol, Z. Methylsulfonylmethane modulates apoptosis of LPS/IFN-γ-activated RAW 264.7 macrophage-like cells by targeting p53, Bax, Bcl-2, cytochrome c and PARP proteins. Immunopharmacol. Immunotoxicol. 2014, 36, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, J.G.; Chen, S.T.; Tafani, M.; Snyder, J.W.; Farber, J.L. The overexpression of Bax produces cell death upon induction of the mitochondrial permeability transition. J. Biol. Chem. 1998, 273, 7770–7775. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, L.; Strasser, A.; O’Reilly, L.A.; Hausmann, G.; Adams, J.M.; Cory, S.; and Huang, D.C. Bim: A novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 1998, 17, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Sionov, R.V.; Vlahopoulos, S.A.; Granot, Z. Regulation of Bim in Health and Disease. Oncotarget 2015, 6, 23058–23134. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, T.; Dass, C.R.; Choong, P.F. Bim-targeted cancer therapy: A link between drug action and underlying molecular changes. Mol. Cancer Ther. 2009, 8, 3173–3180. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.T.; Li, K.K.; Sun, S.S.; Chan, P.K.; Ooi, V.E.; Chiu, L.C. Activation of the JNK pathway promotes phosphorylation and degradation of BimEL—A novel mechanism of chemoresistance in T-cell acute lymphoblastic leukemia. Carcinogenesis 2008, 29, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Hubner, A.; Barrett, T.; Flavell, R.A.; Davis, R.J. Multisite phosphorylation regulates Bim stability and apoptotic activity. Mol. Cell 2008, 30, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, J.; DeGraff, W.G.; Gazdar, A.F.; Minna, J.D.; Mitchell, J.B. Evaluation of a tetrazolium-based semiautomated colorimetric assay: Assessment of chemosensitivity testing. Cancer Res. 1987, 47, 936–942. [Google Scholar] [PubMed]
- Gwak, H.S.; Shingu, T.; Chumbalkar, V.; Hwang, Y.H.; DeJournett, R.; Latha, K.; Koul, D.; Alfred Yung, W.K.; Powis, G.; Farrell, N.P.; et al. Combined action of the dinuclear platinum compound BBR3610 with the PI3-K inhibitor PX-866 in glioblastoma. Int. J. Cancer 2011, 128, 787–796. [Google Scholar] [CrossRef] [PubMed]











© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karabay, A.Z.; Koc, A.; Ozkan, T.; Hekmatshoar, Y.; Sunguroglu, A.; Aktan, F.; Buyukbingol, Z. Methylsulfonylmethane Induces p53 Independent Apoptosis in HCT-116 Colon Cancer Cells. Int. J. Mol. Sci. 2016, 17, 1123. https://doi.org/10.3390/ijms17071123
Karabay AZ, Koc A, Ozkan T, Hekmatshoar Y, Sunguroglu A, Aktan F, Buyukbingol Z. Methylsulfonylmethane Induces p53 Independent Apoptosis in HCT-116 Colon Cancer Cells. International Journal of Molecular Sciences. 2016; 17(7):1123. https://doi.org/10.3390/ijms17071123
Chicago/Turabian StyleKarabay, Arzu Zeynep, Asli Koc, Tulin Ozkan, Yalda Hekmatshoar, Asuman Sunguroglu, Fugen Aktan, and Zeliha Buyukbingol. 2016. "Methylsulfonylmethane Induces p53 Independent Apoptosis in HCT-116 Colon Cancer Cells" International Journal of Molecular Sciences 17, no. 7: 1123. https://doi.org/10.3390/ijms17071123
APA StyleKarabay, A. Z., Koc, A., Ozkan, T., Hekmatshoar, Y., Sunguroglu, A., Aktan, F., & Buyukbingol, Z. (2016). Methylsulfonylmethane Induces p53 Independent Apoptosis in HCT-116 Colon Cancer Cells. International Journal of Molecular Sciences, 17(7), 1123. https://doi.org/10.3390/ijms17071123