
One-Carbon Metabolism in Prostate Cancer: The Role of Androgen Signaling


Abstract
:1. Introduction
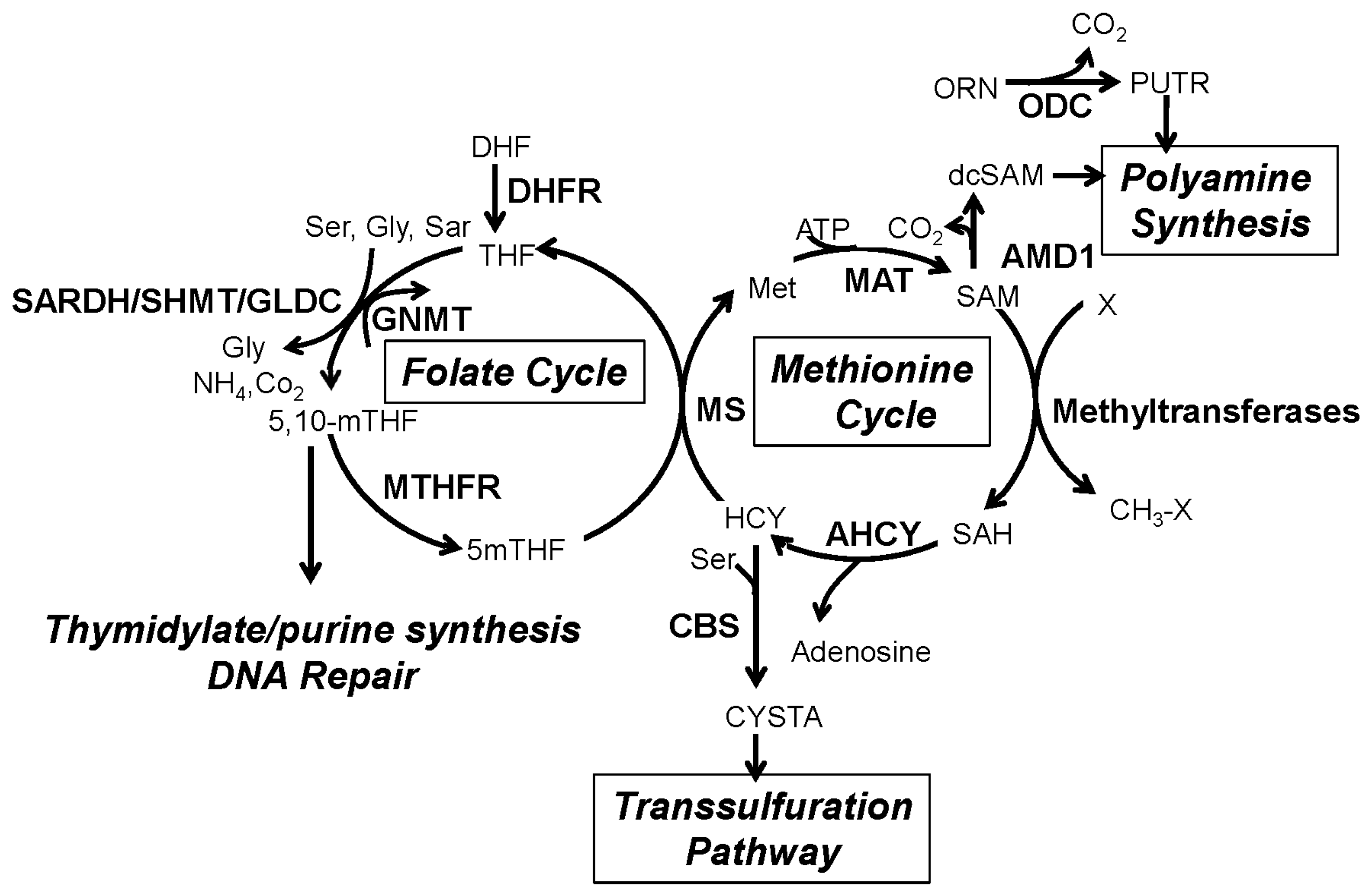
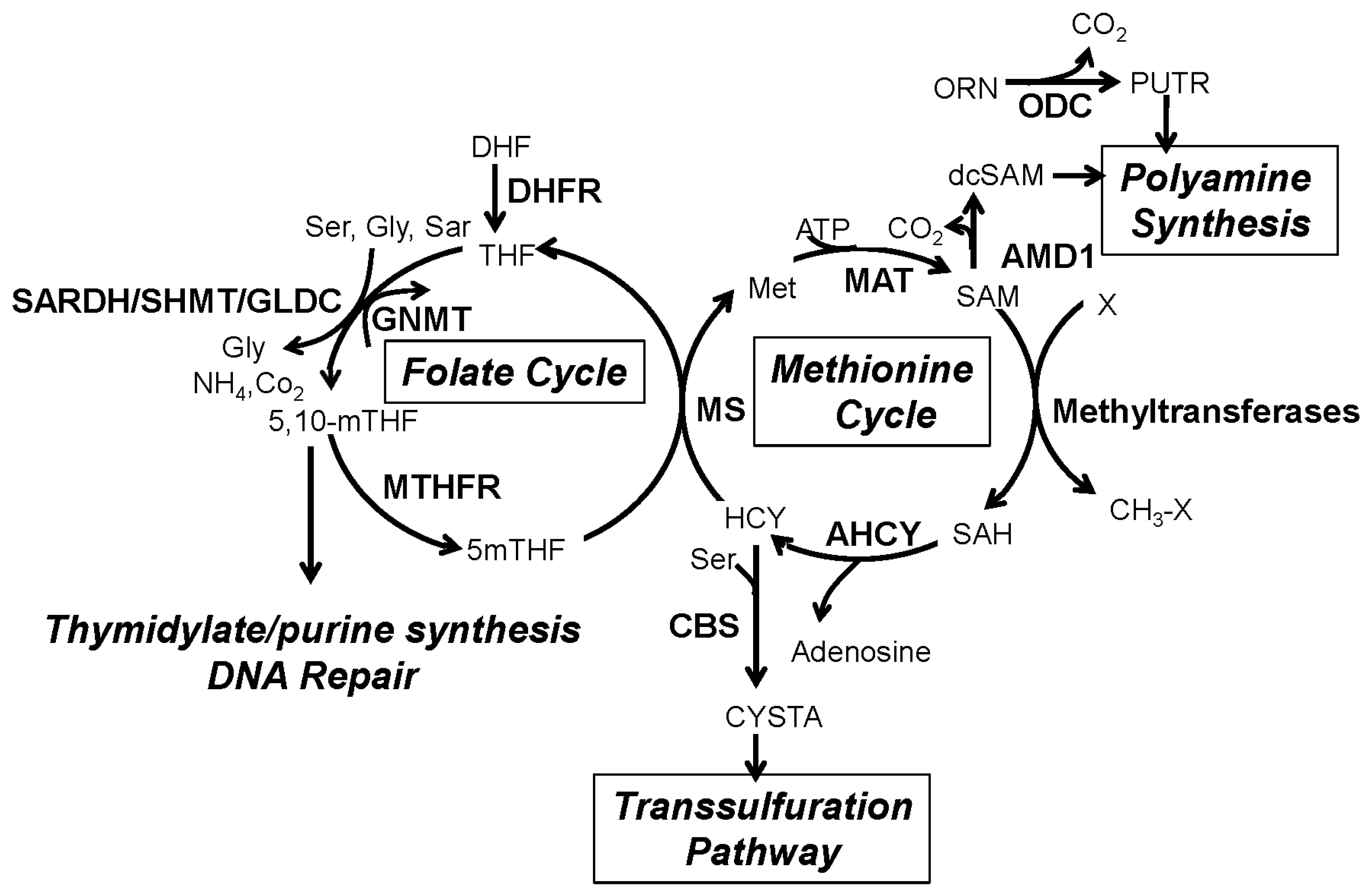
2. The One-Carbon Metabolism Network
3. One-Carbon Metabolism in Cancer
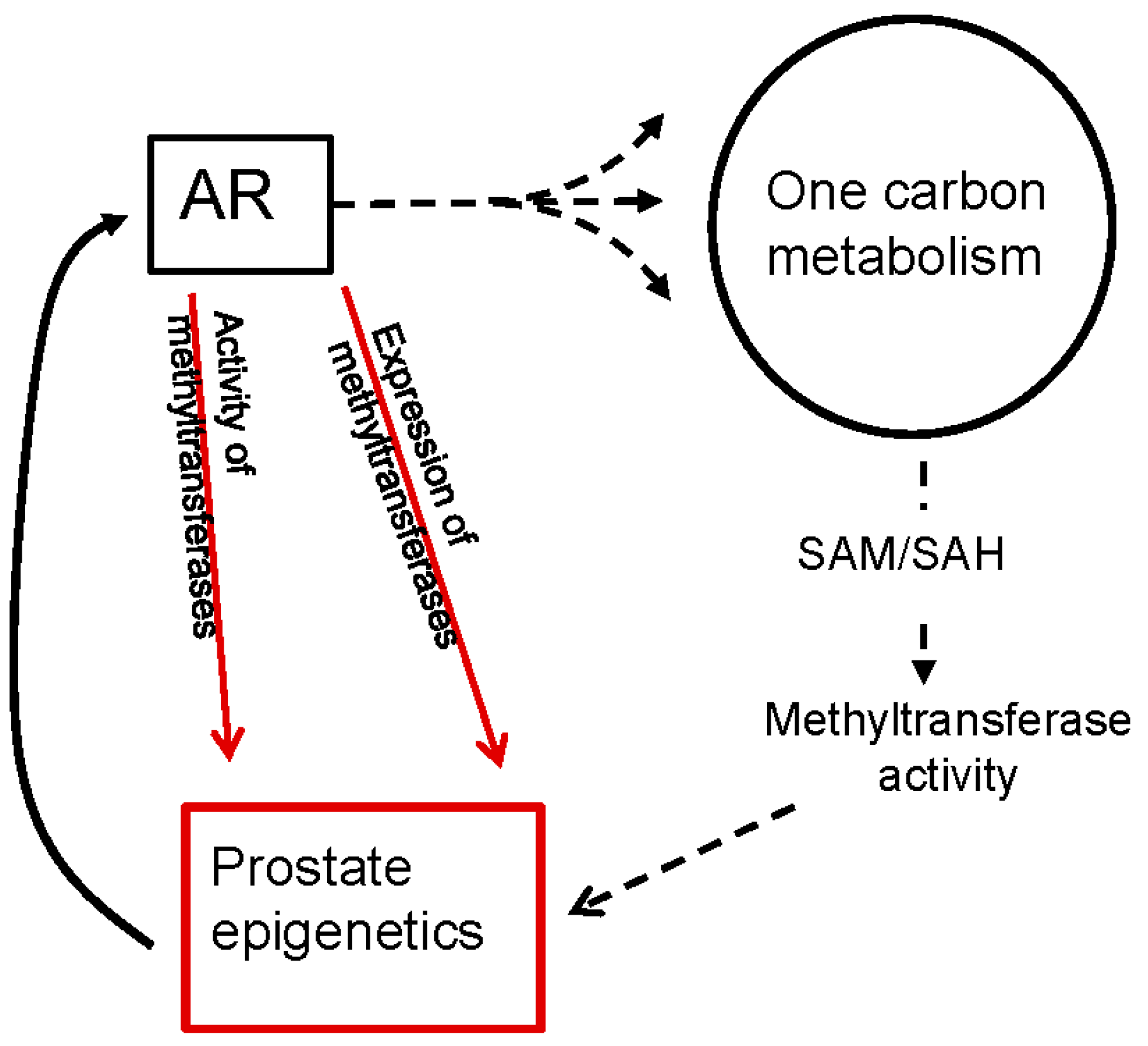
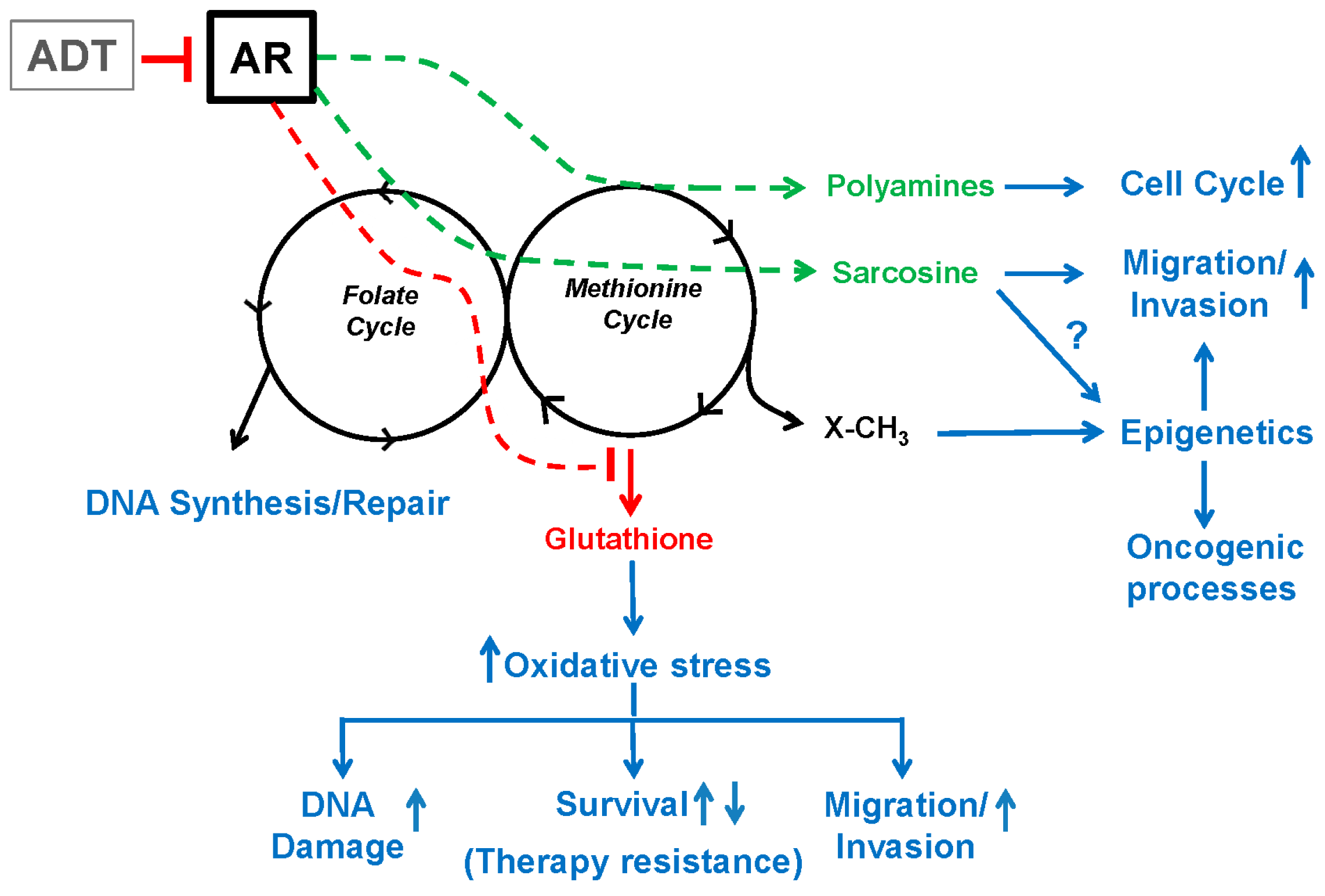
4. Androgen Signaling Modulates One-Carbon Metabolism and Epigenetics
5. Androgen Signaling Regulates the Expression of Enzymes Involved in the One-Carbon Metabolism Network
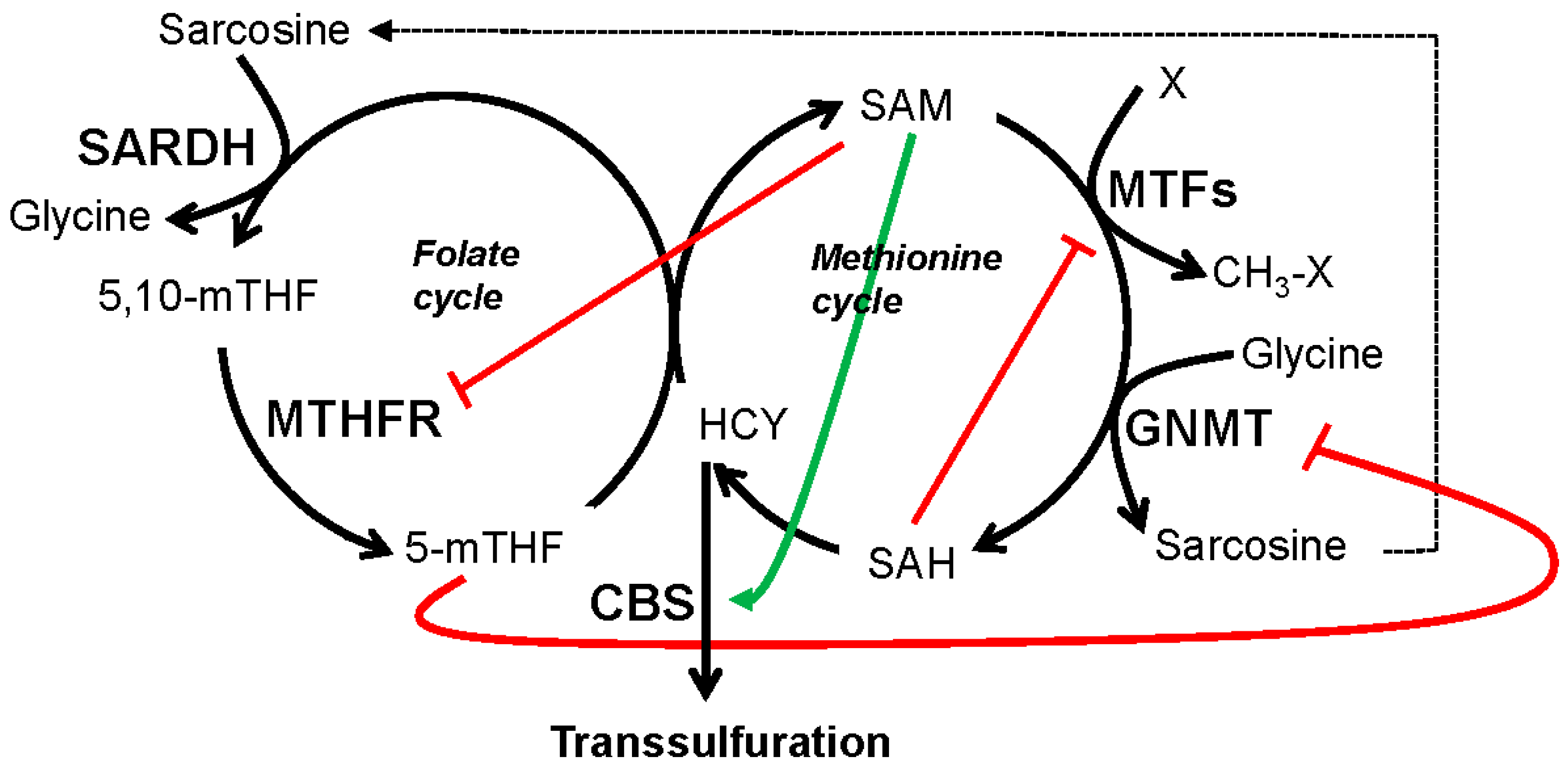
5.1. GNMT, SARDH and Sarcosine Metabolism
5.2. CBS and the Transsulfuration Pathway
5.3. ODC, SAM and Polyamine Synthesis
6. The Role of Androgen Signaling on Methyltransferases and the Epigenetics of PCa
6.1. DNA and Histone Methyltransferases in PCa
6.2. Androgen Signaling Regulates the Expression and Activity of Methyltransferases in PCa
7. Therapeutic Approaches to Prostate Cancer: Targeting the One-Carbon Metabolism
8. Summary and Conclusions
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- De Winter, R.J.A.; Janssen, P.J.; Sleddens, H.M.; Verleun-Mooijman, M.C.; Trapman, J.; Brinkmann, A.O.; Santerse, A.B.; Schroder, F.H.; van der Kwast, T.H. Androgen receptor status in localized and locally progressive hormone refractory human prostate cancer. Am. J. Pathol. 1994, 144, 735–746. [Google Scholar]
- Chodak, G.W.; Kranc, D.M.; Puy, L.A.; Takeda, H.; Johnson, K.; Chang, C. Nuclear localization of androgen receptor in heterogeneous samples of normal, hyperplastic and neoplastic human prostate. J. Urol. 1992, 147, 798–803. [Google Scholar] [PubMed]
- Sadi, M.V.; Walsh, P.C.; Barrack, E.R. Immunohistochemical study of androgen receptors in metastatic prostate cancer. Comparison of receptor content and response to hormonal therapy. Cancer 1991, 67, 3057–3064. [Google Scholar] [CrossRef]
- Hammerer, P.; Madersbacher, S. Landmarks in hormonal therapy for prostate cancer. BJU Int. 2012, 110 (Suppl. 1), 23–29. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Beer, T.M.; Higano, C.S.; Anand, A.; Taplin, M.E.; Efstathiou, E.; Rathkopf, D.; Shelkey, J.; Yu, E.Y.; Alumkal, J.; et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: A phase 1–2 study. Lancet 2010, 375, 1437–1446. [Google Scholar] [CrossRef]
- Albertsen, P.C.; Hanley, J.A.; Fine, J. 20-year outcomes following conservative management of clinically localized prostate cancer. JAMA 2005, 293, 2095–2101. [Google Scholar] [CrossRef] [PubMed]
- Merseburger, A.S.; Haas, G.P.; von Klot, C.A. An update on enzalutamide in the treatment of prostate cancer. Ther. Adv. Urol. 2015, 7, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Pienta, K.J.; Bradley, D. Mechanisms underlying the development of androgen-independent prostate cancer. Clin. Cancer Res. 2006, 12, 1665–1671. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.L.; Massie, C.E.; Ramos-Montoya, A.; Zecchini, V.; Scott, H.E.; Lamb, A.D.; MacArthur, S.; Stark, R.; Warren, A.Y.; Mills, I.G.; et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell 2013, 23, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, W.; Zhang, Y.; Yuan, X.; Xu, K.; Yu, J.; Chen, Z.; Beroukhim, R.; Wang, H.; Lupien, M.; et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 2009, 138, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Barfeld, S.J.; Itkonen, H.M.; Urbanucci, A.; Mills, I.G. Androgen-regulated metabolism and biosynthesis in prostate cancer. Endocr. Relat. Cancer 2014, 21, T57–T66. [Google Scholar] [CrossRef] [PubMed]
- Massie, C.E.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef] [PubMed]
- Shafi, A.A.; Putluri, V.; Arnold, J.M.; Tsouko, E.; Maity, S.; Roberts, J.M.; Coarfa, C.; Frigo, D.E.; Putluri, N.; Sreekumar, A.; et al. Differential regulation of metabolic pathways by androgen receptor (AR) and its constitutively active splice variant, AR-V7, in prostate cancer cells. Oncotarget 2015, 6, 31997–32012. [Google Scholar] [PubMed]
- Locasale, J.W. Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Mentch, S.J.; Locasale, J.W. One-carbon metabolism and epigenetics: Understanding the specificity. Ann. N. Y. Acad. Sci. 2016, 1363, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Tibbetts, A.S.; Appling, D.R. Compartmentalization of mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 2010, 30, 57–81. [Google Scholar] [CrossRef] [PubMed]
- Green, T.; Chen, X.; Ryan, S.; Asch, A.S.; Ruiz-Echevarria, M.J. TMEFF2 and SARDH cooperate to modulate one-carbon metabolism and invasion of prostate cancer cells. Prostate 2013, 73, 1561–1575. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.P.; Rajendiran, T.M.; Ateeq, B.; Asangani, I.A.; Athanikar, J.N.; Yocum, A.K.; Mehra, R.; Siddiqui, J.; Palapattu, G.; Wei, J.T.; et al. The role of sarcosine metabolism in prostate cancer progression. Neoplasia 2013, 15, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Sreekumar, A.; Poisson, L.M.; Rajendiran, T.M.; Khan, A.P.; Cao, Q.; Yu, J.; Laxman, B.; Mehra, R.; Lonigro, R.J.; Li, Y.; et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 2009, 457, 910–914. [Google Scholar] [CrossRef] [PubMed]
- Ottaviani, S.; Brooke, G.N.; O’Hanlon-Brown, C.; Waxman, J.; Ali, S.; Buluwela, L. Characterisation of the androgen regulation of glycine N-methyltransferase in prostate cancer cells. J. Mol. Endocrinol. 2013, 51, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.; van Guelpen, B.; Vollset, S.E.; Hultdin, J.; Bergh, A.; Key, T.; Midttun, O.; Hallmans, G.; Ueland, P.M.; Stattin, P. One-carbon metabolism and prostate cancer risk: Prospective investigation of seven circulating B vitamins and metabolites. Cancer Epidemiol. Biomark. Prev. 2009, 18, 1538–1543. [Google Scholar] [CrossRef] [PubMed]
- Appling, D.R. Compartmentation of folate-mediated one-carbon metabolism in eukaryotes. FASEB J. 1991, 5, 2645–2651. [Google Scholar] [PubMed]
- Fox, J.T.; Stover, P.J. Folate-mediated one-carbon metabolism. Vitam. Horm. 2008, 79, 1–44. [Google Scholar] [PubMed]
- Scotti, M.; Stella, L.; Shearer, E.J.; Stover, P.J. Modeling cellular compartmentation in one-carbon metabolism. Wiley Interdiscip. Rev. Syst. Biol. Med. 2013, 5, 343–365. [Google Scholar] [CrossRef] [PubMed]
- Williams-Ashman, H.G.; Canellakis, Z.N. Polyamines in mammalian biology and medicine. Perspect. Biol. Med. 1979, 22, 421–453. [Google Scholar] [CrossRef] [PubMed]
- Saini, P.; Eyler, D.E.; Green, R.; Dever, T.E. Hypusine-containing protein eIF5A promotes translation elongation. Nature 2009, 459, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Stover, P.J. One-carbon metabolism-genome interactions in folate-associated pathologies. J. Nutr. 2009, 139, 2402–2405. [Google Scholar] [CrossRef] [PubMed]
- Luka, Z. Methyltetrahydrofolate in folate-binding protein glycine N-methyltransferase. Vitam. Horm. 2008, 79, 325–345. [Google Scholar] [PubMed]
- Wagner, C.; Briggs, W.T.; Cook, R.J. Inhibition of glycine N-methyltransferase activity by folate derivatives: Implications for regulation of methyl group metabolism. Biochem. Biophys. Res. Commun. 1985, 127, 746–752. [Google Scholar] [CrossRef]
- Prudova, A.; Bauman, Z.; Braun, A.; Vitvitsky, V.; Lu, S.C.; Banerjee, R. S-adenosylmethionine stabilizes cystathionine β-synthase and modulates redox capacity. Proc. Natl. Acad. Sci. USA 2006, 103, 6489–6494. [Google Scholar] [CrossRef] [PubMed]
- Pey, A.L.; Majtan, T.; Sanchez-Ruiz, J.M.; Kraus, J.P. Human cystathionine β-synthase (CBS) contains two classes of binding sites for S-adenosylmethionine (SAM): Complex regulation of CBS activity and stability by SAM. Biochem. J. 2013, 449, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, G.; Pavlovic, D.; Jevtovic, T.; Stojanovic, I.; Sokolovic, D.; Bjelakovic, G.B.; Nikolic, J.; Basic, J. Vitamin B-12 and folic acid effects on polyamine metabolism in rat liver. Pteridines 2006, 17, 90–94. [Google Scholar] [CrossRef]
- Sun, D.; Wollin, A.; Stephen, A.M. Moderate folate deficiency influences polyamine synthesis in rats. J. Nutr. 2002, 132, 2632–2637. [Google Scholar] [PubMed]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis 2007, 12, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Ozben, T. Oxidative stress and apoptosis: Impact on cancer therapy. J. Pharm. Sci. 2007, 96, 2181–2196. [Google Scholar] [CrossRef] [PubMed]
- Gerner, E.W.; Meyskens, F.L. Polyamines and cancer: Old molecules, new understanding. Nat. Rev. Cancer 2004, 4, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Heby, O. Role of polyamines in the control of cell proliferation and differentiation. Differentiation 1981, 19, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.C.; Shyh-Chang, N.; Yang, H.; Rai, A.; Umashankar, S.; Ma, S.; Soh, B.S.; Sun, L.L.; Tai, B.C.; Nga, M.E.; et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell 2012, 148, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Rahman, L.; Voeller, D.; Rahman, M.; Lipkowitz, S.; Allegra, C.; Barrett, J.C.; Kaye, F.J.; Zajac-Kaye, M. Thymidylate synthase as an oncogene: A novel role for an essential DNA synthesis enzyme. Cancer Cell 2004, 5, 341–351. [Google Scholar] [CrossRef]
- Suzuki, M.; Tsukagoshi, S.; Saga, Y.; Ohwada, M.; Sato, I. Enhanced expression of thymidylate synthase may be of prognostic importance in advanced cervical cancer. Oncology 1999, 57, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Shintani, Y.; Ohta, M.; Hirabayashi, H.; Tanaka, H.; Iuchi, K.; Nakagawa, K.; Maeda, H.; Kido, T.; Miyoshi, S.; Matsuda, H. New prognostic indicator for non-small-cell lung cancer, quantitation of thymidylate synthase by real-time reverse transcription polymerase chain reaction. Int. J. Cancer 2003, 104, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Pestalozzi, B.C.; Peterson, H.F.; Gelber, R.D.; Goldhirsch, A.; Gusterson, B.A.; Trihia, H.; Lindtner, J.; Cortes-Funes, H.; Simmoncini, E.; Byrne, M.J.; et al. Prognostic importance of thymidylate synthase expression in early breast cancer. J. Clin. Oncol. 1997, 15, 1923–1931. [Google Scholar] [PubMed]
- Nomura, T.; Nakagawa, M.; Fujita, Y.; Hanada, T.; Mimata, H.; Nomura, Y. Clinical significance of thymidylate synthase expression in bladder cancer. Int. J. Urol. 2002, 9, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, Y.; Wada, H.; Yoshida, O.; Fukushima, M.; Nonomura, M.; Nakao, M.; Miki, T. Significance of thymidylate synthase activity in renal cell carcinoma. Clin. Cancer Res. 2003, 9, 1453–1460. [Google Scholar] [PubMed]
- Karlberg, M.; Ohrling, K.; Edler, D.; Hallstrom, M.; Ullen, H.; Ragnhammar, P. Prognostic and predictive value of thymidylate synthase expression in primary colorectal cancer. Anticancer Res. 2010, 30, 645–651. [Google Scholar] [PubMed]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-Fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wen, X.; Wu, W.; Guo, Y.; Cui, W. Elevated homocysteine level and folate deficiency associated with increased overall risk of carcinogenesis: Meta-analysis of 83 case-control studies involving 35,758 individuals. PLoS ONE 2015, 10, e0123423. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Gammon, M.D.; Chan, W.; Palomeque, C.; Wetmur, J.G.; Kabat, G.C.; Teitelbaum, S.L.; Britton, J.A.; Terry, M.B.; Neugut, A.I.; et al. One-carbon metabolism, MTHFR polymorphisms, and risk of breast cancer. Cancer Res. 2005, 65, 1606–1614. [Google Scholar] [CrossRef] [PubMed]
- Giovannucci, E. Epidemiologic studies of folate and colorectal neoplasia: A review. J. Nutr. 2002, 132, 2350S–2355S. [Google Scholar] [PubMed]
- Chen, P.; Li, C.; Li, X.; Li, J.; Chu, R.; Wang, H. Higher dietary folate intake reduces the breast cancer risk: A systematic review and meta-analysis. Br. J. Cancer 2014, 110, 2327–2338. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.I.; Pogribny, I.P.; Basnakian, A.G.; Miller, J.W.; Selhub, J.; James, S.J.; Mason, J.B. Folate deficiency in rats induces DNA strand breaks and hypomethylation within the p53 tumor suppressor gene. Am. J. Clin. Nutr. 1997, 65, 46–52. [Google Scholar] [PubMed]
- Blount, B.C.; Mack, M.M.; Wehr, C.M.; MacGregor, J.T.; Hiatt, R.A.; Wang, G.; Wickramasinghe, S.N.; Everson, R.B.; Ames, B.N. Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: Implications for cancer and neuronal damage. Proc. Natl. Acad. Sci. USA 1997, 94, 3290–3295. [Google Scholar] [CrossRef] [PubMed]
- Ergul, E.; Sazci, A.; Utkan, Z.; Canturk, N.Z. Polymorphisms in the MTHFR gene are associated with breast cancer. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2003, 24, 286–290. [Google Scholar] [CrossRef]
- Kumar, P.; Yadav, U.; Rai, V. Methylenetetrahydrofolate reductase gene c677t polymorphism and breast cancer risk: Evidence for genetic susceptibility. Meta Gene 2015, 6, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Jiang, K.; Li, Q.; Ji, Y.J.; Chen, W.L.; Xue, X.H. Polymorphisms in the MTHFR gene are associated with breast cancer risk and prognosis in a chinese population. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 3757–3762. [Google Scholar] [CrossRef] [PubMed]
- Maruti, S.S.; Ulrich, C.M.; Jupe, E.R.; White, E. MTHFR C677T and postmenopausal breast cancer risk by intakes of one-carbon metabolism nutrients: A nested case-control study. Breast Cancer Res. 2009, 11, R91. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, H.; Duan, G. Mthfr gene a1298c polymorphisms are associated with breast cancer risk among chinese population: Evidence based on an updated cumulative meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 20146–20156. [Google Scholar] [PubMed]
- Frosst, P.; Blom, H.J.; Milos, R.; Goyette, P.; Sheppard, C.A.; Matthews, R.G.; Boers, G.J.; den Heijer, M.; Kluijtmans, L.A.; van den Heuvel, L.P.; et al. A candidate genetic risk factor for vascular disease: A common mutation in methylenetetrahydrofolate reductase. Nat. Genet. 1995, 10, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Poirier, L.A. Methyl group deficiency in hepatocarcinogenesis. Drug Metab. Rev. 1994, 26, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Bistulfi, G.; Foster, B.A.; Karasik, E.; Gillard, B.; Miecznikowski, J.; Dhiman, V.K.; Smiraglia, D.J. Dietary folate deficiency blocks prostate cancer progression in the tramp model. Cancer Prev. Res. 2011, 4, 1825–1834. [Google Scholar] [CrossRef] [PubMed]
- Bistulfi, G.; Vandette, E.; Matsui, S.; Smiraglia, D.J. Mild folate deficiency induces genetic and epigenetic instability and phenotype changes in prostate cancer cells. BMC Biol. 2010, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Deghan Manshadi, S.; Ishiguro, L.; Sohn, K.J.; Medline, A.; Renlund, R.; Croxford, R.; Kim, Y.I. Folic acid supplementation promotes mammary tumor progression in a rat model. PLoS ONE 2014, 9, e84635. [Google Scholar]
- Lindzon, G.M.; Medline, A.; Sohn, K.J.; Depeint, F.; Croxford, R.; Kim, Y.I. Effect of folic acid supplementation on the progression of colorectal aberrant crypt foci. Carcinogenesis 2009, 30, 1536–1543. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Medline, A.; Mason, J.B.; Gallinger, S.; Kim, Y.I. Effects of dietary folate on intestinal tumorigenesis in the apcmin mouse. Cancer Res. 2000, 60, 5434–5440. [Google Scholar] [CrossRef]
- Kim, Y.I. Folate: A magic bullet or a double edged sword for colorectal cancer prevention? Gut 2006, 55, 1387–1389. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, C.M.; Potter, J.D. Folate and cancer—Timing is everything. JAMA 2007, 297, 2408–2409. [Google Scholar] [CrossRef] [PubMed]
- Greger, V.; Passarge, E.; Hopping, W.; Messmer, E.; Horsthemke, B. Epigenetic changes may contribute to the formation and spontaneous regression of retinoblastoma. Hum. Genet. 1989, 83, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Yoo, C.B.; Jones, P.A. Epigenetic therapy of cancer: Past, present and future. Nat. Rev. Drug Discov. 2006, 5, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Ellinger, J.; Kahl, P.; von der Gathen, J.; Heukamp, L.C.; Gütgemann, I.; Walter, B.; Hofstädter, F.; Bastian, P.J.; von Ruecker, A.; Müller, S.C.; et al. Global histone H3K27 methylation levels are different in localized and metastatic prostate cancer. Cancer Investig. 2012, 30, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Barlesi, F.; Giaccone, G.; Gallegos-Ruiz, M.I.; Loundou, A.; Span, S.W.; Lefesvre, P.; Kruyt, F.A.; Rodriguez, J.A. Global histone modifications predict prognosis of resected non small-cell lung cancer. J. Clin. Oncol. 2007, 25, 4358–4364. [Google Scholar] [CrossRef] [PubMed]
- Bianco-Miotto, T.; Chiam, K.; Buchanan, G.; Jindal, S.; Day, T.K.; Thomas, M.; Pickering, M.A.; O’Loughlin, M.A.; Ryan, N.K.; Raymond, W.A.; et al. Global levels of specific histone modifications and an epigenetic gene signature predict prostate cancer progression and development. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2611–2622. [Google Scholar] [CrossRef] [PubMed]
- Ellinger, J.; Kahl, P.; von der Gathen, J.; Rogenhofer, S.; Heukamp, L.C.; Gutgemann, I.; Walter, B.; Hofstadter, F.; Buttner, R.; Muller, S.C.; et al. Global levels of histone modifications predict prostate cancer recurrence. Prostate 2010, 70, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Seligson, D.B.; Horvath, S.; McBrian, M.A.; Mah, V.; Yu, H.; Tze, S.; Wang, Q.; Chia, D.; Goodglick, L.; Kurdistani, S.K. Global levels of histone modifications predict prognosis in different cancers. Am. J. Pathol. 2009, 174, 1619–1628. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at LYS16 and trimethylation at LYS20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, I.M.; Halvorsen, O.J.; Collett, K.; Stefansson, I.M.; Straume, O.; Haukaas, S.A.; Salvesen, H.B.; Otte, A.P.; Akslen, L.A. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J. Clin. Oncol. 2006, 24, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.A.; Lange, C.A. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat. Res. 2008, 647, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Van Leenders, G.J.L.H.; Dukers, D.; Hessels, D.; van den Kieboom, S.W.M.; Hulsbergen, C.A.; Witjes, J.A.; Otte, A.P.; Meijer, C.J.; Raaphorst, F.M. Polycomb-group oncogenes EZH2, BMI1, and RING1 are overexpressed in prostate cancer with adverse pathologic and clinical features. Eur. Urol. 2007, 52, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Hyland, P.L.; McDade, S.S.; McCloskey, R.; Dickson, G.J.; Arthur, K.; McCance, D.J.; Patel, D. Evidence for alteration of EZH2, BMI1, and KDM6A and epigenetic reprogramming in human papillomavirus type 16 E6/E7-expressing keratinocytes. J. Virol. 2011, 85, 10999–11006. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Kong, G. Dot1l: A new therapeutic target for aggressive breast cancer. Oncotarget 2015, 6, 30451–30452. [Google Scholar] [PubMed]
- Wong, M.; Polly, P.; Liu, T. The histone methyltransferase DOT1L: Regulatory functions and a cancer therapy target. Am. J. Cancer Res. 2015, 5, 2823–2837. [Google Scholar] [PubMed]
- Dou, Y.; Hess, J.L. Mechanisms of transcriptional regulation by MLL and its disruption in acute leukemia. Int. J. Hematol. 2008, 87, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Krivtsov, A.V.; Armstrong, S.A. MLL translocations, histone modifications and leukaemia stem-cell development. Nat. Rev. Cancer 2007, 7, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Orzan, F.; Pellegatta, S.; Poliani, P.L.; Pisati, F.; Caldera, V.; Menghi, F.; Kapetis, D.; Marras, C.; Schiffer, D.; Finocchiaro, G. Enhancer of zeste 2 (EZH2) is up-regulated in malignant gliomas and in glioma stem-like cells. Neuropathol. Appl. Neurobiol. 2011, 37, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Kerr, S.J. Competing methyltransferase systems. J. Biol. Chem. 1972, 247, 4248–4252. [Google Scholar] [PubMed]
- Shyh-Chang, N.; Locasale, J.W.; Lyssiotis, C.A.; Zheng, Y.; Teo, R.Y.; Ratanasirintrawoot, S.; Zhang, J.; Onder, T.; Unternaehrer, J.J.; Zhu, H.; et al. Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science 2013, 339, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.Y.; Yang, P.M.; Chang, Y.F.; Marquez, V.E.; Chen, C.C. Methotrexate induces apoptosis through p53/p21-dependent pathway and increases e-cadherin expression through downregulation of HDAC/EZH2. Biochem. Pharmacol. 2011, 81, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Mandal, A.; Johansson, H.E.; Orjalo, A.V.; Park, M.H. Depletion of cellular polyamines, spermidine and spermine, causes a total arrest in translation and growth in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 2169–2174. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, T.G.; Megosh, L.C.; Gilliard, G.; Soler, A.P. Ornithine decarboxylase overexpression is a sufficient condition for tumor promotion in mouse skin. Cancer Res. 1997, 57, 2630–2637. [Google Scholar] [PubMed]
- Nilsson, J.A.; Keller, U.B.; Baudino, T.A.; Yang, C.; Norton, S.; Old, J.A.; Nilsson, L.M.; Neale, G.; Kramer, D.L.; Porter, C.W.; et al. Targeting ornithine decarboxylase in MYC-induced lymphomagenesis prevents tumor formation. Cancer Cell 2005, 7, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Lee, O.J.; Schneider-Stock, R.; McChesney, P.A.; Kuester, D.; Roessner, A.; Vieth, M.; Moskaluk, C.A.; El-Rifai, W. Hypermethylation and loss of expression of glutathione peroxidase-3 in barrett’s tumorigenesis. Neoplasia 2005, 7, 854–861. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, J.J.; Kim, Y.S.; Kim, K.Y.; Ahn, W.S.; Yang, S. An 8-gene signature, including methylated and down-regulated glutathione peroxidase 3, of gastric cancer. Int. J. Oncol. 2010, 36, 405–414. [Google Scholar] [PubMed]
- Yu, Y.P.; Yu, G.; Tseng, G.; Cieply, K.; Nelson, J.; Defrances, M.; Zarnegar, R.; Michalopoulos, G.; Luo, J.H. Glutathione peroxidase 3, deleted or methylated in prostate cancer, suppresses prostate cancer growth and metastasis. Cancer Res. 2007, 67, 8043–8050. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.F.; Razvi, M.; Chen, H.; Washington, K.; Roessner, A.; Schneider-Stock, R.; El-Rifai, W. DNA hypermethylation regulates the expression of members of the Mu-class glutathione S-transferases and glutathione peroxidases in barrett’s adenocarcinoma. Gut 2009, 58, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, M.S.; Morrow, C.S. Methylation-mediated regulation of the glutathione S-transferase p1 gene in human breast cancer cells. Gene 1998, 210, 1–7. [Google Scholar] [CrossRef]
- Lee, W.H.; Morton, R.A.; Epstein, J.I.; Brooks, J.D.; Campbell, P.A.; Bova, G.S.; Hsieh, W.S.; Isaacs, W.B.; Nelson, W.G. Cytidine methylation of regulatory sequences near the Pi-class glutathione S-transferase gene accompanies human prostatic carcinogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 11733–11737. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef] [PubMed]
- Godwin, A.K.; Meister, A.; O’Dwyer, P.J.; Huang, C.S.; Hamilton, T.C.; Anderson, M.E. High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. Proc. Natl. Acad. Sci. USA 1992, 89, 3070–3074. [Google Scholar] [CrossRef] [PubMed]
- Kramer, R.A.; Zakher, J.; Kim, G. Role of the glutathione redox cycle in acquired and de novo multidrug resistance. Science 1988, 241, 694–697. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Liu, Y.; Greiner, C.D.; Holtzman, J.L. Physiologic concentrations of homocysteine inhibit the human plasma gsh peroxidase that reduces organic hydroperoxides. J. Lab. Clin. Med. 2000, 136, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Durmaz, A.; Dikmen, N. Homocysteine effects on cellular glutathione peroxidase (GPX-1) activity under in vitro conditions. J. Enzym. Inhib. Med. Chem. 2007, 22, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Zhang, Y.; Loscalzo, J. Homocysteine down-regulates cellular glutathione peroxidase (GPX1) by decreasing translation. J. Biol. Chem. 2005, 280, 15518–15525. [Google Scholar] [CrossRef] [PubMed]
- Tastekin, D.; Erturk, K.; Bozbey, H.U.; Olmuscelik, O.; Kiziltan, H.; Tuna, S.; Tas, F. Plasma homocysteine, folate and vitamin B12 levels in patients with lung cancer. Exp. Oncol. 2015, 37, 218–222. [Google Scholar] [PubMed]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Homocysteine and glutathione peroxidase-1. Antioxid. Redox Signal. 2007, 9, 1923–1940. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Lee, I.M.; Song, Y.; Cook, N.R.; Selhub, J.; Manson, J.E.; Buring, J.E.; Zhang, S.M. Plasma homocysteine and cysteine and risk of breast cancer in women. Cancer Res. 2010, 70, 2397–2405. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Sakari, M.; Okada, M.; Yokoyama, A.; Takahashi, S.; Kouzmenko, A.; Kato, S. The androgen receptor in health and disease. Annu. Rev. Physiol. 2013, 75, 201–224. [Google Scholar] [CrossRef] [PubMed]
- Tsouko, E.; Khan, A.S.; White, M.A.; Han, J.J.; Shi, Y.; Merchant, F.A.; Sharpe, M.A.; Xin, L.; Frigo, D.E. Regulation of the pentose phosphate pathway by an androgen receptor-mtor-mediated mechanism and its role in prostate cancer cell growth. Oncogenesis 2014, 3, e103. [Google Scholar] [CrossRef] [PubMed]
- Tennakoon, J.B.; Shi, Y.; Han, J.J.; Tsouko, E.; White, M.A.; Burns, A.R.; Zhang, A.; Xia, X.; Ilkayeva, O.R.; Xin, L.; et al. Androgens regulate prostate cancer cell growth via an AMPK-PGC-1α-mediated metabolic switch. Oncogene 2014, 33, 5251–5261. [Google Scholar] [CrossRef] [PubMed]
- Luka, Z.; Mudd, S.H.; Wagner, C. Glycine N-methyltransferase and regulation of S-adenosylmethionine levels. J. Biol. Chem. 2009, 284, 22507–22511. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.C.; Lee, C.M.; Chen, M.; Chung, M.Y.; Chang, Y.H.; Huang, W.J.; Ho, D.M.; Pan, C.C.; Wu, T.T.; Yang, S.; et al. Haplotypes, loss of heterozygosity, and expression levels of glycine N-methyltransferase in prostate cancer. Clin. Cancer Res. 2007, 13, 1412–1420. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.H.; Shiota, M.; Kuroiwa, K.; Naito, S.; Oda, Y. The important role of glycine N-methyltransferase in the carcinogenesis and progression of prostate cancer. Mod. Pathol. 2011, 24, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Putluri, N.; Shojaie, A.; Vasu, V.T.; Nalluri, S.; Vareed, S.K.; Putluri, V.; Vivekanandan-Giri, A.; Byun, J.; Pennathur, S.; Sana, T.R.; et al. Metabolomic profiling reveals a role for androgen in activating amino acid metabolism and methylation in prostate cancer cells. PLoS ONE 2011, 6, e21417. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.K.; Kim, D.H.; Koo, J.S. Implications of differences in expression of sarcosine metabolism-related proteins according to the molecular subtype of breast cancer. J. Transl. Med. 2014, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ulanovskaya, O.A.; Zuhl, A.M.; Cravatt, B.F. NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nat. Chem. Biol. 2013, 9, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, Y.; Li, G.; Yu, H.; Xie, X. Down-regulation of nicotinamide N-methyltransferase induces apoptosis in human breast cancer cells via the mitochondria-mediated pathway. PLoS ONE 2014, 9, e89202. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Overcash, R.; Green, T.; Hoffman, D.; Asch, A.S.; Ruiz-Echevarria, M.J. The tumor suppressor activity of the transmembrane protein with epidermal growth factor and two follistatin motifs 2 (TMEFF2) correlates with its ability to modulate sarcosine levels. J. Biol. Chem. 2011, 286, 16091–16100. [Google Scholar] [CrossRef] [PubMed]
- Corbin, J.M.; Overcash, R.F.; Wren, J.D.; Coburn, A.; Tipton, G.J.; Ezzell, J.A.; McNaughton, K.K.; Fung, K.M.; Kosanke, S.D.; Ruiz-Echevarria, M.J. Analysis of TMEFF2 allografts and transgenic mouse models reveals roles in prostate regeneration and cancer. Prostate 2016, 76, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Reed, M.C.; Thomas, R.L.; Pavisic, J.; James, S.J.; Ulrich, C.M.; Nijhout, H.F. A mathematical model of glutathione metabolism. Theor. Biol. Med. Model. 2008, 5, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Li, Q.; Du, H.P.; Wang, Y.L.; You, S.J.; Wang, F.; Xu, X.S.; Cheng, J.; Cao, Y.J.; Liu, C.F.; et al. Homocysteine triggers inflammatory responses in macrophages through inhibiting CSE-H2s signaling via DNA hypermethylation of CSE promoter. Int. J. Mol. Sci. 2015, 16, 12560–12577. [Google Scholar] [CrossRef] [PubMed]
- Kabil, O.; Banerjee, R. Enzymology of h2s biogenesis, decay and signaling. Antioxid. Redox Signal. 2014, 20, 770–782. [Google Scholar] [CrossRef] [PubMed]
- Kajimura, M.; Fukuda, R.; Bateman, R.M.; Yamamoto, T.; Suematsu, M. Interactions of multiple gas-transducing systems: Hallmarks and uncertainties of Co, No, and H2S gas biology. Antioxid. Redox Signal. 2010, 13, 157–192. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, A.K.; Gadalla, M.M.; Snyder, S.H. Signaling by gasotransmitters. Sci. Signal. 2009, 2, re2. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Li, S.; Wu, L.; Lai, C.; Yang, G. Hydrogen sulfide represses androgen receptor transactivation by targeting at the second zinc finger module. J. Biol. Chem. 2014, 289, 20824–20835. [Google Scholar] [CrossRef] [PubMed]
- Schalinske, K.L.; Smazal, A.L. Homocysteine imbalance: A pathological metabolic marker. Adv. Nutr. 2012, 3, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Saha, S.; Giri, K.; Lanza, I.R.; Nair, K.S.; Jennings, N.B.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Basal, E.; Weaver, A.L. Cystathionine β-synthase (CBS) contributes to advanced ovarian cancer progression and drug resistance. PLoS ONE 2013, 8, e79167. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C.; Coletta, C.; Chao, C.; Módis, K.; Szczesny, B.; Papapetropoulos, A.; Hellmich, M.R. Tumor-derived hydrogen sulfide, produced by cystathionine-β-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 12474–12479. [Google Scholar] [CrossRef] [PubMed]
- Janošík, M.; Kery, V.; Gaustadnes, M.; Maclean, K.N.; Kraus, J.P. Regulation of human cystathionine β-synthase by S-adenosyl-l-methionine: Evidence for two catalytically active conformations involving an autoinhibitory domain in the C-terminal region. Biochemistry 2001, 40, 10625–10633. [Google Scholar] [CrossRef] [PubMed]
- Prudova, A.; Albin, M.; Bauman, Z.; Lin, A.; Vitvitsky, V.; Banerjee, R. Testosterone regulation of homocysteine metabolism modulates redox status in human prostate cancer cells. Antioxid. Redox Signal. 2007, 9, 1875–1881. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Gai, J.-W.; Wang, Y.; Jin, H.-F.; Du, J.-B.; Jin, J. Characterization of hydrogen sulfide and its synthases, cystathionine β-synthase and cystathionine γ-lyase, in human prostatic tissue and cells. Urology 2012, 79, 483.e481–483.e485. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Braun, A.; Bauman, Z.; Olteanu, H.; Madzelan, P.; Banerjee, R. Expression profiling of homocysteine junction enzymes in the nci60 panel of human cancer cell lines. Cancer Res. 2005, 65, 1554–1560. [Google Scholar] [CrossRef] [PubMed]
- Al-Awadi, F.; Yang, M.; Tan, Y.; Han, Q.; Li, S.; Hoffman, R.M. Human tumor growth in nude mice is associated with decreased plasma cysteine and homocysteine. Anticancer Res. 2008, 28, 2541–2544. [Google Scholar] [PubMed]
- Stabler, S.; Koyama, T.; Zhao, Z.; Martinez-Ferrer, M.; Allen, R.H.; Luka, Z.; Loukachevitch, L.V.; Clark, P.E.; Wagner, C.; Bhowmick, N.A. Serum methionine metabolites are risk factors for metastatic prostate cancer progression. PLoS ONE 2011, 6, e22486. [Google Scholar] [CrossRef] [PubMed]
- Chwatko, G.; Forma, E.; Wilkosz, J.; Głowacki, R.; Jóźwiak, P.; Różański, W.; Bryś, M.; Krześlak, A. Thiosulfate in urine as a facilitator in the diagnosis of prostate cancer for patients with prostate-specific antigen less or equal 10 ng/mL. Clin. Chem. Lab. Med. 2013, 51, 1825–1831. [Google Scholar] [CrossRef] [PubMed]
- Hatayama, I.; Satoh, K.; Sato, K. Developmental and hormonal regulation of the major form of hepatic glutathione s-transferase in male mice. Biochem. Biophys. Res. Commun. 1986, 140, 581–588. [Google Scholar] [CrossRef]
- Ikeda, H.; Serria, M.S.; Kakizaki, I.; Hatayama, I.; Satoh, K.; Tsuchida, S.; Muramatsu, M.; Nishi, S.; Sakai, M. Activation of mouse pi-class glutathione S-transferase gene by Nrf2(Nf-E2-related factor 2) and androgen. Biochem. J. 2002, 364, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Imperlini, E.; Mancini, A.; Spaziani, S.; Martone, D.; Alfieri, A.; Gemei, M.; del Vecchio, L.; Buono, P.; Orru, S. Androgen receptor signaling induced by supraphysiological doses of dihydrotestosterone in human peripheral blood lymphocytes. Proteomics 2010, 10, 3165–3175. [Google Scholar] [CrossRef] [PubMed]
- Khandrika, L.; Kumar, B.; Koul, S.; Maroni, P.; Koul, H.K. Oxidative stress in prostate cancer. Cancer Lett. 2009, 282, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Pendeville, H.; Carpino, N.; Marine, J.C.; Takahashi, Y.; Muller, M.; Martial, J.A.; Cleveland, J.L. The ornithine decarboxylase gene is essential for cell survival during early murine development. Mol. Cell. Biol. 2001, 21, 6549–6558. [Google Scholar] [CrossRef] [PubMed]
- Casero, R.A.; Pegg, A.E. Polyamine catabolism and disease. Biochem. J. 2009, 421, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Nowotarski, S.L.; Woster, P.M.; Casero, R.A., Jr. Polyamines and cancer: Implications for chemotherapy and chemoprevention. Expert Rev. Mol. Med. 2013, 15, e3. [Google Scholar] [CrossRef] [PubMed]
- Casero, R.A.; Marton, L.J. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat. Rev. Drug Discov. 2007, 6, 373–390. [Google Scholar] [CrossRef] [PubMed]
- Giardiello, F.M.; Hamilton, S.R.; Hylind, L.M.; Yang, V.W.; Tamez, P.; Casero, R.A., Jr. Ornithine decarboxylase and polyamines in familial adenomatous polyposis. Cancer Res. 1997, 57, 199–201. [Google Scholar] [PubMed]
- Pegg, A.E.; Lockwood, D.H.; Williams-Ashman, H.G. Concentrations of putrescine and polyamines and their enzymic synthesis during androgen-induced prostatic growth. Biochem. J. 1970, 117, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Blackshear, P.J.; Manzella, J.M.; Stumpo, D.J.; Wen, L.; Huang, J.K.; Oyen, O.; Young, W.S., 3rd. High level, cell-specific expression of ornithine decarboxylase transcripts in rat genitourinary tissues. Mol. Endocrinol. 1989, 3, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Pegg, A.E. Polyamine metabolism and its importance in neoplastic growth and a target for chemotherapy. Cancer Res. 1988, 48, 759–774. [Google Scholar] [PubMed]
- Janne, J.; Alhonen, L.; Leinonen, P. Polyamines: From molecular biology to clinical applications. Ann. Med. 1991, 23, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Mohan, R.R.; Challa, A.; Gupta, S.; Bostwick, D.G.; Ahmad, N.; Agarwal, R.; Marengo, S.R.; Amini, S.B.; Paras, F.; MacLennan, G.T.; et al. Overexpression of ornithine decarboxylase in prostate cancer and prostatic fluid in humans. Clin. Cancer Res. 1999, 5, 143–147. [Google Scholar] [PubMed]
- Rhodes, D.R.; Barrette, T.R.; Rubin, M.A.; Ghosh, D.; Chinnaiyan, A.M. Meta-analysis of microarrays: Interstudy validation of gene expression profiles reveals pathway dysregulation in prostate cancer. Cancer Res. 2002, 62, 4427–4433. [Google Scholar] [PubMed]
- Cipolla, B.G.; Havouis, R.; Moulinoux, J.P. Polyamine reduced diet (PRD) nutrition therapy in hormone refractory prostate cancer patients. Biomed. Pharmacother. 2010, 64, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Janne, O.A.; Crozat, A.; Palvimo, J.; Eisenberg, L.M. Androgen-regulation of ornithine decarboxylase and S-adenosylmethionine decarboxylase genes. J. Steroid Biochem. Mol. Biol. 1991, 40, 307–315. [Google Scholar] [CrossRef]
- Fjosne, H.E.; Strand, H.; Sunde, A. Dose-dependent induction of ornithine decarboxylase and S-adenosyl-methionine decarboxylase activity by testosterone in the accessory sex organs of male rats. Prostate 1992, 21, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Cyriac, J.; Haleem, R.; Cai, X.; Wang, Z. Androgen regulation of spermidine synthase expression in the rat prostate. Prostate 2002, 50, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Crozat, A.; Palvimo, J.J.; Julkunen, M.; Janne, O.A. Comparison of androgen regulation of ornithine decarboxylase and S-adenosylmethionine decarboxylase gene expression in rodent kidney and accessory sex organs. Endocrinology 1992, 130, 1131–1144. [Google Scholar] [PubMed]
- Fjosne, H.E.; Strand, H.; Ostensen, M.A.; Sunde, A. Ornithine decarboxylase and S-adenosylmethionine decarboxylase activity in the accessory sex organs of intact, castrated, and androgen-stimulated castrated rats. Prostate 1988, 12, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Bai, G.; Kasper, S.; Matusik, R.J.; Rennie, P.S.; Moshier, J.A.; Krongrad, A. Androgen regulation of the human ornithine decarboxylase promoter in prostate cancer cells. J. Androl. 1998, 19, 127–135. [Google Scholar] [PubMed]
- Cohen, R.J.; Fujiwara, K.; Holland, J.W.; McNeal, J.E. Polyamines in prostatic epithelial cells and adenocarcinoma: The effects of androgen blockade. Prostate 2001, 49, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, S.M.; Arnold, J.; Sreekumar, A. Metabolomic profiling of hormone-dependent cancers: A bird’s eye view. Trends Endocrinol. Metab. 2015, 26, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Bistulfi, G.; Diegelman, P.; Foster, B.A.; Kramer, D.L.; Porter, C.W.; Smiraglia, D.J. Polyamine biosynthesis impacts cellular folate requirements necessary to maintain S-adenosylmethionine and nucleotide pools. FASEB J. 2009, 23, 2888–2897. [Google Scholar] [CrossRef] [PubMed]
- Yegnasubramanian, S.; Haffner, M.C.; Zhang, Y.; Gurel, B.; Cornish, T.C.; Wu, Z.; Irizarry, R.A.; Morgan, J.; Hicks, J.; DeWeese, T.L.; et al. DNA hypomethylation arises later in prostate cancer progression than CPG island hypermethylation and contributes to metastatic tumor heterogeneity. Cancer Res. 2008, 68, 8954–8967. [Google Scholar] [CrossRef] [PubMed]
- Jerónimo, C.; Bastian, P.J.; Bjartell, A.; Carbone, G.M.; Catto, J.W.F.; Clark, S.J.; Henrique, R.; Nelson, W.G.; Shariat, S.F. Epigenetics in prostate cancer: Biologic and clinical relevance. Eur. Urol. 2011, 60, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Labbe, D.P.; Zadra, G.; Ebot, E.M.; Mucci, L.A.; Kantoff, P.W.; Loda, M.; Brown, M. Role of diet in prostate cancer: The epigenetic link. Oncogene 2015, 34, 4683–4691. [Google Scholar] [CrossRef] [PubMed]
- Seligson, D.B.; Horvath, S.; Shi, T.; Yu, H.; Tze, S.; Grunstein, M.; Kurdistani, S.K. Global histone modification patterns predict risk of prostate cancer recurrence. Nature 2005, 435, 1262–1266. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.S.; Watson, R.W.; Lawler, M.; Hollywood, D. The epigenome as a therapeutic target in prostate cancer. Nat. Rev. Urol. 2010, 7, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Nelson, W.G.; de Marzo, A.M.; Yegnasubramanian, S. Epigenetic alterations in human prostate cancers. Endocrinology 2009, 150, 3991–4002. [Google Scholar] [CrossRef] [PubMed]
- Yegnasubramanian, S.; Kowalski, J.; Gonzalgo, M.L.; Zahurak, M.; Piantadosi, S.; Walsh, P.C.; Bova, G.S.; de Marzo, A.M.; Isaacs, W.B.; Nelson, W.G. Hypermethylation of CPG islands in primary and metastatic human prostate cancer. Cancer Res. 2004, 64, 1975–1986. [Google Scholar] [CrossRef] [PubMed]
- Jeronimo, C.; Henrique, R.; Hoque, M.O.; Mambo, E.; Ribeiro, F.R.; Varzim, G.; Oliveira, J.; Teixeira, M.R.; Lopes, C.; Sidransky, D. A quantitative promoter methylation profile of prostate cancer. Clin. Cancer Res. 2004, 10, 8472–8478. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, R.; Toyooka, S.; Toyooka, K.O.; Virmani, A.K.; Zöchbauer-Müller, S.; Farinas, A.J.; Minna, J.D.; McConnell, J.; Frenkel, E.P.; Gazdar, A.F. Aberrant promoter methylation profile of prostate cancers and its relationship to clinicopathological features. Clin. Cancer Res. 2002, 8, 514–519. [Google Scholar] [PubMed]
- Florl, A.R.; Steinhoff, C.; Müller, M.; Seifert, H.H.; Hader, C.; Engers, R.; Ackermann, R.; Schulz, W.A. Coordinate hypermethylation at specific genes in prostate carcinoma precedes line-1 hypomethylation. Br. J. Cancer 2004, 91, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Padar, A.; Sathyanarayana, U.G.; Suzuki, M.; Maruyama, R.; Hsieh, J.T.; Frenkel, E.P.; Minna, J.D.; Gazdar, A.F. Inactivation of cyclin D2 gene in prostate cancers by aberrant promoter methylation. Clin. Cancer Res. 2003, 9, 4730–4734. [Google Scholar] [PubMed]
- Yamanaka, M.; Watanabe, M.; Yamada, Y.; Takagi, A.; Murata, T.; Takahashi, H.; Suzuki, H.; Ito, H.; Tsukino, H.; Katoh, T.; et al. Altered methylation of multiple genes in carcinogenesis of the prostate. Int. J. Cancer 2003, 106, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Ellinger, J.; Bastian, P.J.; Jurgan, T.; Biermann, K.; Kahl, P.; Heukamp, L.C.; Wernert, N.; Müller, S.C.; von Ruecker, A. Cpg island hypermethylation at multiple gene sites in diagnosis and prognosis of prostate cancer. Urology 2008, 71, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Lodygin, D.; Epanchintsev, A.; Menssen, A.; Diebold, J.; Hermeking, H. Functional epigenomics identifies genes frequently silenced in prostate cancer. Cancer Res. 2005, 65, 4218–4227. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.S.; Foley, R.; Woodson, K.; Lawler, M. The emerging roles of DNA methylation in the clinical management of prostate cancer. Endocr. Relat. Cancer 2006, 13, 357–377. [Google Scholar] [CrossRef] [PubMed]
- Li, L.C. Epigenetics of prostate cancer. Front. Biosci. J. Virtual Libr. 2007, 12, 3377–3397. [Google Scholar] [CrossRef]
- Sasaki, M.; Tanaka, Y.; Perinchery, G.; Dharia, A.; Kotcherguina, I.; Fujimoto, S.; Dahiya, R. Methylation and inactivation of estrogen, progesterone, and androgen receptors in prostate cancer. J. Natl. Cancer Inst. 2002, 94, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Reibenwein, J.; Pils, D.; Horak, P.; Tomicek, B.; Goldner, G.; Worel, N.; Elandt, K.; Krainer, M. Promoter hypermethylation of GSTP1, AR, and 14–3-3σ in serum of prostate cancer patients and its clinical relevance. Prostate 2007, 67, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.F.; Chen, W.C.; Chang, Y.J.; Wu, C.F.; Wu, C.T. Role of DNA methyltransferase 1 in hormone-resistant prostate cancer. J. Mol. Med. 2010, 88, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Patra, S.K.; Patra, A.; Zhao, H.; Dahiya, R. DNA methyltransferase and demethylase in human prostate cancer. Mol. Carcinog. 2002, 33, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Jiao, H.; Zhang, X.; Zhao, R.; Wang, F.; He, W.; Zong, H.; Fan, Q.; Wang, L. Correlation between the expression of DNMT1, and GSTP1 and APC, and the methylation status of GSTP1 and APC in association with their clinical significance in prostate cancer. Mol. Med. Rep. 2015, 12, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Valdez, C.D.; Kunju, L.; Daignault, S.; Wojno, K.J.; Day, M.L. The e2f1/dnmt1 axis is associated with the development of ar negative castration resistant prostate cancer. Prostate 2013, 73, 1776–1785. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chen, L.; Helfand, B.T.; Jang, T.L.; Sharma, V.; Kozlowski, J.; Kuzel, T.M.; Zhu, L.J.; Yang, X.J.; Javonovic, B.; et al. Tgf-beta regulates DNA methyltransferase expression in prostate cancer, correlates with aggressive capabilities, and predicts disease recurrence. PLoS ONE 2011, 6, e25168. [Google Scholar]
- Kinney, S.R.; Moser, M.T.; Pascual, M.; Greally, J.M.; Foster, B.A.; Karpf, A.R. Opposing roles of DNMT1 in early- and late-stage murine prostate cancer. Mol. Cell. Biol. 2010, 30, 4159–4174. [Google Scholar] [CrossRef] [PubMed]
- McCabe, M.T.; Low, J.A.; Daignault, S.; Imperiale, M.J.; Wojno, K.J.; Day, M.L. Inhibition of DNA methyltransferase activity prevents tumorigenesis in a mouse model of prostate cancer. Cancer Res. 2006, 66, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Absher, D.M.; Gulzar, Z.G.; Young, S.R.; McKenney, J.K.; Peehl, D.M.; Brooks, J.D.; Myers, R.M.; Sherlock, G. DNA methylation profiling reveals novel biomarkers and important roles for DNA methyltransferases in prostate cancer. Genome Res. 2011, 21, 1017–1027. [Google Scholar] [CrossRef] [PubMed]
- Brothman, A.R.; Swanson, G.; Maxwell, T.M.; Cui, J.; Murphy, K.J.; Herrick, J.; Speights, V.O.; Isaac, J.; Rohr, L.R. Global hypomethylation is common in prostate cancer cells: A quantitative predictor for clinical outcome? Cancer Gen. Cytogenet. 2005, 156, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Fuso, A.; Nicolia, V.; Cavallaro, R.A.; Scarpa, S. DNA methylase and demethylase activities are modulated by one-carbon metabolism in Alzheimer’s disease models. J. Nut. Biochem. 2011, 22, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.R.; Sanda, M.G.; Otte, A.P.; Chinnaiyan, A.M.; Rubin, M.A. Multiplex biomarker approach for determining risk of prostate-specific antigen-defined recurrence of prostate cancer. J. Natl. Cancer Inst. 2003, 95, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.J.; Engers, R.; Florl, A.R.; Otte, A.P.; Müller, M.; Schulz, W.A. Expression changes in EZH2, but not in BMI-1, SIRT1, DNMT1 or DNMT3b, are associated with DNA methylation changes in prostate cancer. Cancer Biol. Ther. 2007, 6, 1403–1412. [Google Scholar] [CrossRef] [PubMed]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.A.B.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Laitinen, S.; Martikainen, P.M.; Tolonen, T.; Isola, J.; Tammela, T.L.J.; Visakorpi, T. EZH2, Ki-67 and MCM7 are prognostic markers in prostatectomy treated patients. Int. J. Cancer 2008, 122, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Rhodes, D.R.; Tomlins, S.A.; Cao, X.; Chen, G.; Mehra, R.; Wang, X.; Ghosh, D.; Shah, R.B. A polycomb repression signature in metastatic prostate cancer predicts cancer outcome. Cancer Res. 2007, 67, 10657–10663. [Google Scholar] [CrossRef] [PubMed]
- Viré, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; van Eynde, A.; Bernard, D.; Vanderwinden, J.M.; et al. The polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, Y.; Straussman, R.; Keshet, I.; Farkash, S.; Hecht, M.; Zimmerman, J.; Eden, E.; Yakhini, Z.; Ben-Shushan, E.; Reubinoff, B.E.; et al. Polycomb-mediated methylation on LYS27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet. 2007, 39, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Crea, F.; Sun, L.; Mai, A.; Chiang, Y.T.; Farrar, W.L.; Danesi, R.; Helgason, C.D. The emerging role of histone lysine demethylases in prostate cancer. Mol. Cancer 2012, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Berry, W.L.; Janknecht, R. KDM4/JMJD2 histone demethylases: Epigenetic regulators in cancer cells. Cancer Res. 2013, 73, 2936–2942. [Google Scholar] [CrossRef] [PubMed]
- Franci, G.; Ciotta, A.; Altucci, L. The Jumonji family: Past, present and future of histone demethylases in cancer. Biomol. Concepts 2014, 5, 209–224. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, T.; Barchas, J. Inhibition of transmethylations of biogenic amines by S-adenosylhomocysteine. Enhancement of transmethylation by adenosylhomocysteinase. J. Biol. Chem. 1971, 246, 3175–3181. [Google Scholar] [PubMed]
- Wang, Y.-C.; Tang, F.-Y.; Chen, S.-Y.; Chen, Y.-M.; Chiang, E.-P.I. Glycine-N methyltransferase expression in HepG2 cells is involved in methyl group homeostasis by regulating transmethylation kinetics and DNA methylation. J. Nutr. 2011, 141, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.C.; Yu, J.; Runkle, C.; Wu, L.; Hu, M.; Wu, D.; Liu, J.S.; Wang, Q.; Qin, Z.S.; Yu, J. Cooperation between polycomb and androgen receptor during oncogenic transformation. Genome Res. 2012, 22, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Chng, K.R.; Chang, C.W.; Tan, S.K.; Yang, C.; Hong, S.Z.; Sng, N.Y.; Cheung, E. A transcriptional repressor co-regulatory network governing androgen response in prostate cancers. EMBO J. 2012, 31, 2810–2823. [Google Scholar] [CrossRef] [PubMed]
- Malik, R.; Khan, A.P.; Asangani, I.A.; Cieślik, M.; Prensner, J.R.; Wang, X.; Iyer, M.K.; Jiang, X.; Borkin, D.; Escara-Wilke, J.; et al. Targeting the MLL complex in castration resistant prostate cancer. Nat. Med. 2015, 21, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Yamane, K.; Toumazou, C.; Tsukada, Y.; Erdjument-Bromage, H.; Tempst, P.; Wong, J.; Zhang, Y. JHDM2A, a JMJC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell 2006, 125, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; He, H.H.; Chen, S.; Coleman, I.; Wang, H.; Fang, Z.; Chen, S.; Nelson, P.S.; Liu, X.S.; Brown, M.; et al. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell 2011, 20, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Gaughan, L.; Stockley, J.; Wang, N.; McCracken, S.R.C.; Treumann, A.; Armstrong, K.; Shaheen, F.; Watt, K.; McEwan, I.J.; Wang, C.; et al. Regulation of the androgen receptor by set9-mediated methylation. Nucleic Acids Res. 2011, 39, 1266–1279. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Wu, Z.J.; Groner, A.C.; He, H.H.; Cai, C.; Lis, R.T.; Wu, X.; Stack, E.C.; Loda, M.; Liu, T.; et al. Ezh2 oncogenic activity in castration-resistant prostate cancer cells is polycomb-independent. Science 2012, 338, 1465–1469. [Google Scholar] [CrossRef] [PubMed]
- Bohrer, L.R.; Chen, S.; Hallstrom, T.C.; Huang, H. Androgens suppress ezh2 expression via retinoblastoma (RB) and p130-dependent pathways: A potential mechanism of androgen-refractory progression of prostate cancer. Endocrinology 2010, 151, 5136–5145. [Google Scholar] [CrossRef] [PubMed]
- Hofman, K.; Swinnen, J.V.; Verhoeven, G.; Heyns, W. E2f activity is biphasically regulated by androgens in lncap cells. Biochem. Biophys. Res. Commun. 2001, 283, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Deng, Z.; Wan, M.; Huang, W.; Cramer, S.D.; Xu, J.; Lei, M.; Sui, G. MicroRNA-101 negatively regulates EZH2 and its expression is modulated by androgen receptor and HIF-1α/HIF-1β. Mol. Cancer 2010, 9, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varambally, S.; Cao, Q.; Mani, R.S.; Shankar, S.; Wang, X.; Ateeq, B.; Laxman, B.; Cao, X.; Jing, X.; Ramnarayanan, K.; et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science 2008, 322, 1695–1699. [Google Scholar] [CrossRef] [PubMed]
- Farber, S.; Diamond, L.K. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N. Engl. J. Med. 1948, 238, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Loening, S.A.; Beckley, S.; Brady, M.F.; Chu, T.M.; deKernion, J.B.; Dhabuwala, C.; Gaeta, J.F.; Gibbons, R.P.; McKiel, C.F.; McLeod, D.G.; et al. Comparison of estramustine phosphate, methotrexate and cis-platinum in patients with advanced, hormone refractory prostate cancer. J. Urol. 1983, 129, 1001–1006. [Google Scholar] [PubMed]
- Saxman, S.; Ansari, R.; Drasga, R.; Miller, M.; Wheeler, B.; McClean, J.; Einhorn, L. Phase III trial of cyclophosphamide versus cyclophosphamide, doxorubicin, and methotrexate in hormone-refractory prostatic cancer. A hoosier oncology group study. Cancer 1992, 70, 2488–2492. [Google Scholar] [CrossRef]
- Jones, W.G.; Fossa, S.D.; Verbaeys, A.C.; Droz, J.P.; Klijn, J.G.; Boven, E.; de Pauw, M.; Sylvester, R. Low-dose fortnightly methotrexate in advanced prostate cancer. The eortc genito-urinary tract cancer cooperative group. Eur. J. Cancer 1990, 26, 646. [Google Scholar] [CrossRef]
- Linsalata, M.; Caruso, M.G.; Leo, S.; Guerra, V.; D’Attoma, B.; di Leo, A. Prognostic value of tissue polyamine levels in human colorectal carcinoma. Anticancer Res. 2002, 22, 2465–2469. [Google Scholar] [PubMed]
- Canizares, F.; Salinas, J.; de las Heras, M.; Diaz, J.; Tovar, I.; Martinez, P.; Penafiel, R. Prognostic value of ornithine decarboxylase and polyamines in human breast cancer: Correlation with clinicopathologic parameters. Clin. Cancer Res. 1999, 5, 2035–2041. [Google Scholar] [PubMed]
- Heston, W.D. Prostatic polyamines and polyamine targeting as a new approach to therapy of prostatic cancer. Cancer Surv. 1991, 11, 217–238. [Google Scholar] [PubMed]
- Durie, B.G.; Salmon, S.E.; Russell, D.H. Polyamines as markers of response and disease activity in cancer chemotherapy. Cancer Res. 1977, 37, 214–221. [Google Scholar] [PubMed]
- Kubota, S.; Okada, M.; Yoshimoto, M.; Murata, N.; Yamasaki, Z.; Wada, T.; Imahori, K.; Ohsawa, N.; Takaku, F. Urinary polyamines as a tumor marker. Cancer Detect. Prev. 1985, 8, 189–192. [Google Scholar] [PubMed]
- Russell, D.H. Clinical relevance of polyamines. Crit. Rev. Clin. Lab. Sci. 1983, 18, 261–311. [Google Scholar] [CrossRef] [PubMed]
- Sakai, S.; Ito, Y.; Koide, T.; Tei, K.; Hara, A.; Sawada, H. Detection of urinary polyamine by a new enzymatic differential assay. (III). Studies on urinary polyamines in patients with malignant genitourinary diseases. Hinyokika Kiyo 1986, 32, 343–350. [Google Scholar] [PubMed]
- Chatel, M.; Darcel, F.; Quemener, V.; Hercouet, H.; Moulinoux, J.P. Red blood cell polyamines as biochemical markers of supratentorial malignant gliomas. Anticancer Res. 1987, 7, 33–38. [Google Scholar] [PubMed]
- Cipolla, B.; Guille, F.; Moulinoux, J.P.; Bansard, J.Y.; Roth, S.; Staerman, F.; Corbel, L.; Quemener, V.; Lobel, B. Erythrocyte polyamines and prognosis in stage D2 prostatic carcinoma patients. J. Urol. 1994, 151, 629–633. [Google Scholar] [PubMed]
- Cipolla, B.; Guille, F.; Moulinoux, J.P.; Quemener, V.; Staerman, F.; Corbel, L.; Lobel, B. Polyamines and prostatic carcinoma: Clinical and therapeutic implications. Eur. Urol. 1993, 24, 124–131. [Google Scholar] [PubMed]
- Weiss, T.S.; Bernhardt, G.; Buschauer, A.; Thasler, W.E.; Dolgner, D.; Zirngibl, H.; Jauch, K.W. Polyamine levels of human colorectal adenocarcinomas are correlated with tumor stage and grade. Int. J. Colorectal Dis. 2002, 17, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, C.; Bansard, J.Y.; Le Moine, P.; Bouet, F.; Goasguen, J.E.; Moulinoux, J.P.; Le Gall, E.; Catros-Quemener, V. Erythrocyte spermine levels: A prognostic parameter in childhood common acute lymphoblastic leukemia. Leukemia 1997, 11, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Soda, K. The mechanisms by which polyamines accelerate tumor spread. J. Exp. Clin. Cancer Res. 2011, 30, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cipolla, B.; Moulinoux, J.P.; Quemener, V.; Havouis, R.; Martin, L.A.; Guille, F.; Lobel, B. Erythrocyte polyamine levels in human prostatic carcinoma. J. Urol. 1990, 144, 1164–1166. [Google Scholar] [PubMed]
- Cheng, L.L.; Wu, C.-L.; Smith, M.R.; Gonzalez, R.G. Non-destructive quantitation of spermine in human prostate tissue samples using HRMAS 1 H NMR spectroscopy at 9.4 T. FEBS Lett. 2001, 494, 112–116. [Google Scholar] [CrossRef]
- McDunn, J.E.; Li, Z.; Adam, K.P.; Neri, B.P.; Wolfert, R.L.; Milburn, M.V.; Lotan, Y.; Wheeler, T.M. Metabolomic signatures of aggressive prostate cancer. Prostate 2013, 73, 1547–1560. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Ahmad, N.; Marengo, S.R.; MacLennan, G.T.; Greenberg, N.M.; Mukhtar, H. Chemoprevention of prostate carcinogenesis by α-difluoromethylornithine in tramp mice. Cancer Res. 2000, 60, 5125–5133. [Google Scholar] [PubMed]
- Kee, K.; Foster, B.A.; Merali, S.; Kramer, D.L.; Hensen, M.L.; Diegelman, P.; Kisiel, N.; Vujcic, S.; Mazurchuk, R.V.; Porter, C.W. Activated polyamine catabolism depletes acetyl-coa pools and suppresses prostate tumor growth in tramp mice. J. Biol. Chem. 2004, 279, 40076–40083. [Google Scholar] [CrossRef] [PubMed]
- Devens, B.H.; Weeks, R.S.; Burns, M.R.; Carlson, C.L.; Brawer, M.K. Polyamine depletion therapy in prostate cancer. Prostate Cancer Prostatic Dis. 2000, 3, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Kadmon, D. Chemoprevention in prostate cancer: The role of difluoromethylornithine (DFMO). J. Cell. Biochem. Suppl. 1992, 16H, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Moulinoux, J.P.; Quemener, V.; Cipolla, B.; Guille, F.; Havouis, R.; Martin, C.; Lobel, B.; Seiler, N. The growth of MAT-LyLu rat prostatic adenocarcinoma can be prevented in vivo by polyamine deprivation. J. Urol. 1991, 146, 1408–1412. [Google Scholar] [PubMed]
- Danzin, C.; Jung, M.J.; Grove, J.; Bey, P. Effect of alpha-difluoromethylornithine, an enzyme-activated irreversible inhibitor of ornithine decarboxylase, on polyamine levels in rat tissues. Life Sci. 1979, 24, 519–524. [Google Scholar] [CrossRef]
- Meyskens, F.L., Jr.; Simoneau, A.R.; Gerner, E.W. Chemoprevention of prostate cancer with the polyamine synthesis inhibitor difluoromethylornithine. Recent Results Cancer Res. 2014, 202, 115–120. [Google Scholar] [PubMed]
- Simoneau, A.R.; Gerner, E.W.; Nagle, R.; Ziogas, A.; Fujikawa-Brooks, S.; Yerushalmi, H.; Ahlering, T.E.; Lieberman, R.; McLaren, C.E.; Anton-Culver, H.; et al. The effect of difluoromethylornithine on decreasing prostate size and polyamines in men: Results of a year-long phase IIB randomized placebo-controlled chemoprevention trial. Cancer Epidemiol. Biomarkers Prev. 2008, 17, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Streiff, R.R.; Bender, J.F. Phase I study of N1-N11-diethylnorspermine (DENSPM) administered TID for 6 days in patients with advanced malignancies. Investig. New Drugs 2001, 19, 29–39. [Google Scholar] [CrossRef]
- Wagner, T.; Jung, M. New lysine methyltransferase drug targets in cancer. Nat. Biotechnol. 2012, 30, 622–623. [Google Scholar] [CrossRef] [PubMed]
- Kaminskas, E.; Farrell, A.; Abraham, S.; Baird, A.; Hsieh, L.S.; Lee, S.L.; Leighton, J.K.; Patel, H.; Rahman, A.; Sridhara, R.; et al. Approval summary: Azacitidine for treatment of myelodysplastic syndrome subtypes. Clin. Cancer Res. 2005, 11, 3604–3608. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Hu, W.; Iyer, R.; Kavanagh, J.J.; Coleman, R.L.; Levenback, C.F.; Sood, A.K.; Wolf, J.K.; Gershenson, D.M.; Markman, M.; et al. Phase 1b–2a study to reverse platinum resistance through use of a hypomethylating agent, azacitidine, in patients with platinum-resistant or platinum-refractory epithelial ovarian cancer. Cancer 2011, 117, 1661–1669. [Google Scholar] [CrossRef] [PubMed]
- Iwata, H.; Sato, H.; Suzuki, R.; Yamada, R.; Ichinomiya, S.; Yanagihara, M.; Okabe, H.; Sekine, Y.; Yano, T.; Ueno, K. A demethylating agent enhances chemosensitivity to vinblastine in a xenograft model of renal cell carcinoma. Int. J. Oncol. 2011, 38, 1653–1661. [Google Scholar] [CrossRef] [PubMed]
- Kiziltepe, T.; Hideshima, T.; Catley, L.; Raje, N.; Yasui, H.; Shiraishi, N.; Okawa, Y.; Ikeda, H.; Vallet, S.; Pozzi, S.; et al. 5-Azacytidine, a DNA methyltransferase inhibitor, induces ATR-mediated DNA double-strand break responses, apoptosis, and synergistic cytotoxicity with doxorubicin and bortezomib against multiple myeloma cells. Mol. Cancer Ther. 2007, 6, 1718–1727. [Google Scholar] [CrossRef] [PubMed]
- Mao, M.; Tian, F.; Mariadason, J.M.; Tsao, C.C.; Lemos, R., Jr.; Dayyani, F.; Gopal, Y.N.; Jiang, Z.Q.; Wistuba, II; Tang, X.M.; et al. Resistance to braf inhibition in braf-mutant colon cancer can be overcome with pi3k inhibition or demethylating agents. Clin. Cancer Res. 2013, 19, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, K.; Gordian, E.; Singal, R. 5-Azacytidine reverses drug resistance in bladder cancer cells. Anticancer Res. 2011, 31, 3757–3766. [Google Scholar] [PubMed]
- Singal, R.; Ramachandran, K.; Gordian, E.; Quintero, C.; Zhao, W.; Reis, I.M. Phase I/II study of azacitidine, docetaxel, and prednisone in patients with metastatic castration-resistant prostate cancer previously treated with docetaxel-based therapy. Clin. Genitourin. Cancer 2015, 13, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lin, C.; Jin, C.; Yang, J.C.; Tanasa, B.; Li, W.; Merkurjev, D.; Ohgi, K.A.; Meng, D.; Zhang, J.; et al. Lncrna-dependent mechanisms of androgen-receptor-regulated gene activation programs. Nature 2013, 500, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Wee, Z.N.; Li, Z.; Lee, P.L.; Lee, S.T.; Lim, Y.P.; Yu, Q. EZH2-mediated inactivation of IFN-γ-JAK-STAT1 signaling is an effective therapeutic target in MYC-driven prostate cancer. Cell Rep. 2014, 8, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Androgen Responsive Genes * | |||
|---|---|---|---|
| Folate | Methionine | Polyamine | Transulfuration |
| SHMT | MAT | ODC | CBS |
| SARDH | AHCY | SMS | CTH |
| GLDC | SAT | GPX | |
| DHFR | AMD1 | GSR | |
| MTHFD | |||
| MTHFR | |||
| GNMT | |||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corbin, J.M.; Ruiz-Echevarría, M.J. One-Carbon Metabolism in Prostate Cancer: The Role of Androgen Signaling. Int. J. Mol. Sci. 2016, 17, 1208. https://doi.org/10.3390/ijms17081208
Corbin JM, Ruiz-Echevarría MJ. One-Carbon Metabolism in Prostate Cancer: The Role of Androgen Signaling. International Journal of Molecular Sciences. 2016; 17(8):1208. https://doi.org/10.3390/ijms17081208
Chicago/Turabian StyleCorbin, Joshua M., and Maria J. Ruiz-Echevarría. 2016. "One-Carbon Metabolism in Prostate Cancer: The Role of Androgen Signaling" International Journal of Molecular Sciences 17, no. 8: 1208. https://doi.org/10.3390/ijms17081208
APA StyleCorbin, J. M., & Ruiz-Echevarría, M. J. (2016). One-Carbon Metabolism in Prostate Cancer: The Role of Androgen Signaling. International Journal of Molecular Sciences, 17(8), 1208. https://doi.org/10.3390/ijms17081208