
T315 Decreases Acute Myeloid Leukemia Cell Viability through a Combination of Apoptosis Induction and Autophagic Cell Death

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
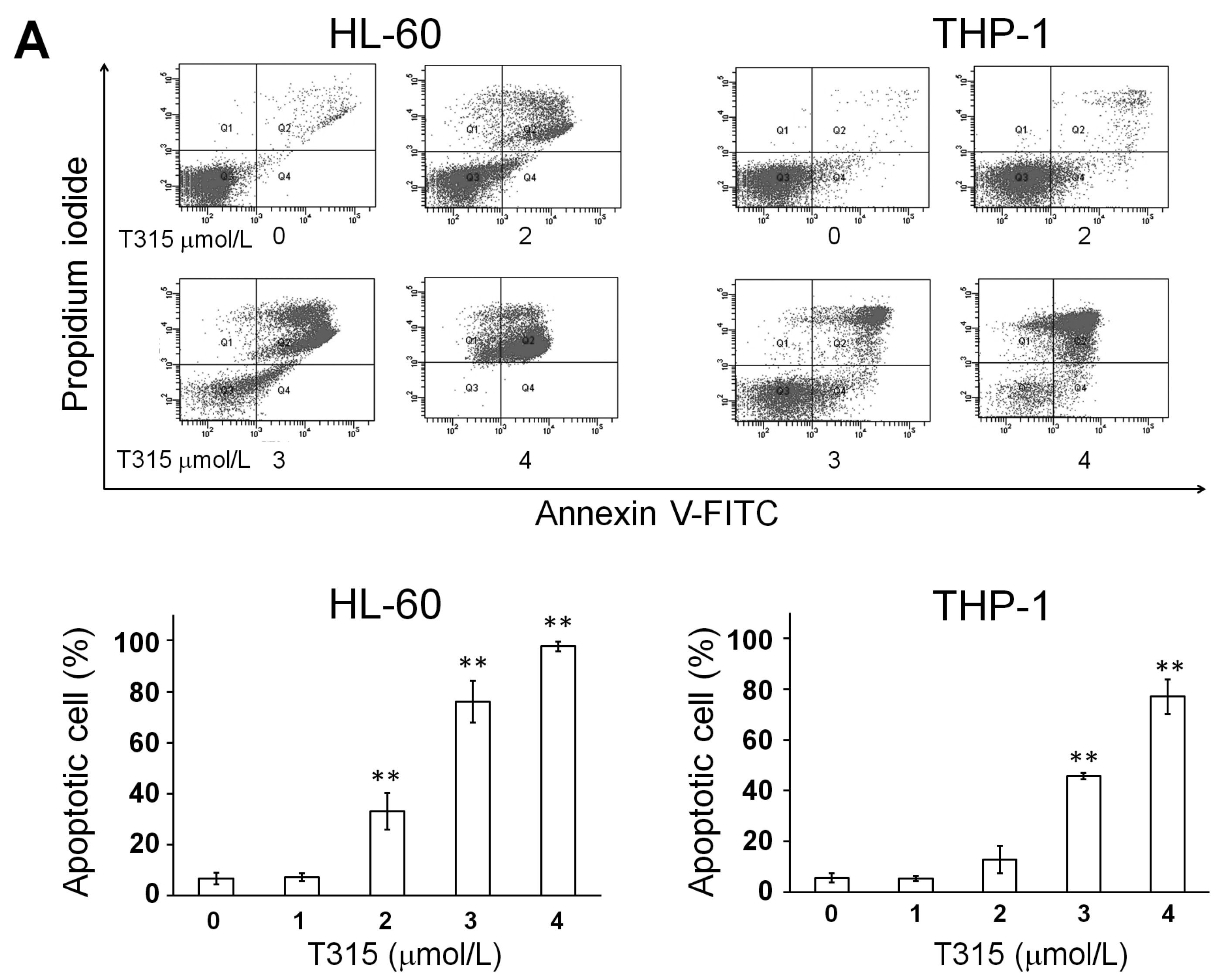
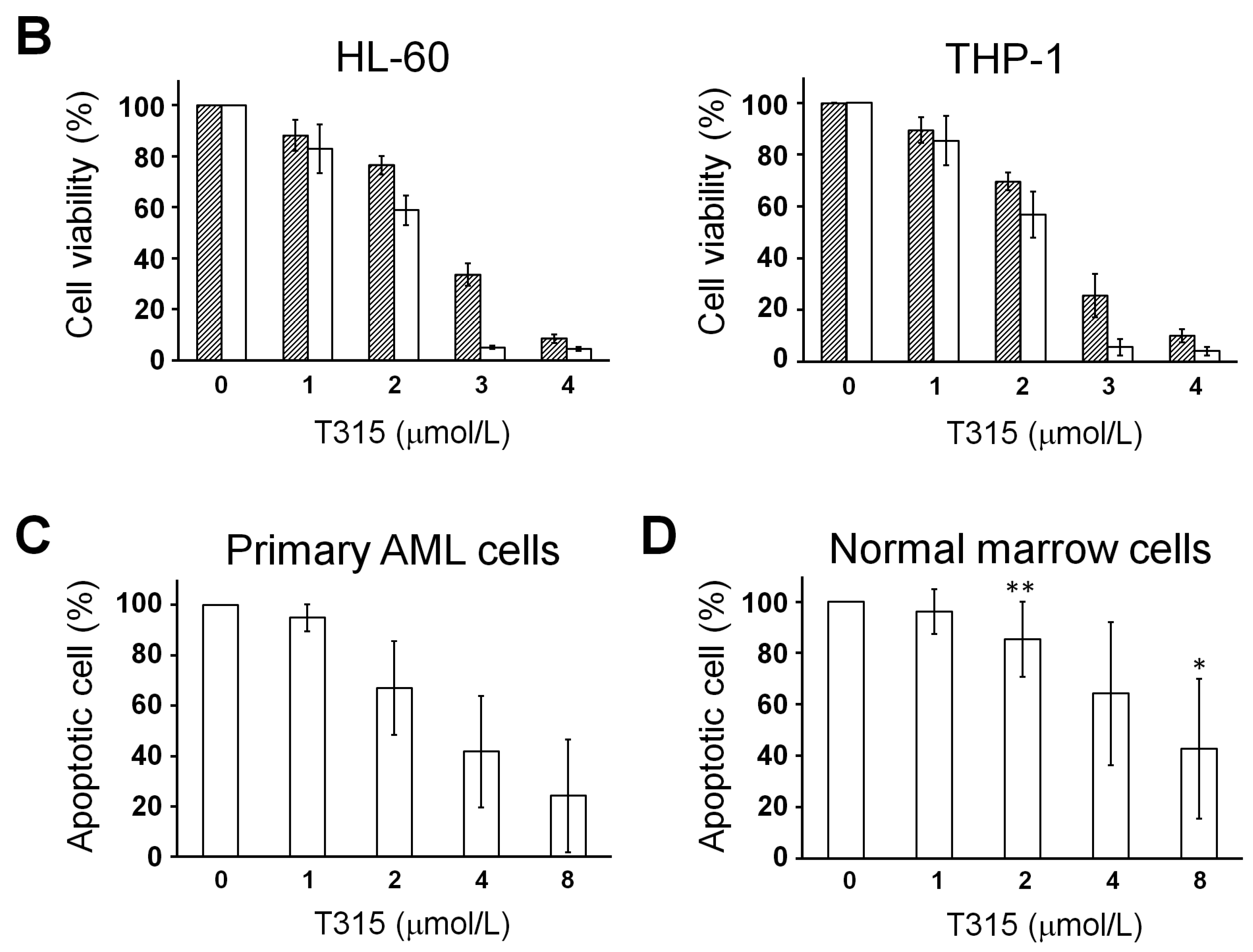
2.1. T315 Increases Apoptotic Cells and Reduces Viability of Acute Myeloid Leukemia (AML) Cell Lines and Primary Leukemia Cells from AML Patients
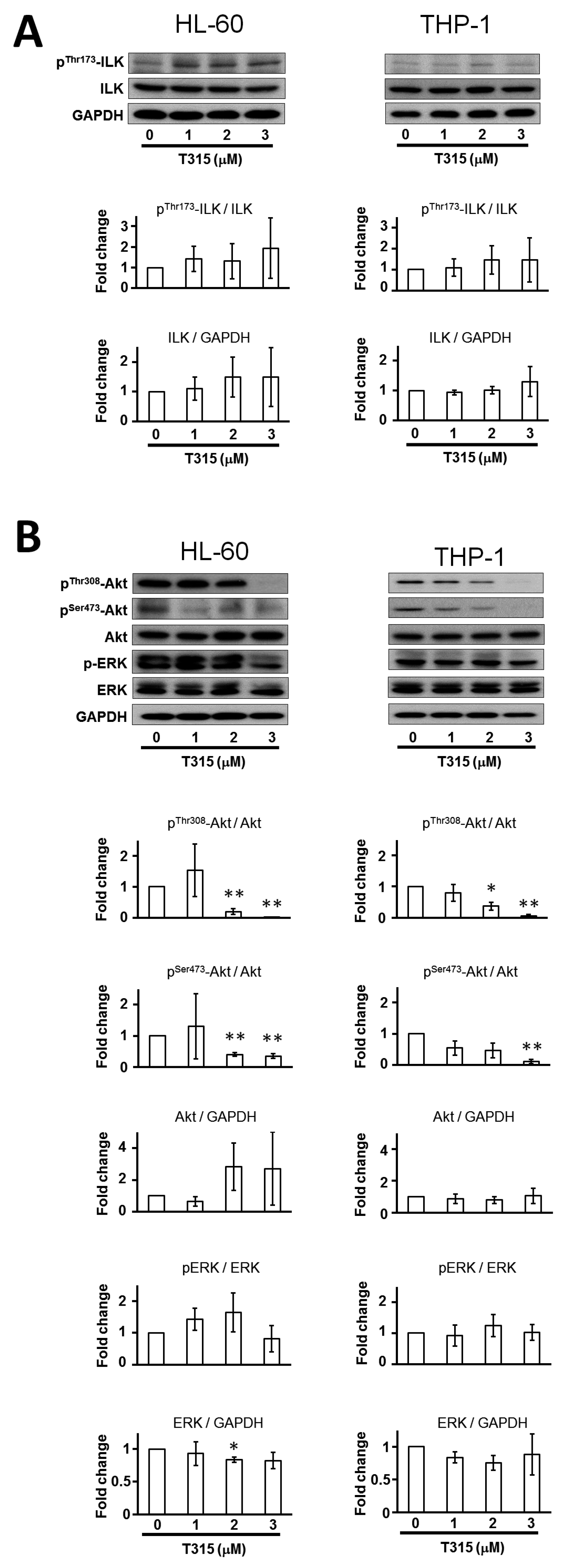
2.2. T315 Induces Down-Regulation of Protein Kinase B (Akt) and Phosphorylated Akt in AML Cell Lines
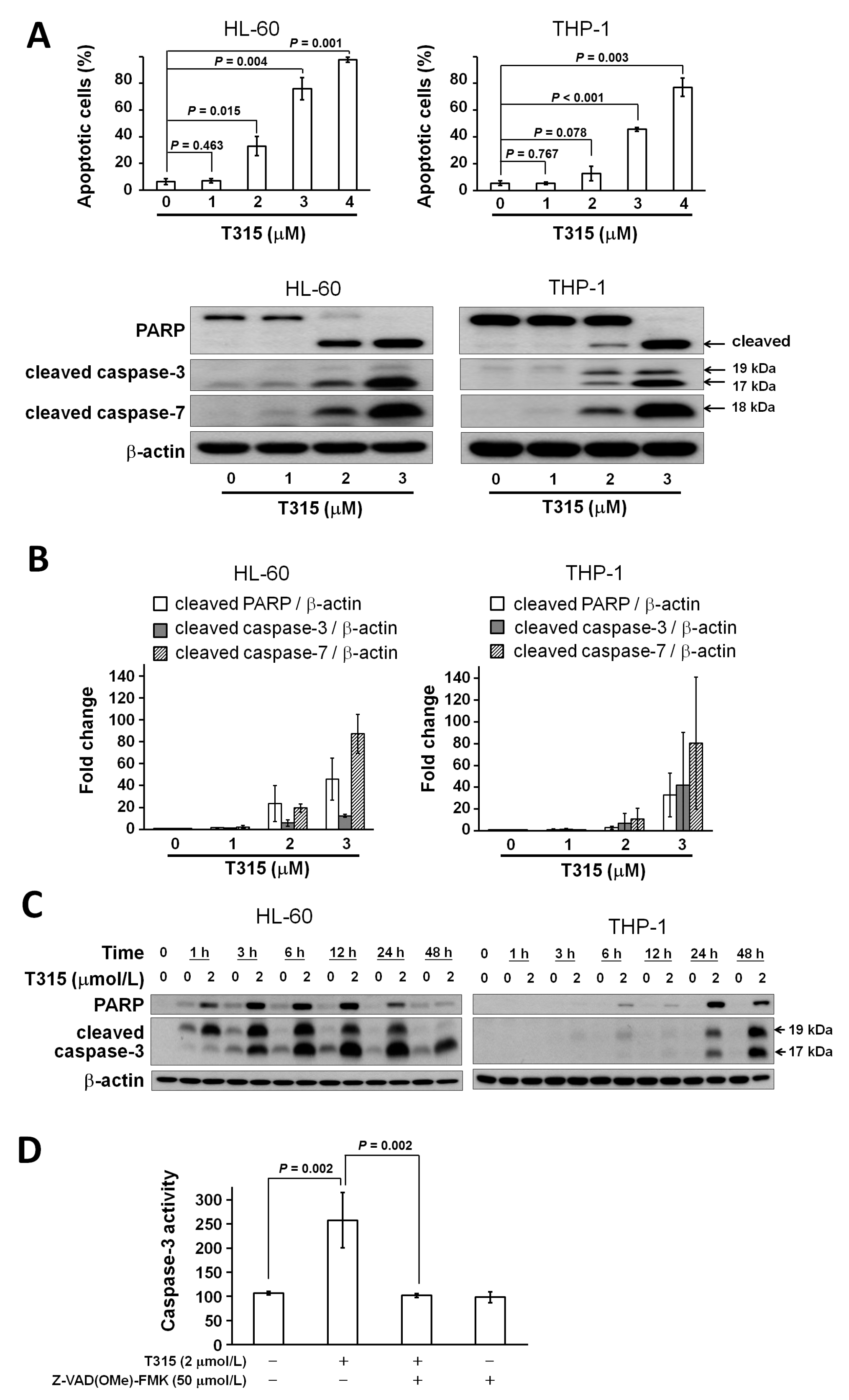
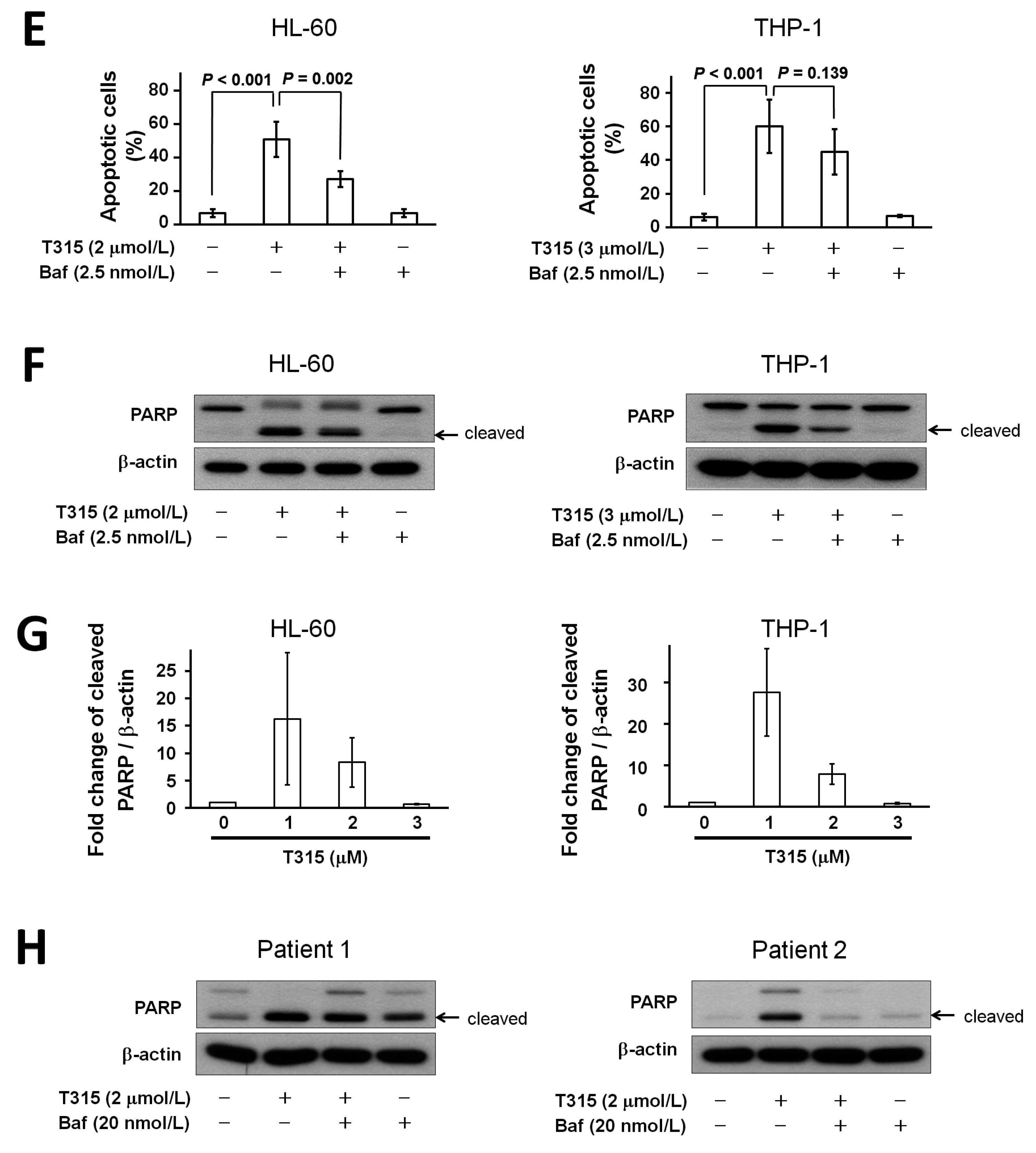
2.3. T315 Induces Apoptosis, Caspase Activation and Poly-ADP-Ribose Polymerase (PARP) Cleavage in AML Cell Lines
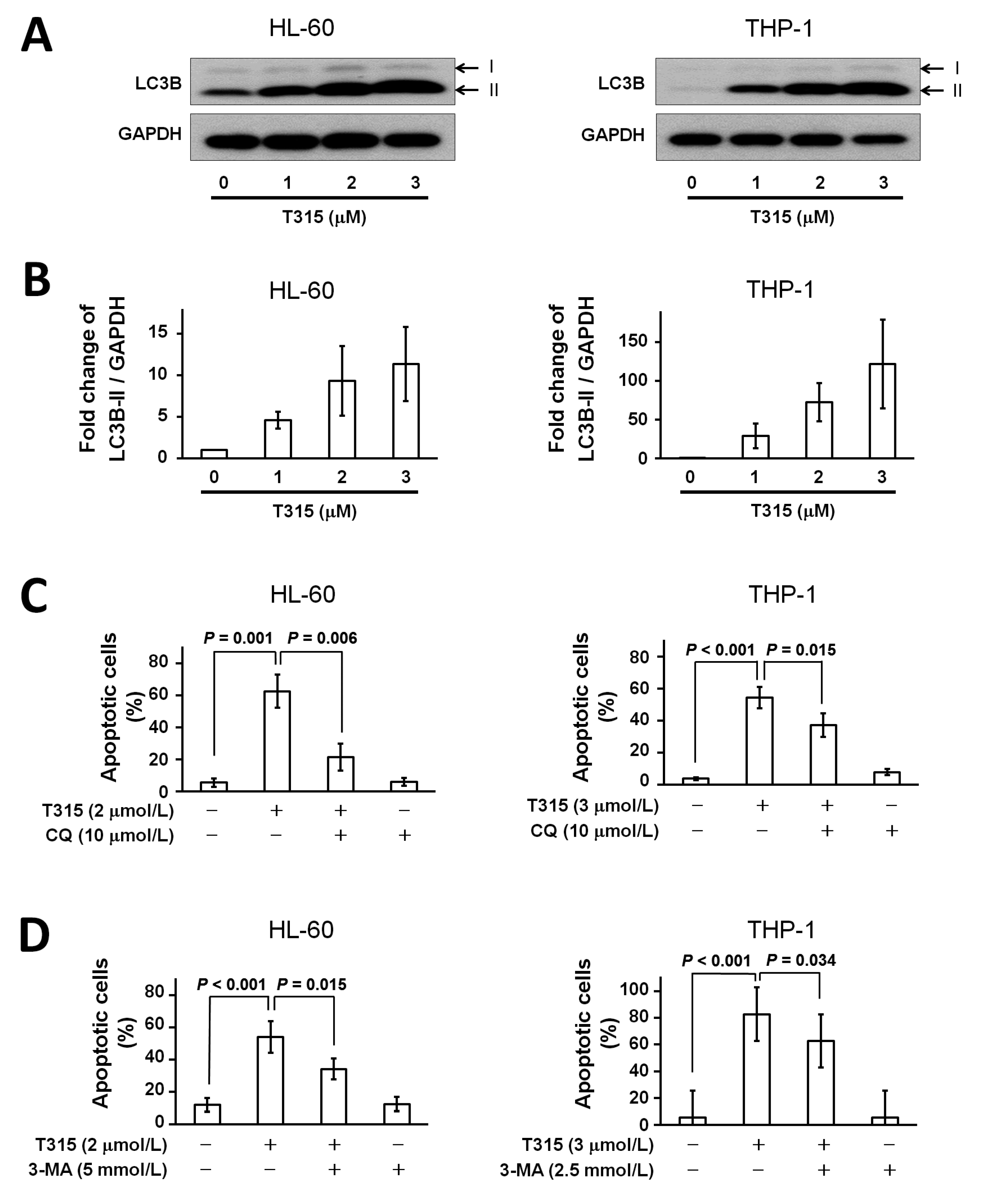
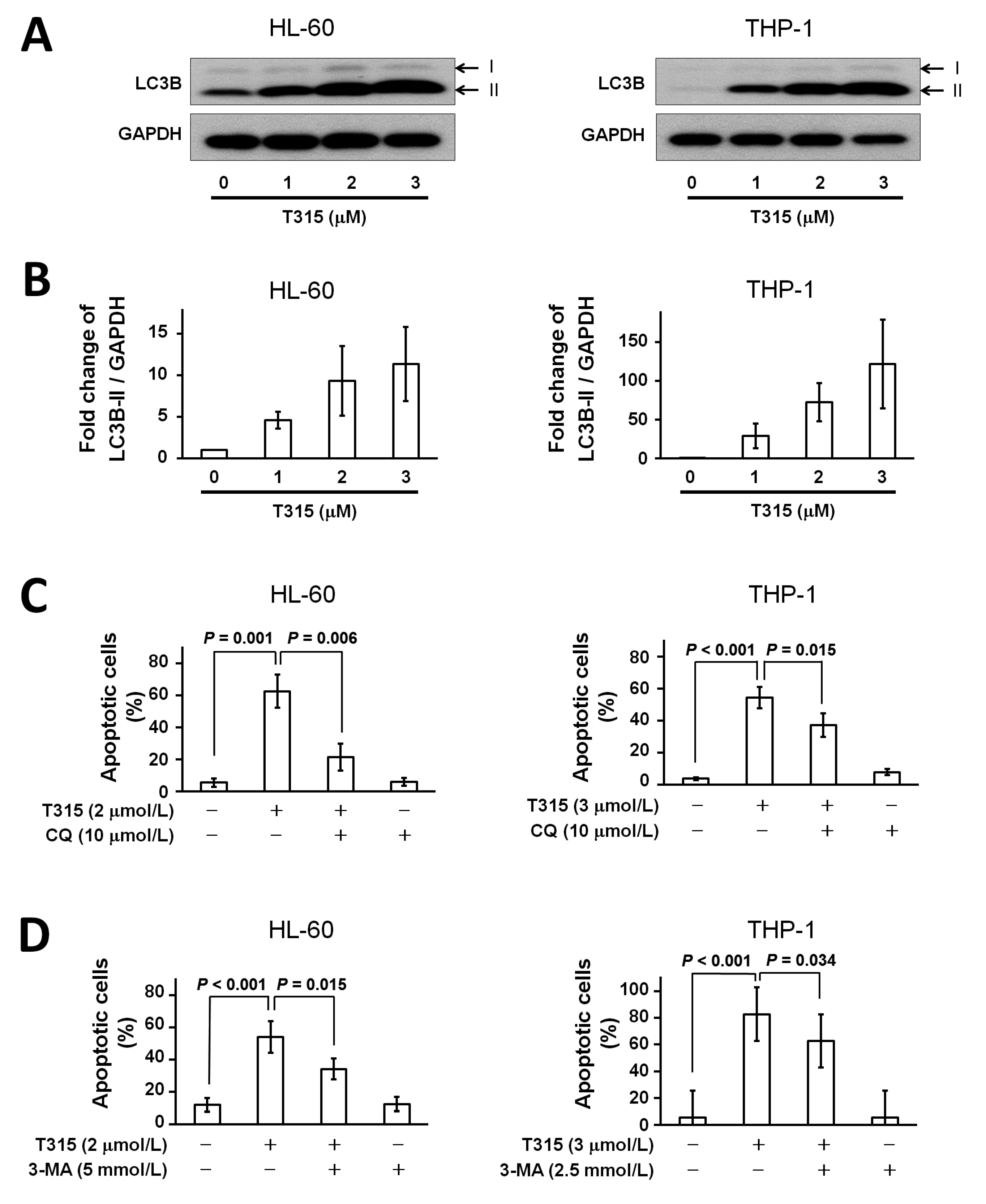
2.4. T315 Induces Autophagic Cell Death in AML Cell Lines
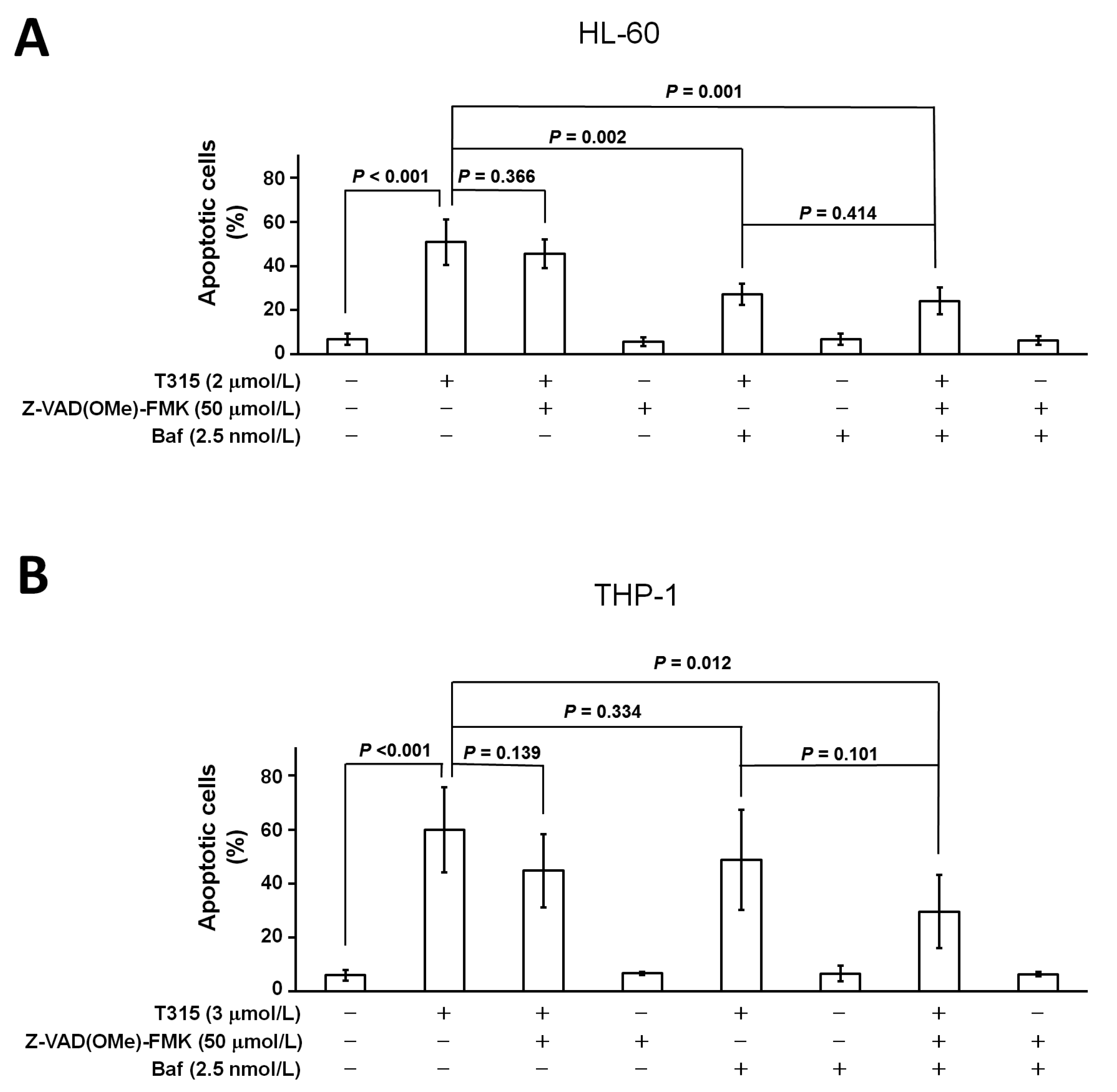
2.5. T315-Mediated Cytotoxicity Is Rescued by Combination of an Apoptosis Inhibitor and an Autophagy Inhibitor
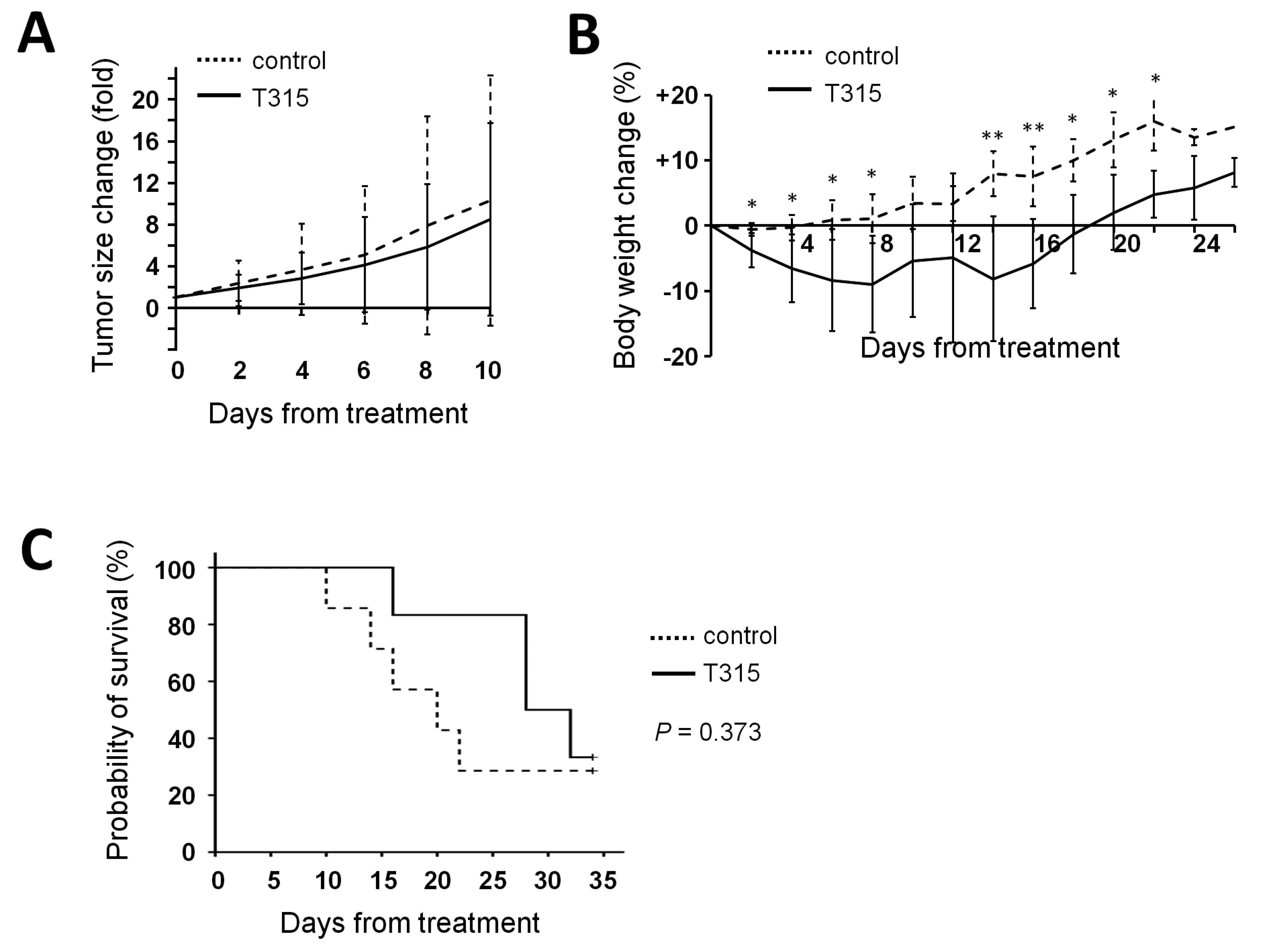
2.6. T315 Slows the Growth of THP-1 Xenografts and Prolongs the Survival of Tumor-Bearing Athymic Nude Mice
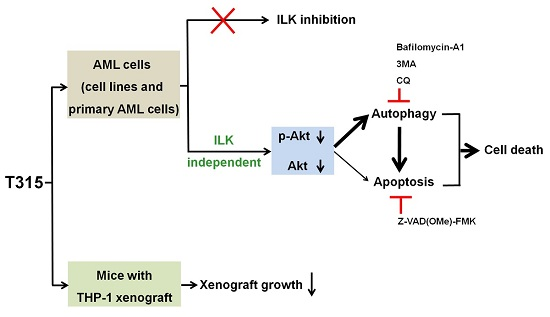
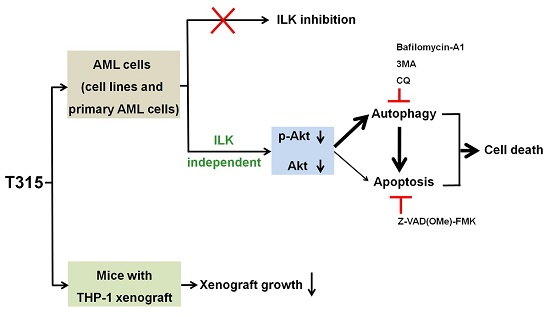
3. Discussion
4. Materials and Methods
4.1. Cells and Culture Conditions
4.2. Reagents
4.3. MTS Assay
4.4. Cell Viability and Apoptosis Assay by Flow Cytometry
4.5. Western Blotting
4.6. Analysis of Caspase-3 Activity
4.7. In Vivo Therapeutic Efficacy Evaluation of T315 in the THP-1 Xenograft Model
4.8. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AML | acute myeloid leukemia |
| DMSO | dimethyl sulfoxide |
| FITC | fluorescein isothiocyanate |
| HRP | horseradish peroxidase |
| ILK | integrin-linked kinase |
| PARP | poly-ADP-ribose polymerase |
| PI | propidium iodide |
References
- Tallman, M.S.; Gilliland, D.G.; Rowe, J.M. Drug therapy for acute myeloid leukemia. Blood 2005, 106, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.L.; Hsu, E.C.; Chou, C.C.; Chuang, H.C.; Bai, L.Y.; Kulp, S.K.; Chen, C.S. Identification and characterization of a novel integrin-linked kinase inhibitor. J. Med. Chem. 2011, 54, 6364–6374. [Google Scholar] [CrossRef] [PubMed]
- Hsu, E.C.; Kulp, S.K.; Huang, H.L.; Tu, H.J.; Salunke, S.B.; Sullivan, N.J.; Sun, D.; Wicha, M.S.; Shapiro, C.L.; Chen, C.S. Function of integrin-linked kinase in modulating the stemness of IL-6-abundant breast cancer cells by regulating γ-secretase-mediated NOTCH1 activation in caveolae. Neoplasia 2015, 17, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Tseng, P.C.; Chen, C.L.; Shan, Y.S.; Chang, W.T.; Liu, H.S.; Hong, T.M.; Hsieh, C.Y.; Lin, S.H.; Lin, C.F. An increase in integrin-linked kinase non-canonically confers NF-κB-mediated growth advantages to gastric cancer cells by activating ERK1/2. Cell Commun. Signal. 2014, 12, 69. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.M.; Ling, Y.; Woyach, J.A.; Beckwith, K.; Yeh, Y.Y.; Hertlein, E.; Zhang, X.; Lehman, A.; Awan, F.; Jones, J.A.; et al. OSU-T315: A novel targeted therapeutic that antagonizes AKT membrane localization and activation of chronic lymphocytic leukemia cells. Blood 2015, 125, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Ekiz, H.A.; Can, G.; Baran, Y. Role of autophagy in the progression and suppression of leukemias. Crit. Rev. Oncol. Hematol. 2012, 81, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Chaabane, W.; User, S.D.; El-Gazzah, M.; Jaksik, R.; Sajjadi, E.; Rzeszowska-Wolny, J.; Los, M.J. Autophagy, apoptosis, mitoptosis and necrosis: Interdependence between those pathways and effects on cancer. Arch. Immunol. Ther. Exp. (Warsz) 2013, 61, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Mei, Y.; Sinha, S. Role of the crosstalk between autophagy and apoptosis in cancer. J. Oncol. 2013, 2013, 102735. [Google Scholar] [CrossRef] [PubMed]
- Bredholt, T.; Dimba, E.A.; Hagland, H.R.; Wergeland, L.; Skavland, J.; Fossan, K.O.; Tronstad, K.J.; Johannessen, A.C.; Vintermyr, O.K.; Gjertsen, B.T. Camptothecin and khat (Catha edulis Forsk.) induced distinct cell death phenotypes involving modulation of c-FLIPL, Mcl-1, procaspase-8 and mitochondrial function in acute myeloid leukemia cell lines. Mol. Cancer 2009, 8, 101. [Google Scholar] [CrossRef] [PubMed]
- Crazzolara, R.; Bradstock, K.F.; Bendall, L.J. RAD001 (Everolimus) induces autophagy in acute lymphoblastic leukemia. Autophagy 2009, 5, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Aoki, H.; Kuhnel, F.; Kondo, Y.; Kubicka, S.; Wirth, T.; Iwado, E.; Iwamaru, A.; Fujiwara, K.; Hess, K.R.; et al. Autophagic cell death of malignant glioma cells induced by a conditionally replicating adenovirus. J. Natl. Cancer Inst. 2006, 98, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Peng, P.L.; Kuo, W.H.; Tseng, H.C.; Chou, F.P. Synergistic tumor-killing effect of radiation and berberine combined treatment in lung cancer: The contribution of autophagic cell death. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D′Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, E.; Maiuri, M.C.; Orhon, I.; Kepp, O.; Morselli, E.; Criollo, A.; Kroemer, G. p53 represses autophagy in a cell cycle-dependent fashion. Cell Cycle 2008, 7, 3006–3011. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.Y.; Weng, J.R.; Chiu, C.F.; Wu, C.Y.; Yeh, S.P.; Sargeant, A.M.; Lin, P.H.; Liao, Y.M. OSU-A9, an indole-3-carbinol derivative, induces cytotoxicity in acute myeloid leukemia through reactive oxygen species-mediated apoptosis. Biochem. Pharmacol. 2013, 86, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.Y.; Weng, J.R.; Lo, W.J.; Yeh, S.P.; Wu, C.Y.; Wang, C.Y.; Chiu, C.F.; Lin, C.W. Inhibition of hedgehog signaling induces monocytic differentiation of HL-60 cells. Leuk. Lymphoma 2012, 53, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.Y.; Chiu, C.F.; Chu, P.C.; Lin, W.Y.; Chiu, S.J.; Weng, J.R. A triterpenoid from wild bitter gourd inhibits breast cancer cells. Sci. Rep. 2016, 6, 22419. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.Y.; Chiu, C.F.; Kapuriya, N.P.; Shieh, T.M.; Tsai, Y.C.; Wu, C.Y.; Sargeant, A.M.; Weng, J.R. BX795, a TBK1 inhibitor, exhibits antitumor activity in human oral squamous cell carcinoma through apoptosis induction and mitotic phase arrest. Eur. J. Pharmacol. 2015, 769, 287–296. [Google Scholar] [CrossRef] [PubMed]
 ) or 48 h (□). The cells were analyzed by MTS assay, as described in Materials and Methods; (C) Primary AML cells (0.25 × 106 cells/mL) were incubated with T315 or DMSO for 24 h. The cells were stained with annexin V-FITC and PI to assess apoptotic cells percentage (n = 26); (D) Normal bone marrow nucleated cells (0.25 × 106 cells/mL) were incubated with T315 or DMSO for 24 h. The cells were stained with annexin V-FITC and PI to assess apoptotic cells percentage (n = 16). * denotes p < 0.05; ** denotes p < 0.01 compared to the control group (in panel A) or compared to the primary AML cells at the same concentration of T315 (in panel D).
) or 48 h (□). The cells were analyzed by MTS assay, as described in Materials and Methods; (C) Primary AML cells (0.25 × 106 cells/mL) were incubated with T315 or DMSO for 24 h. The cells were stained with annexin V-FITC and PI to assess apoptotic cells percentage (n = 26); (D) Normal bone marrow nucleated cells (0.25 × 106 cells/mL) were incubated with T315 or DMSO for 24 h. The cells were stained with annexin V-FITC and PI to assess apoptotic cells percentage (n = 16). * denotes p < 0.05; ** denotes p < 0.01 compared to the control group (in panel A) or compared to the primary AML cells at the same concentration of T315 (in panel D).
) or 48 h (□). The cells were analyzed by MTS assay, as described in Materials and Methods; (C) Primary AML cells (0.25 × 106 cells/mL) were incubated with T315 or DMSO for 24 h. The cells were stained with annexin V-FITC and PI to assess apoptotic cells percentage (n = 26); (D) Normal bone marrow nucleated cells (0.25 × 106 cells/mL) were incubated with T315 or DMSO for 24 h. The cells were stained with annexin V-FITC and PI to assess apoptotic cells percentage (n = 16). * denotes p < 0.05; ** denotes p < 0.01 compared to the control group (in panel A) or compared to the primary AML cells at the same concentration of T315 (in panel D).
) or 48 h (□). The cells were analyzed by MTS assay, as described in Materials and Methods; (C) Primary AML cells (0.25 × 106 cells/mL) were incubated with T315 or DMSO for 24 h. The cells were stained with annexin V-FITC and PI to assess apoptotic cells percentage (n = 26); (D) Normal bone marrow nucleated cells (0.25 × 106 cells/mL) were incubated with T315 or DMSO for 24 h. The cells were stained with annexin V-FITC and PI to assess apoptotic cells percentage (n = 16). * denotes p < 0.05; ** denotes p < 0.01 compared to the control group (in panel A) or compared to the primary AML cells at the same concentration of T315 (in panel D).





© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiu, C.-F.; Weng, J.-R.; Jadhav, A.; Wu, C.-Y.; Sargeant, A.M.; Bai, L.-Y. T315 Decreases Acute Myeloid Leukemia Cell Viability through a Combination of Apoptosis Induction and Autophagic Cell Death. Int. J. Mol. Sci. 2016, 17, 1337. https://doi.org/10.3390/ijms17081337
Chiu C-F, Weng J-R, Jadhav A, Wu C-Y, Sargeant AM, Bai L-Y. T315 Decreases Acute Myeloid Leukemia Cell Viability through a Combination of Apoptosis Induction and Autophagic Cell Death. International Journal of Molecular Sciences. 2016; 17(8):1337. https://doi.org/10.3390/ijms17081337
Chicago/Turabian StyleChiu, Chang-Fang, Jing-Ru Weng, Appaso Jadhav, Chia-Yung Wu, Aaron M. Sargeant, and Li-Yuan Bai. 2016. "T315 Decreases Acute Myeloid Leukemia Cell Viability through a Combination of Apoptosis Induction and Autophagic Cell Death" International Journal of Molecular Sciences 17, no. 8: 1337. https://doi.org/10.3390/ijms17081337
APA StyleChiu, C.-F., Weng, J.-R., Jadhav, A., Wu, C.-Y., Sargeant, A. M., & Bai, L.-Y. (2016). T315 Decreases Acute Myeloid Leukemia Cell Viability through a Combination of Apoptosis Induction and Autophagic Cell Death. International Journal of Molecular Sciences, 17(8), 1337. https://doi.org/10.3390/ijms17081337