
Comparative Genomics of the Extreme Acidophile Acidithiobacillus thiooxidans Reveals Intraspecific Divergence and Niche Adaptation

Abstract
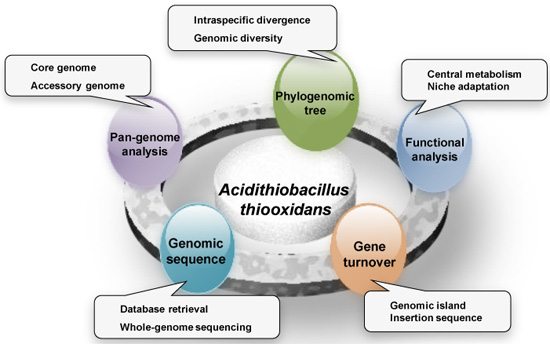
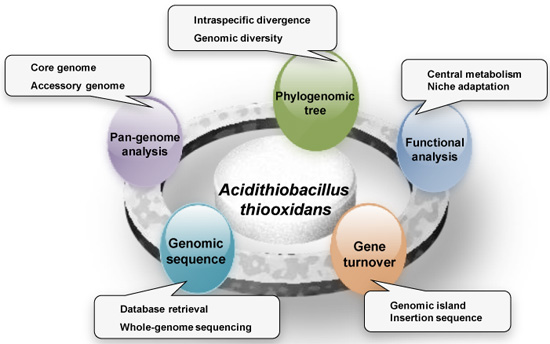
:
1. Introduction
2. Results and Discussion
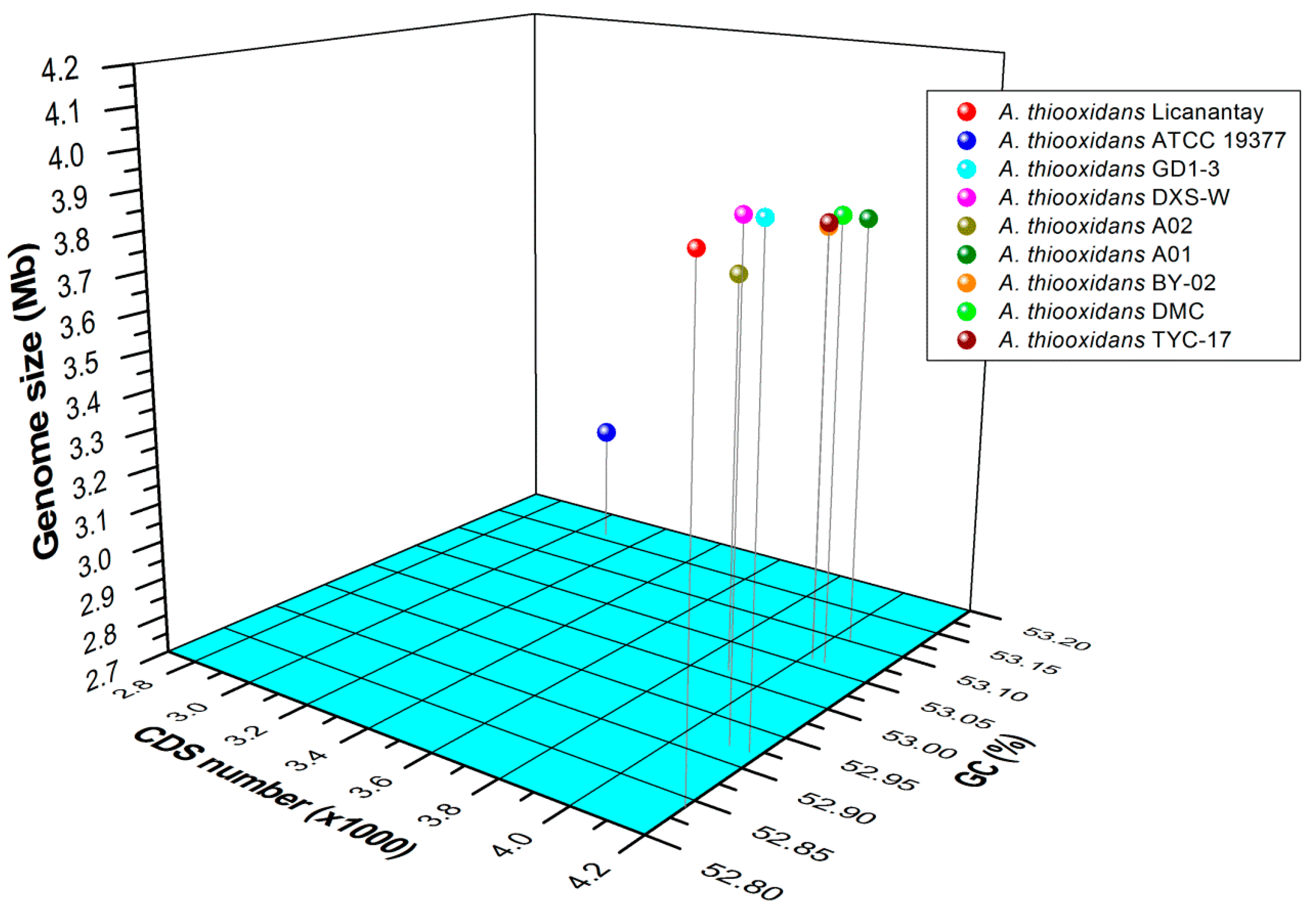
2.1. General Features of Acidithiobacillus thiooxidans (A. thiooxidans) Genomes
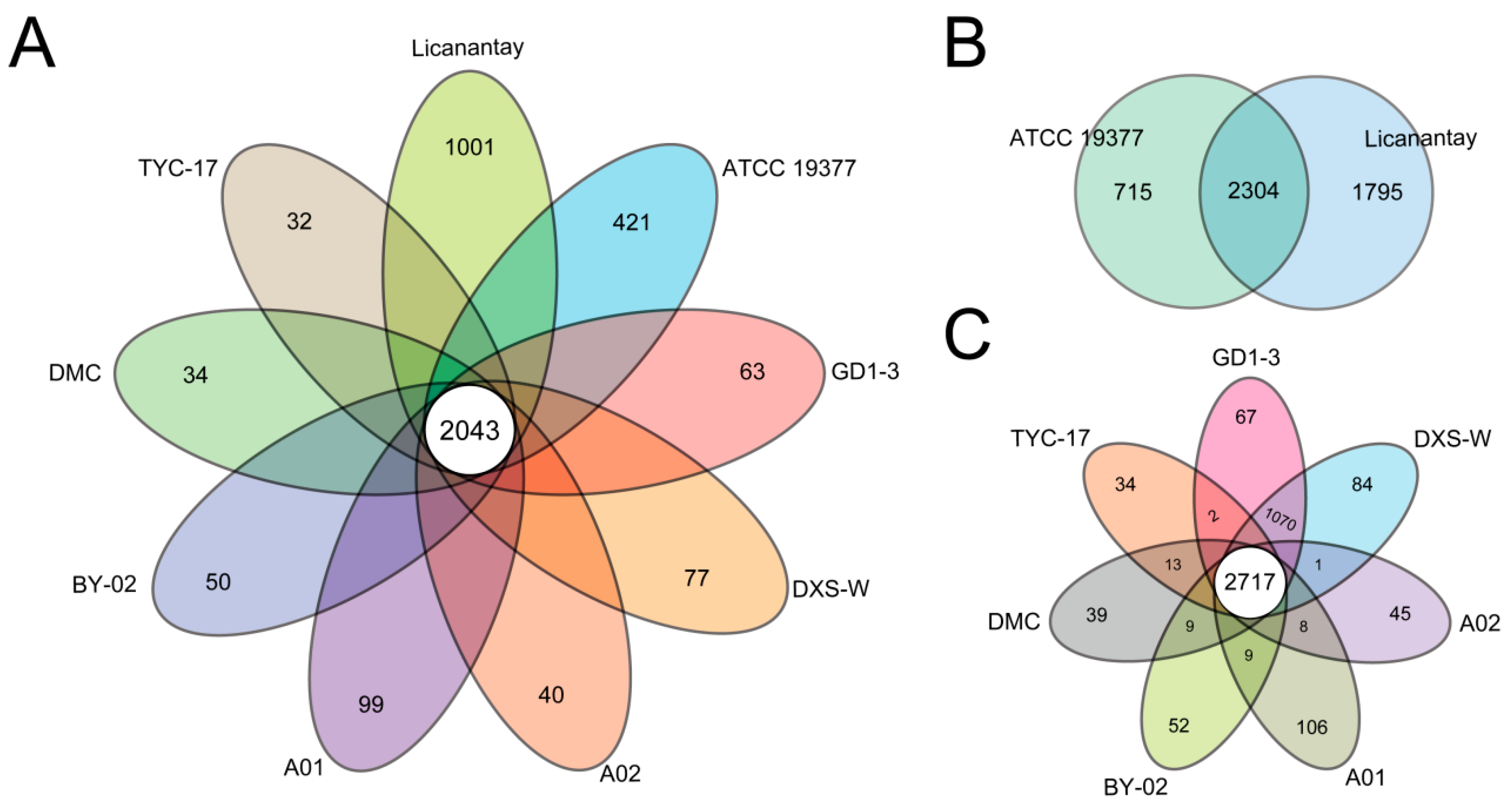
2.2. Pan-Genome Analysis of A. thiooxidans Strains
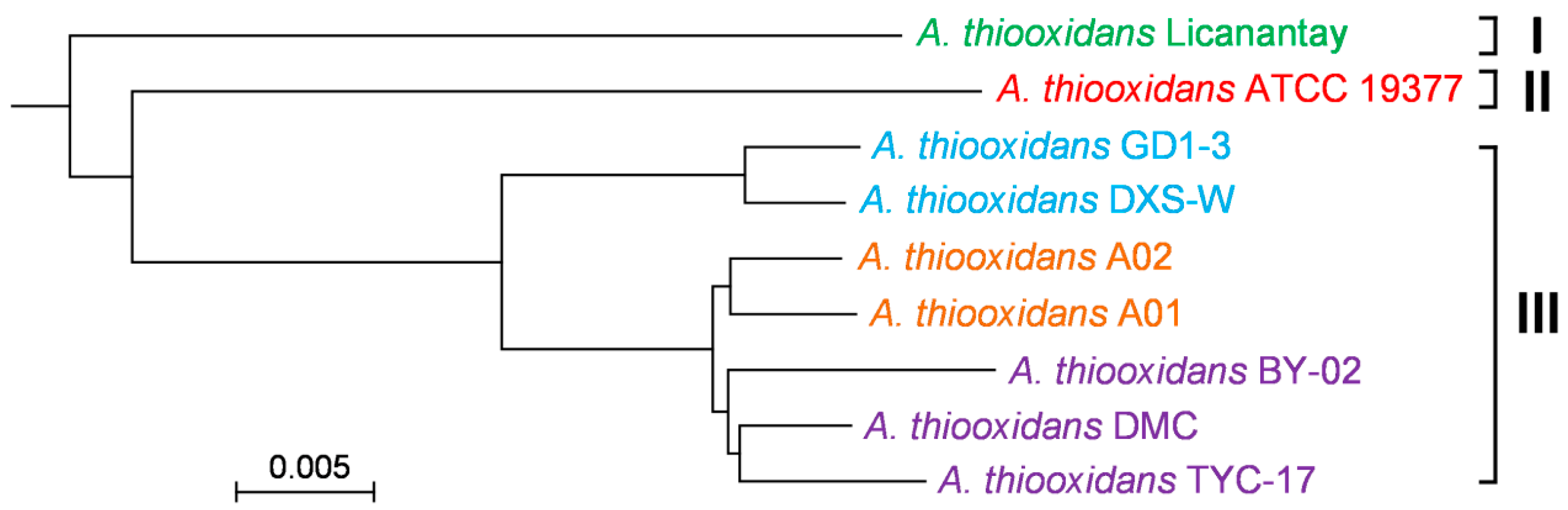
2.3. Phylogenomic Tree Based on Core Genome
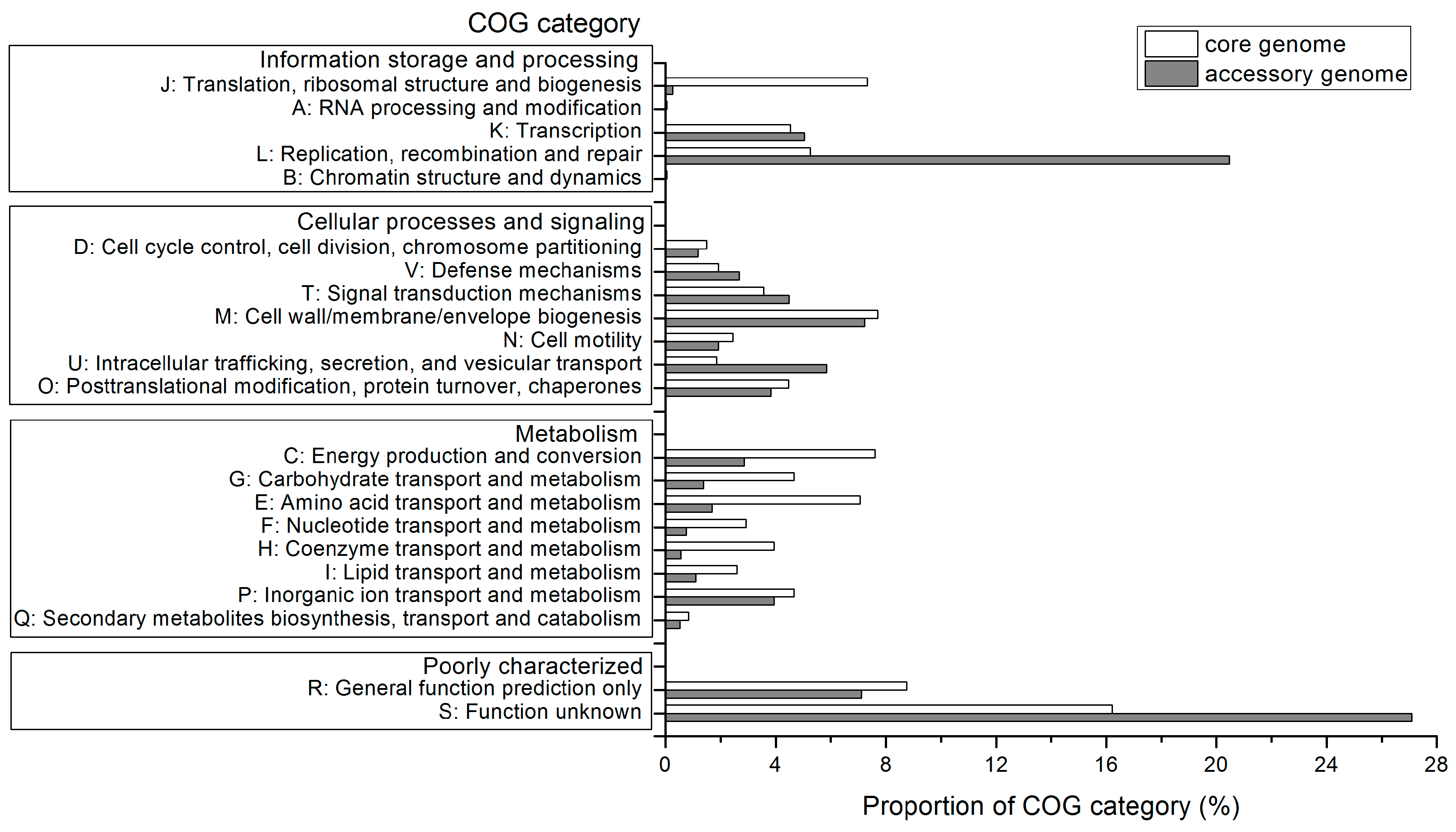
2.4. Functional Features of the Pan-Genome
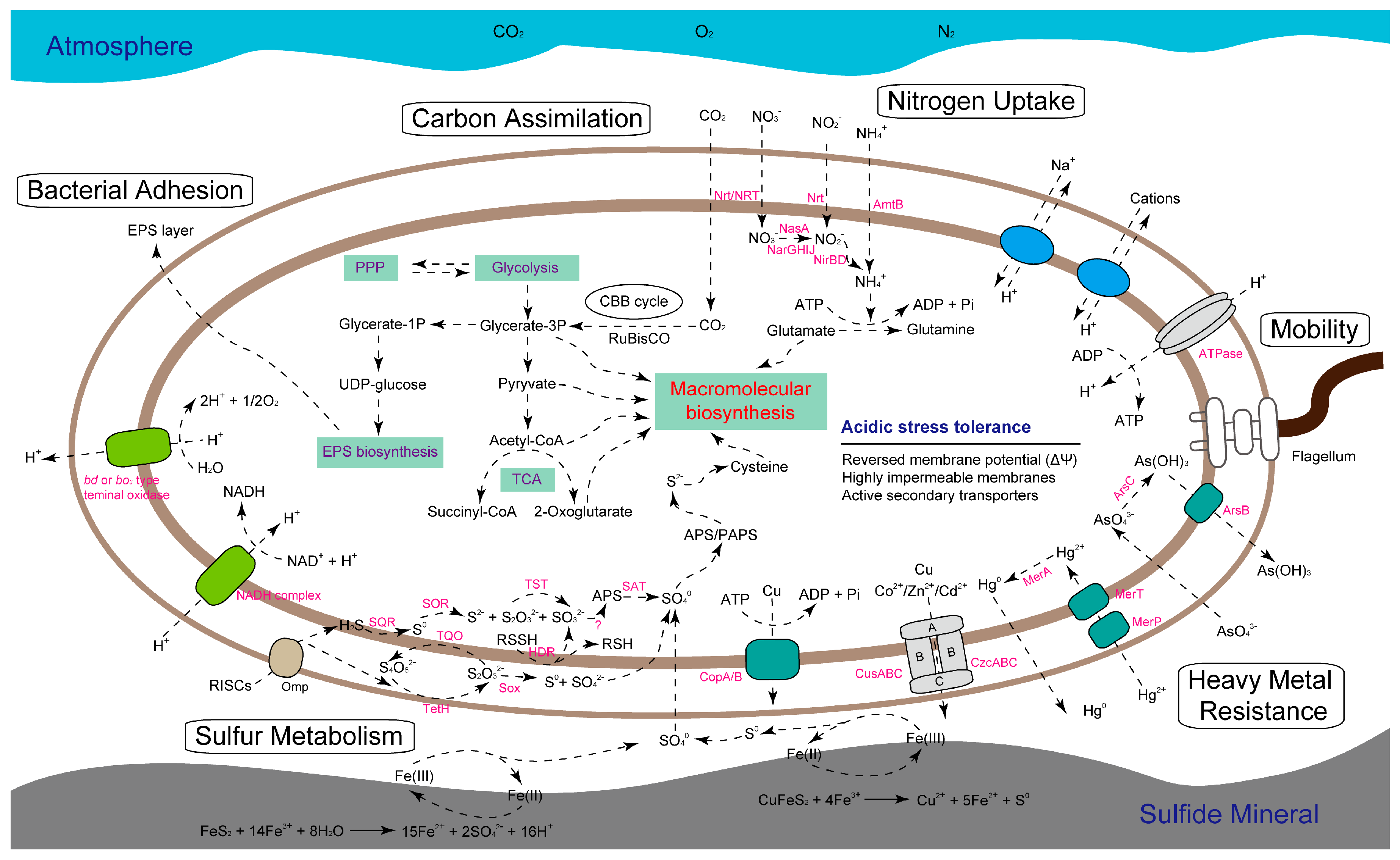
2.5. Identification of Metabolic Traits and Management Strategies to Environmental Stress
2.5.1. Feature of Central Metabolism
2.5.2. Predicted Stress Tolerance Mechanisms
2.6. Gene Turnover Analysis
3. Materials and Methods
3.1. Bacterial Strains Used in This Study
3.2. Genome Sequencing and Bioinformatics Analyses
3.3. Pan-Genome Analysis
3.4. Phylogenomic and Phylogenetic Analyses
3.5. Data Deposition
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kennedy, S.P.; Ng, W.V.; Salzberg, S.L.; Hood, L.; DasSarma, S. Understanding the adaptation of Halobacterium species NRC-1 to its extreme environment through computational analysis of its genome sequence. Genome Res. 2001, 11, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Takami, H.; Takaki, Y.; Uchiyama, I. Genome sequence of Oceanobacillus iheyensis isolated from the Iheya Ridge and its unexpected adaptive capabilities to extreme environments. Nucleic Acids Res. 2002, 30, 3927–3935. [Google Scholar] [CrossRef] [PubMed]
- Falb, M.; Pfeiffer, F.; Palm, P.; Rodewald, K.; Hickmann, V.; Tittor, J.; Oesterhelt, D. Living with two extremes: Conclusions from the genome sequence of Natronomonas pharaonis. Genome Res. 2005, 15, 1336–1343. [Google Scholar] [CrossRef] [PubMed]
- Rothschild, L.J.; Mancinelli, R.L. Life in extreme environments. Nature 2001, 409, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Yin, H.; Liang, Y.; Hu, Q.; Zhou, X.; Xiao, Y.; Ma, L.; Zhang, X.; Qiu, G.; Liu, X. Comparative genome analysis reveals metabolic versatility and environmental adaptations of Sulfobacillus thermosulfidooxidans strain ST. PLoS ONE 2014, 9, e99417. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Niu, J.; Liang, Y.; Liu, X.; Yin, H. Metagenome-scale analysis yields insights into the structure and function of microbial communities in a copper bioleaching heap. BMC Genet. 2016, 17, 21. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B. Biodiversity and ecology of acidophilic microorganisms. FEMS Microbiol. Ecol. 1998, 27, 307–317. [Google Scholar] [CrossRef]
- Jacobsen, A.; Hendriksen, R.S.; Aaresturp, F.M.; Ussery, D.W.; Friis, C. The Salmonella enterica pan-genome. Microb. Ecol. 2011, 62, 487–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyson, G.W.; Chapman, J.; Hugenholtz, P.; Allen, E.; Ram, R.; Richardson, P.; Solovyev, V.; Rubin, E.; Rokhsar, D.; Banfield, J. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 2004, 428, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Zhang, X.; Li, X.; He, Z.; Liang, Y.; Guo, X.; Hu, Q.; Xiao, Y.; Cong, J.; Ma, L.; et al. Whole-genome sequencing reveals novel insights into sulfur oxidation in the extremophile Acidithiobacillus thiooxidans. BMC Microbiol. 2014, 14, 179. [Google Scholar] [CrossRef] [PubMed]
- Pallen, M.J.; Wren, B.W. Bacterial pathogenomics. Nature 2007, 449, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative genomics: The bacterial pan-genome. Curr. Opin. Microbiol. 2008, 11, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Medini, D.; Donati, C.; Tettelin, H.; Masignani, V.; Rappuoli, R. The microbial pan-genome. Curr. Opin. Genet. Dev. 2005, 15, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, X.; Liang, Y.; Fan, F.; Zhang, X.; Yin, H. Metabolic diversity and adaptive mechanisms of iron- and/or sulfur-oxidizing autotrophic acidophiles in extremely acidic environments. Environ. Microbiol. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Valdes, J.; Ossandon, F.; Quatrini, R.; Dopson, M.; Holmes, D.S. Draft genome sequence of the extremely acidophilic biomining bacterium Acidithiobacillus thiooxidans ATCC 19377 provides insights into the evolution of the Acidithiobacillus genus. J. Bacteriol. 2011, 193, 7003–7004. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Zhang, X.; Liang, Y.; Xiao, Y.; Niu, J.; Liu, X. Draft genome sequence of the extremophile Acidithiobacillus thiooxidans A01, isolated from the wastewater of a coal dump. Genome Announc. 2014, 2, e00222-14. [Google Scholar] [CrossRef] [PubMed]
- Travisany, D.; Cortés, M.P.; Latorre, M.; Di Genova, A.; Budinich, M.; Bobadilla-Fazzini, R.; Parada, P.; González, M.; Maass, A. A new genome of Acidithiobacillus thiooxidans provides insights into adaptation to a bioleaching environment. Res. Microbiol. 2014, 165, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Nuñez, P.A.; Romero, H.; Farber, M.D.; Rocha, E.P.C. Natural selection for operons depends on genome size. Genome Biol. Evol. 2013, 5, 2242–2254. [Google Scholar] [CrossRef] [PubMed]
- Levicán, G.; Ugalde, J.A.; Ehrenfeld, N.; Maass, A.; Parada, P. Comparative genomic analysis of carbon and nitrogen assimilation mechanisms in three indigenous bioleaching bacteria: Predictions and validations. BMC Genom. 2008, 9, 581. [Google Scholar] [CrossRef] [PubMed]
- Douillard, F.P.; Ribbera, A.; Kant, R.; Pietilä, T.; Järvinen, H.; Messing, M.; Randazzo, C.; Paulin, L.; Laine, P.; Ritari, J.; et al. Comparative genomic and functional analysis of 100 Lactobacillus rhamnosus strains and their comparison with strain GG. PLoS Genet. 2013, 9, e1003683. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, M.; Epstein, B.; Badgley, B.D.; Unno, T.; Xu, L.; Reese, J.; Gyaneshwar, P.; Denny, R.; Mudge, J.; Bharti, A.; et al. Comparative genomics of the core and accessory genomes of 48 Sinorhizobium strains comprising five genospecies. Genome Biol. 2013, 14, R17. [Google Scholar] [CrossRef] [PubMed]
- Kittichotirat, W.; Bumgarner, R.E.; Asikainen, S.; Chen, C. Identification of the pangenome and its components in 14 distinct Aggregatibacter actinomycetemcomitans strains by comparative genomic analysis. PLoS ONE 2011, 6, e22420. [Google Scholar] [CrossRef] [PubMed]
- Baker-Austin, C.; Dopson, M. Life in acid: pH homeostasis in acidophiles. Trends Microbiol. 2007, 15, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.X.; Li, J.T.; Chen, Y.T.; Huang, L.N.; Hua, Z.S.; Hu, M.; Shu, W.S. Shifts in microbial community composition and function in the acidification of a lead/zinc mine tailings. Environ. Microbiol. 2013, 15, 2431–2444. [Google Scholar] [CrossRef] [PubMed]
- Silver, S.; Phung, L.T. Bacterial heavy metal resistance: New surprises. Annu. Rev. Microbiol. 1996, 50, 753–789. [Google Scholar] [CrossRef] [PubMed]
- Ramos, P.I.P.; Picão, R.C.; de Almeidal, L.G.P.; Lima, N.C.B.; Girardello, R.; Vivan, A.C.P.; Xavier, D.E.; Barcellos, F.G.; Pelisson, M.; Vespero, E.C.; et al. Comparative analysis of the complete genome of KPC-2-producing Klebsiella pneumoniae Kp13 reveals remarkable genome plasticity and a wide repertoire of virulence and resistance mechanisms. BMC Genom. 2014, 15, 54. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, P.; Gogarten, J.P. Estimating the size of the bacterial pan-genome. Trends Genet. 2009, 25, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Fondi, M.; Rizzi, E.; Emiliani, G.; Orlandini, V.; Berna, L.; Papaleo, M.C.; Perrin, E.; Maida, I.; Corti, G.; de Bellis, G.; et al. The genome sequence of the hydrocarbon-degrading Acinetobacter venetianus VE-C3. Res. Microbiol. 2013, 164, 439–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Wong, C.; Chung, K.M.; Jiang, J.; Leung, F.C. Comparative genome analysis of Enterobacter cloacae. PLoS ONE 2013, 8, e74487. [Google Scholar] [CrossRef] [PubMed]
- Cannon, G.C.; Bradburne, C.E.; Aldrich, H.C.; Baker, S.H.; Heinhorst, S.; Shively, J.M. Microcompartments in prokaryotes: Carboxysomes and related polyhedra. Appl. Environ. Microbiol. 2001, 67, 5351–5361. [Google Scholar] [CrossRef] [PubMed]
- Barreto, M.; Jedlicki, E.; Holmes, D.S. Identification of a gene cluster for the formation of extracellular polysaccharide precursors in the chemolithoautotroph Acidithiobacillus ferrooxidans. Appl. Environ. Microbiol. 2005, 71, 2902–2909. [Google Scholar] [CrossRef] [PubMed]
- González, A.; Bellenberg, S.; Mamani, S.; Ruiz, L.; Echeverría, A.; Soulère, L.; Doutheau, A.; Demergasso, C.; Sand, W.; Queneau, Y.; et al. AHL signaling molecules with a large acyl chain enhance biofilm formation on sulfur and metal sulfides by the bioleaching bacterium Acidithiobacillus ferrooxidans. Appl. Microbiol. Biotechnol. 2013, 97, 3729–3737. [Google Scholar] [CrossRef] [PubMed]
- Watling, H.R. The bioleaching of sulphide minerals with emphasis on copper sulphides—A review. Hydrometallurgy 2006, 84, 81–108. [Google Scholar] [CrossRef]
- Valdés, J.; Pedroso, I.; Quatrini, R.; Dodson, R.J.; Tettelin, H.; Blake, R.; Eisen, J.A.; Holmes, D.S. Acidithiobacillus ferrooxidans metabolism: From genome sequence to industrial applications. BMC Genom. 2008, 9, 597. [Google Scholar] [CrossRef] [PubMed]
- Bobadilla Fazzini, R.A.; Cortés, M.Z.; Padilla, L.; Maturana, D.; Budinich, M.; Maass, A.; Parada, P. Stoichiometric modeling of oxidation of reduced inorganic sulfur compounds (Riscs) in Acidithiobacillus thiooxidans. Biotechnol. Bioeng. 2013, 110, 2242–2251. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Huaqun, Y.; Yili, L.; Qiu, G.; Liu, X. Theoretical model of the structure and the reaction mechanisms of sulfur oxygenase reductase in Acidithiobacillus thiooxidans. Adv. Mater. Res. 2015, 1130, 67–70. [Google Scholar] [CrossRef]
- Krulwich, T.A.; Sachs, G.; Padan, E. Molecular aspects of bacterial pH sensing and homeostasis. Nat. Rev. Microbiol. 2011, 9, 330–343. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Fang, Y.; Li, Q.; Yin, H.; Qin, W.; Liang, Y.; Li, Q.; Li, N.; Liu, X.; Qiu, G.; et al. Global transcriptional analysis of stress-response strategies in Acidithiobacillus ferrooxidans ATCC 23270 exposed to organic extractant-Lix984n. World J. Microbiol. Biotechnol. 2012, 28, 1045–1055. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Qiu, G.; Ge, Y.; Xu, J. Extraction of copper from bacterial leach solution using Lix984. Trans. Nonferrous Met. Soc. China 2002, 12, 313–316. [Google Scholar]
- Acuña, L.G.; Cárdenas, J.P.; Covarrubias, P.C.; Haristoy, J.J.; Flores, R.; Nuñez, H.; Riadi, G.; Shmaryahu, A.; Valdés, J.; Dopson, M.; et al. Architecture and gene repertoire of the flexible genome of the extreme acidophile Acidithiobacillus caldus. PLoS ONE 2013, 8, e78237. [Google Scholar]
- Mira, A.; Ochman, H.; Moran, N.A. Deletional bias and the evolution of bacterial genomes. Trends Genet. 2001, 17, 589–596. [Google Scholar] [CrossRef]
- Wu, X.; Monchy, S.; Taghavi, S.; Zhu, W.; Ramos, J.; van der Lelie, D. Comparative genomics and functional analysis of niche-specific adaptation in Pseudomonas putida. FEMS Microbiol. Rev. 2011, 35, 299–323. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Zhao, L.; Gao, J.; Fei, Z. iAssembler: A package for de novo assembly of Roche-454/Sanger transcriptome sequences. BMC Bioinform. 2011, 12, 453. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, H.; Taniguchi, T.; Itoh, T. MetaGeneAnnotator: Detecting species-specific patterns of ribosomal binding site for precise gene prediction in anonymous prokaryotic and phage genomes. DNA Res. 2008, 15, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Punta, M.; Coggill, P.C.; Lestrade, L.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; et al. The Pfam protein families database. Nucleic Acids Res. 2012, 40, D290–D301. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 34, D32–D36. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, B.K.; Laird, M.R.; Shay, J.A.; Winsor, G.L.; Lo, R.; Nizam, F.; Pereira, S.K.; Waglechner, N.; McArthur, A.G.; Langille, M.G.; et al. IslandViewer 3: More flexible, interactive genomic island discovery, visualization and analysis. Nucleic Acids Res. 2015, 43, W104–W108. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Fouts, D.E.; Brinkac, L.; Beck, E.; Inman, J.; Sutton, G. PanOCT: Automated clustering of orthologs using conserved gene neighborhood for pan-genomic analysis of bacterial strains and closely related species. Nucleic Acids Res. 2012, 40, e172. [Google Scholar] [CrossRef] [PubMed]
- Zuo, G.; Hao, B. CVTree3 web server for whole-genome-based and alignment-free prokaryotic phylogeny and taxonomy. Genom. Proteom. Bioinform. 2015, 13, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Geographic Origin | Reference |
|---|---|---|
| Licanantay | Copper mine, Atacama, Chile | [17] |
| ATCC 19377 | Kimmeridge clay, UK | [15] |
| GD1-3 | Copper Mine, Shaoguan, Guangdong, China | This study |
| DXS-W | Copper Mine, Dongxiang Mountain, Hami, Xinjiang, China | This study |
| A02 | Coal heap drainage, Pingxiang, Jiangxi, China | This study |
| A01 | Coal dump, Pingxiang, Jiangxi, China | [16] |
| BY-02 | Copper Mine, Baiyin, Gansu, China | This study |
| DMC | Coal heap drainage, Chenzhou, Hunan, China | This study |
| TYC-17 | Copper Mine, Baiyin, Gansu, China | This study |
| A. The Putative Insertion Sequences | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| IS Family | DXS-W | Licanantay | A01 | ATCC 19377 | GD1-3 | DMC | A02 | BY-02 | TYC-17 |
| IS110 | 9 | 11 | 9 | 4 | 8 | 9 | 9 | 9 | 10 |
| IS1182 | 2 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 |
| IS1380 | 3 | 2 | 0 | 0 | 3 | 0 | 0 | 0 | 0 |
| IS1595 | 5 | 8 | 8 | 4 | 5 | 9 | 7 | 9 | 8 |
| IS1634 | 2 | 1 | 3 | 9 | 1 | 3 | 3 | 3 | 3 |
| IS200/IS605 | 6 | 15 | 4 | 3 | 5 | 4 | 2 | 4 | 4 |
| IS21 | 6 | 8 | 11 | 2 | 6 | 8 | 9 | 9 | 9 |
| IS256 | 1 | 8 | 2 | 15 | 1 | 3 | 3 | 2 | 2 |
| IS3 | 11 | 12 | 4 | 1 | 10 | 6 | 5 | 5 | 5 |
| IS30 | 1 | 2 | 0 | 3 | 1 | 2 | 2 | 2 | 2 |
| IS4 | 7 | 6 | 3 | 12 | 7 | 9 | 7 | 7 | 7 |
| IS481 | 8 | 1 | 2 | 1 | 8 | 2 | 2 | 1 | 2 |
| IS5 | 4 | 4 | 5 | 5 | 4 | 6 | 5 | 5 | 6 |
| IS51 | 0 | 2 | 0 | 2 | 0 | 0 | 0 | 0 | 0 |
| IS605 | 1 | 0 | 1 | 0 | 1 | 1 | 1 | 1 | 1 |
| IS607 | 4 | 4 | 1 | 0 | 4 | 4 | 1 | 4 | 1 |
| IS630 | 11 | 18 | 6 | 9 | 12 | 14 | 12 | 11 | 9 |
| IS66 | 11 | 0 | 1 | 0 | 9 | 2 | 1 | 1 | 1 |
| IS91 | 20 | 16 | 16 | 8 | 19 | 15 | 16 | 16 | 15 |
| ISAs1 | 1 | 2 | 1 | 0 | 1 | 1 | 0 | 1 | 0 |
| ISAzo13 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| ISKra4 | 7 | 2 | 4 | 2 | 7 | 1 | 4 | 1 | 4 |
| ISL3 | 43 | 44 | 39 | 30 | 47 | 41 | 41 | 40 | 42 |
| ISNCY | 2 | 1 | 1 | 0 | 2 | 1 | 2 | 2 | 2 |
| Tn3 | 21 | 16 | 17 | 16 | 20 | 19 | 19 | 20 | 21 |
| Total | 187 | 184 | 138 | 126 | 184 | 160 | 151 | 153 | 154 |
| B. The Predicted Genomic Islands | |||||||||
| Strain | DXS-W | Licanantay | A01 | ATCC 19377 | GD1-3 | DMC | A02 | BY-02 | TYC-17 |
| GI Number | 65 | 54 | 39 | 36 | 56 | 53 | 51 | 50 | 44 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Feng, X.; Tao, J.; Ma, L.; Xiao, Y.; Liang, Y.; Liu, X.; Yin, H. Comparative Genomics of the Extreme Acidophile Acidithiobacillus thiooxidans Reveals Intraspecific Divergence and Niche Adaptation. Int. J. Mol. Sci. 2016, 17, 1355. https://doi.org/10.3390/ijms17081355
Zhang X, Feng X, Tao J, Ma L, Xiao Y, Liang Y, Liu X, Yin H. Comparative Genomics of the Extreme Acidophile Acidithiobacillus thiooxidans Reveals Intraspecific Divergence and Niche Adaptation. International Journal of Molecular Sciences. 2016; 17(8):1355. https://doi.org/10.3390/ijms17081355
Chicago/Turabian StyleZhang, Xian, Xue Feng, Jiemeng Tao, Liyuan Ma, Yunhua Xiao, Yili Liang, Xueduan Liu, and Huaqun Yin. 2016. "Comparative Genomics of the Extreme Acidophile Acidithiobacillus thiooxidans Reveals Intraspecific Divergence and Niche Adaptation" International Journal of Molecular Sciences 17, no. 8: 1355. https://doi.org/10.3390/ijms17081355
APA StyleZhang, X., Feng, X., Tao, J., Ma, L., Xiao, Y., Liang, Y., Liu, X., & Yin, H. (2016). Comparative Genomics of the Extreme Acidophile Acidithiobacillus thiooxidans Reveals Intraspecific Divergence and Niche Adaptation. International Journal of Molecular Sciences, 17(8), 1355. https://doi.org/10.3390/ijms17081355