
How Many Conformations of Enzymes Should Be Sampled for DFT/MM Calculations? A Case Study of Fluoroacetate Dehalogenase

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
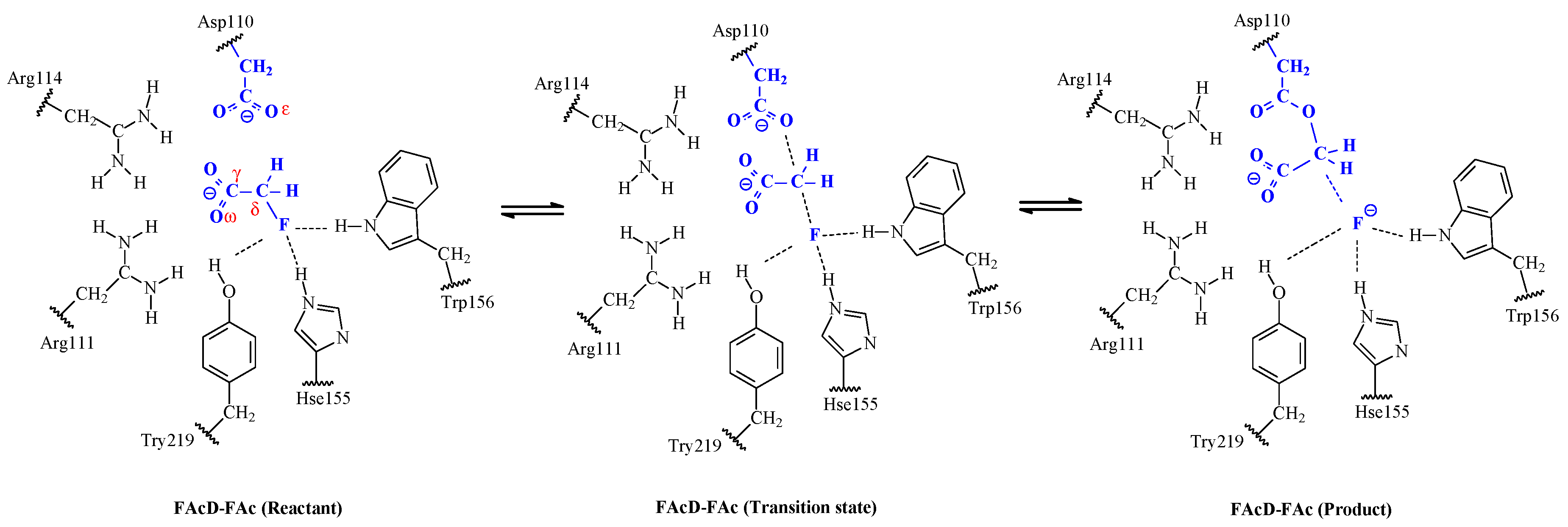
2. Results and Discussion
2.1. Convergence Criteria
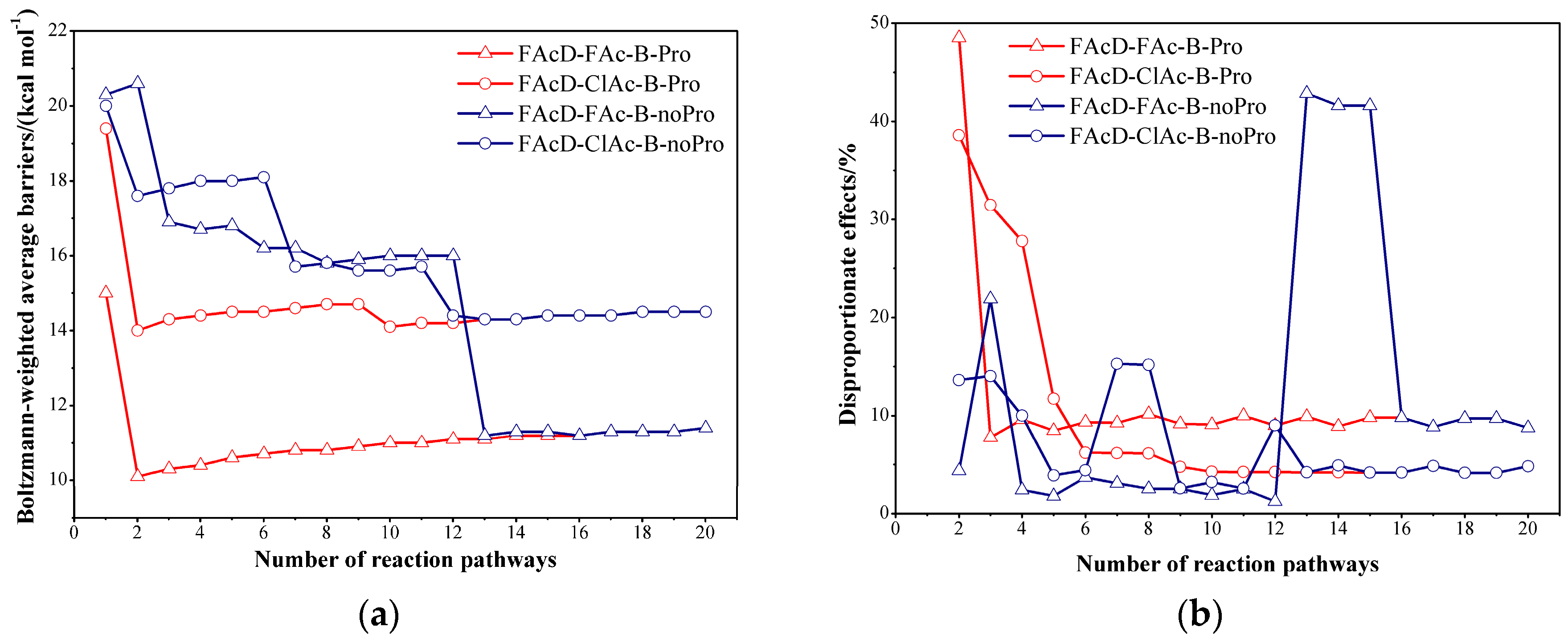
2.2. Small QM Region versus Big QM Region
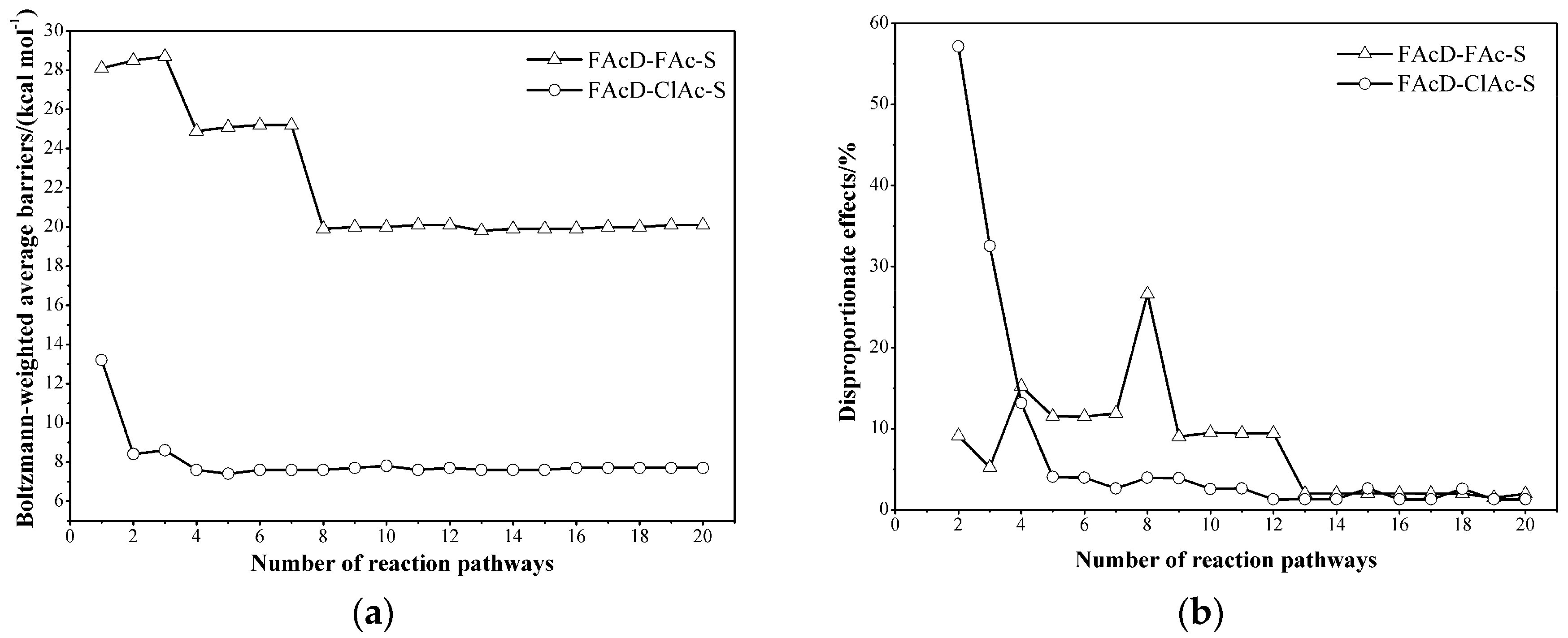
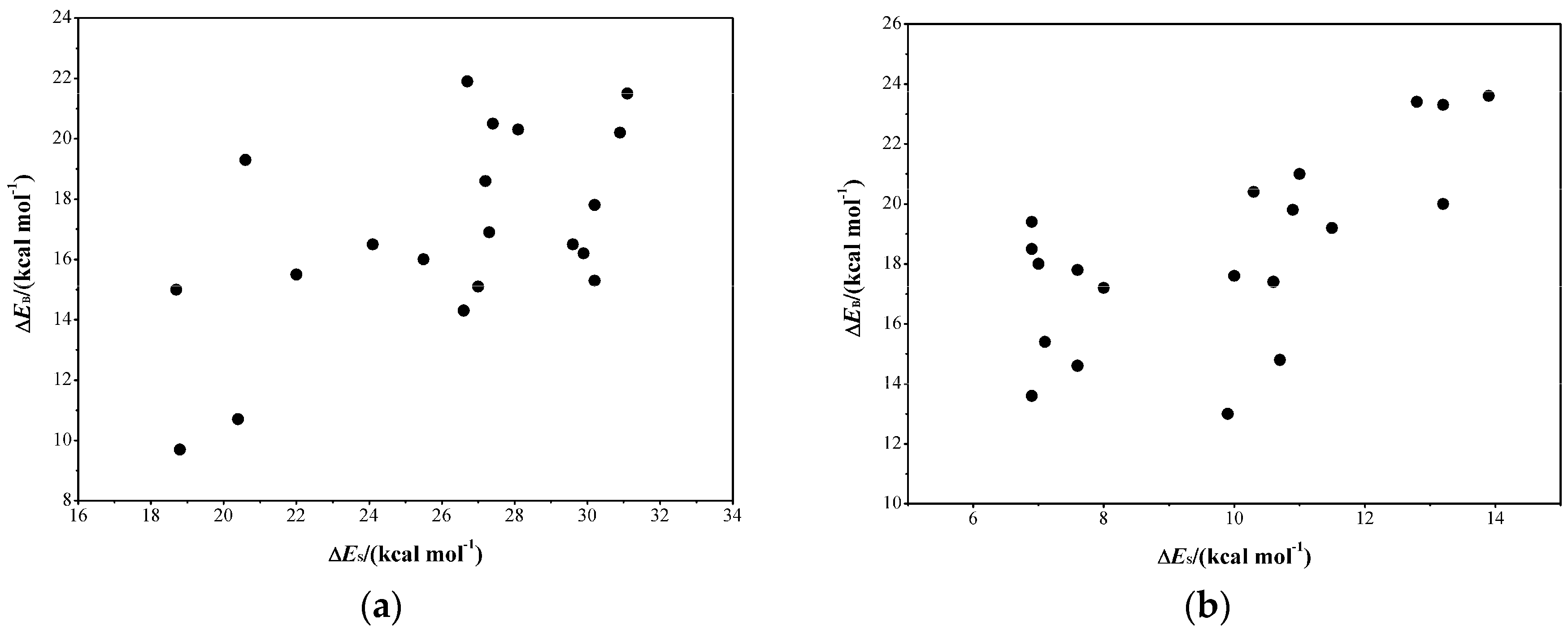
2.3. Determine the Minimum Number of Conformations to Be Sampled
3. Materials and Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lonsdale, R.; Ranaghan, K.E.; Mulholland, A.J. Computational enzymology. Chem. Commun. 2010, 46, 2354–2372. [Google Scholar] [CrossRef] [PubMed]
- Brunk, E.; Rothlisberger, U. Mixed quantum mechanical/molecular mechanical molecular dynamics simulations of biological systems in ground and electronically excited states. Chem. Rev. 2015, 115, 6217–6263. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Althoff, E.A.; Clemente, F.R.; Doyle, L.; Rothlisberger, D.; Zanghellini, A.; Gallaher, J.L.; Betker, J.L.; Tanaka, F.; Barbas, C.F., III; et al. De novo computational design of retro-aldol enzymes. Science 2008, 319, 1387–1391. [Google Scholar] [PubMed]
- Martí, S.; Andrés, J.; Moliner, V.; Silla, E.; Tuñón, I.; Bertrán, J. Computational design of biological catalysts. Chem. Soc. Rev. 2008, 37, 2634–2643. [Google Scholar] [CrossRef] [PubMed]
- Van der Kamp, M.W.; Mulholland, A.J. Combined quantum mechanics/molecular mechanics (QM/MM) methods in computational enzymology. Biochemistry 2013, 52, 2708–2728. [Google Scholar] [CrossRef] [PubMed]
- Warshel, A.; Levitt, M. Theoretical studies of enzymic reactions: Dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Biol. 1976, 103, 227–249. [Google Scholar] [CrossRef]
- Kästner, J. Umbrella sampling. WIREs Comput. Mol. Sci. 2011, 1, 932–942. [Google Scholar] [CrossRef]
- Acevedo, O.; Jorgensen, W.L. Advances in quantum and molecular mechanical (QM/MM) simulations for organic and enzymatic reactions. Acc. Chem. Res. 2010, 43, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Doubleday, C.; Houk, K.N. Molecular dynamics of dimethyldioxirane C–H oxidation. J. Chem. Theory Comput. 2015, 11, 5060–5612. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, R.; Harvey, J.N.; Mulholland, A.J. A practical guide to modelling enzyme-catalysed reactions. Chem. Soc. Rev. 2012, 41, 3025–3038. [Google Scholar] [CrossRef] [PubMed]
- Senn, H.M.; Thiel, W. QM/MM methods for biomolecular systems. Angew. Chem. Int. Ed. 2009, 48, 1198–1229. [Google Scholar] [CrossRef] [PubMed]
- Wallrapp, F.H.; Guallar, V. Mixed quantum mechanics and molecular mechanics methods: Looking inside proteins. WIREs Comput. Mol. Sci. 2011, 1, 315–322. [Google Scholar] [CrossRef]
- Oláh, J.; Mulholland, A.J.; Harvey, J.N. Understanding the determinants of selectivity in drug metabolism through modeling of dextromethorphan oxidation by cytochrome P450. Proc. Natl. Acad. Sci. USA 2011, 108, 6050–6055. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, R.; Du, L.; Zhang, Q.; Wang, W. Insight into the catalytic mechanism of meta-cleavage product hydrolase BphD: A quantum mechanics/molecular mechanics study. RSC Adv. 2015, 5, 66591–66597. [Google Scholar] [CrossRef]
- Lonsdale, R.; Reetz, M.T. Reduction of α,β-unsaturated ketones by old yellow enzymes: Mechanistic insights from quantum mechanics/molecular mechanics calculations. J. Am. Chem. Soc. 2015, 137, 14733–14742. [Google Scholar] [CrossRef] [PubMed]
- Abad, E.; Zenn, R.K.; Kästner, J. Reaction mechanism of monoamine oxidase from QM/MM calculations. J. Phys. Chem. B 2013, 117, 14238–14246. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Martinez, M.; Marcos, E.; Tauler, R.; Field, M.; Crehuet, R. Conformational compression and barrier height heterogeneity in the N-acetylglutamate kinase. J. Phys. Chem. B 2013, 117, 14261–14272. [Google Scholar] [CrossRef] [PubMed]
- Rommel, J.B.; Kästner, J. The fragmentation-recombination mechanism of the enzyme glutamate mutase studied by QM/MM simulations. J. Am. Chem. Soc. 2011, 133, 10195–10203. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, R.; Houghton, K.T.; Żurek, J.; Bathelt, C.M.; Foloppe, N.; de Groot, M.J.; Harvey, J.N.; Mulholland, A.J. Quantum mechanics/molecular mechanics modeling of regioselectivity of drug metabolism in cytochrome P450 2C9. J. Am. Chem. Soc. 2013, 135, 8001–8015. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Shi, X.; Zhang, Q.; Hu, J.; Chen, J.; Wang, W. Computational evidence for the detoxifying mechanism of epsilon class glutathione transferase toward the insecticide DDT. Environ. Sci. Technol. 2014, 48, 5008–5016. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, R.; Harvey, J.N.; Mulholland, A.J. Compound I reactivity defines alkene oxidation selectivity in cytochrome P450cam. J. Phys. Chem. B 2010, 114, 1156–1162. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.M.; Kästner, J. Averaging techniques for reaction barriers in QM/MM simulations. ChemPhysChem 2014, 15, 3264–3269. [Google Scholar] [CrossRef] [PubMed]
- Logunov, I.; Schulten, K. Quantum chemistry: Molecular dynamics study of the dark-adaptation process in bacteriorhodopsin. J. Am. Chem. Soc. 1996, 118, 9727–9735. [Google Scholar] [CrossRef]
- Shaik, S.; Cohen, S.; Wang, Y.; Chen, H.; Kumar, D.; Thiel, W. P450 enzymes: Their structure, reactivity, and selectivity modeled by QM/MM calculations. Chem. Rev. 2010, 110, 949–1017. [Google Scholar] [CrossRef] [PubMed]
- Sokkar, P.; Boulanger, E.; Thiel, W.; Sanchez-Garcia, E. Hybrid quantum mechanics/molecular mechanics/coarse grained modeling: A triple-resolution approach for biomolecular systems. J. Chem. Theory. Comput. 2015, 11, 1809–1818. [Google Scholar] [CrossRef] [PubMed]
- Saura, P.; Suardíaz, R.; Masgrau, L.; Lluch, J.M.; González-Lafont, À. Unraveling how enzymes can use bulky residues to drive site-selective C–H activation: The case of mammalian lipoxygenases catalyzing arachidonic acid oxidation. ACS Catal. 2014, 4, 4351–4363. [Google Scholar] [CrossRef]
- Ribeiro, A.J.M.; Santos-Martins, D.; Russo, N.; Ramos, M.J.; Fernandes, P.A. Enzymatic flexibility and reaction rate: A QM/MM study of HIV-1 protease. ACS Catal. 2015, 5, 5617–5626. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, R.; Du, L.; Zhang, Q.; Wang, W. Catalytic mechanism of C–F bond cleavage: Insights from QM/MM analysis of fluoroacetate dehalogenase. Catal. Sci. Technol. 2016, 6, 73–80. [Google Scholar] [CrossRef]
- Muller, K.; Faeh, C.; Diederich, F. Fluorine in pharmaceuticals: Looking beyond intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- Douvris, C.; Ozerov, O.V. Hydrodefluorination of perfluoroalkyl groups using silylium-carborane catalysts. Science 2008, 321, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.W.Y.; Yakunin, A.F.; Edwards, E.A.; Pai, E.F. Mapping the reaction coordinates of enzymatic defluorination. J. Am. Chem. Soc. 2011, 133, 7461–7468. [Google Scholar] [CrossRef] [PubMed]
- Liao, R.Z.; Thiel, W. Comparison of QM-only and QM/MM models for the mechanism of tungsten-dependent acetylene hydratase. J. Chem. Theory. Comput. 2012, 8, 3793–3803. [Google Scholar] [CrossRef] [PubMed]
- Sumner, S.; Söderhjelm, P.; Ryde, U. Effect of geometry optimizations on QM-cluster and QM/MM studies of reaction energies in proteins. J. Chem. Theory Comput. 2013, 9, 4205–4214. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, P.; Vries, A.H.D.; Guest, M.F.; Schreckenbach, G.; Catlow, C.R.A.; French, S.A.; Sokol, A.A.; Bromley, S.T.; Thiel, W.; Turner, A.J.; et al. QUASI: A general purpose implementation of the QM/MM approach and its application to problems in catalysis. J. Mol. Struct. THEOCHEM 2003, 632, 1–28. [Google Scholar] [CrossRef]
- Metz, S.; Kästner, J.; Sokol, A.A.; Keal, T.W.; Sherwood, P. ChemShell-a modular software package for QM/MM simulations. WIREs Comput. Mol. Sci. 2014, 4, 101–110. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kölmel, C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Smith, W.; Forester, T.R. DL_POLY_2.0: A general-purpose parallel molecular dynamics simulation package. J. Mol. Graph. 1996, 14, 136–141. [Google Scholar] [CrossRef]
- Billeter, S.R.; Turner, A.J.; Thiel, W. Linear scaling geometry optimisation and transition state search in hybrid delocalised internal coordinates. Phys. Chem. Chem. Phys. 2000, 2, 2177–2186. [Google Scholar] [CrossRef]




© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Zhang, R.; Du, L.; Zhang, Q.; Wang, W. How Many Conformations of Enzymes Should Be Sampled for DFT/MM Calculations? A Case Study of Fluoroacetate Dehalogenase. Int. J. Mol. Sci. 2016, 17, 1372. https://doi.org/10.3390/ijms17081372
Li Y, Zhang R, Du L, Zhang Q, Wang W. How Many Conformations of Enzymes Should Be Sampled for DFT/MM Calculations? A Case Study of Fluoroacetate Dehalogenase. International Journal of Molecular Sciences. 2016; 17(8):1372. https://doi.org/10.3390/ijms17081372
Chicago/Turabian StyleLi, Yanwei, Ruiming Zhang, Likai Du, Qingzhu Zhang, and Wenxing Wang. 2016. "How Many Conformations of Enzymes Should Be Sampled for DFT/MM Calculations? A Case Study of Fluoroacetate Dehalogenase" International Journal of Molecular Sciences 17, no. 8: 1372. https://doi.org/10.3390/ijms17081372
APA StyleLi, Y., Zhang, R., Du, L., Zhang, Q., & Wang, W. (2016). How Many Conformations of Enzymes Should Be Sampled for DFT/MM Calculations? A Case Study of Fluoroacetate Dehalogenase. International Journal of Molecular Sciences, 17(8), 1372. https://doi.org/10.3390/ijms17081372