
Development of Novel Immunotherapies for Multiple Myeloma

 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Current Treatment Options for Multiple Myeloma (MM)
3. Monoclonal Antibody (mAb) Therapy
4. Adoptive Cell Therapy
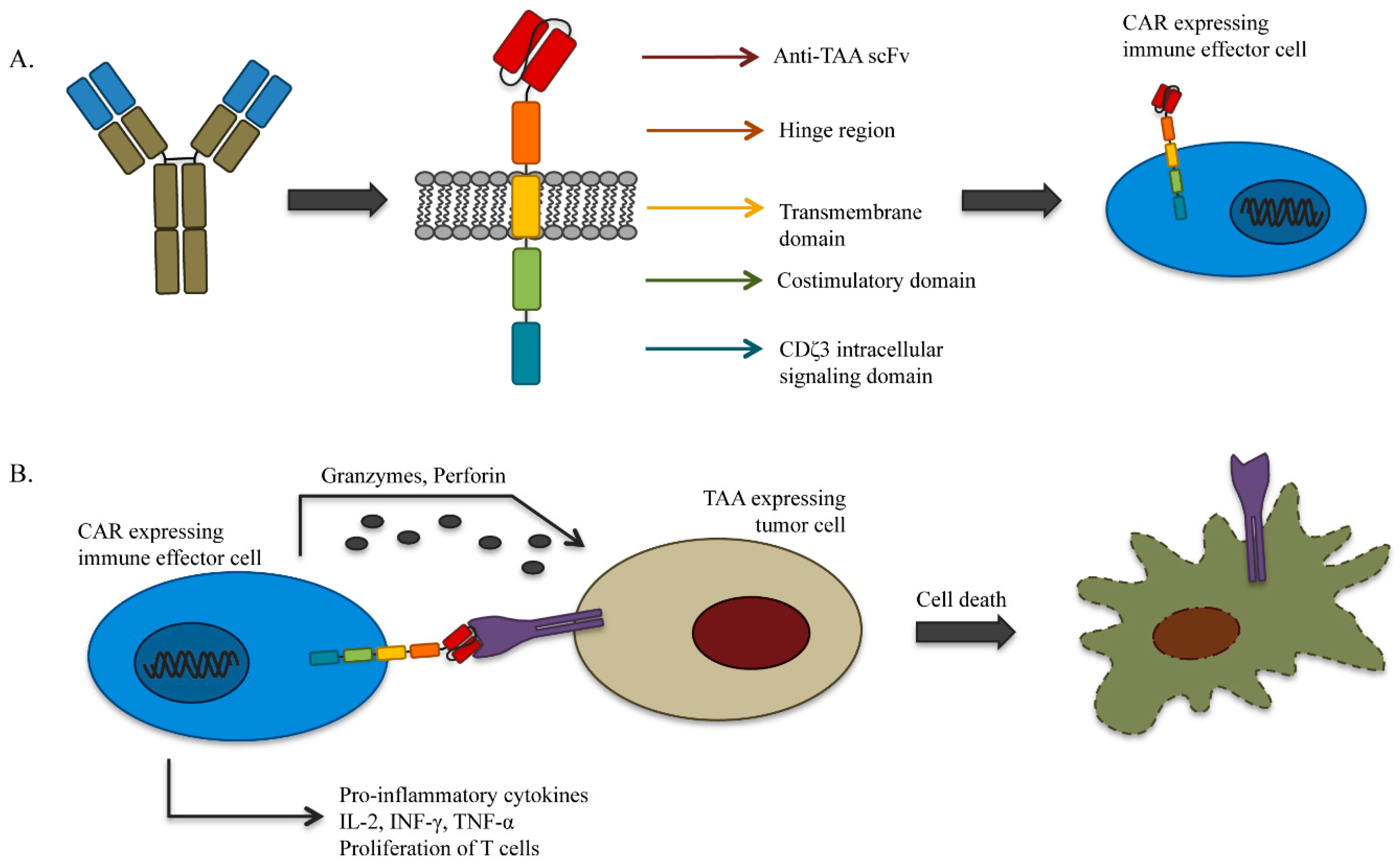
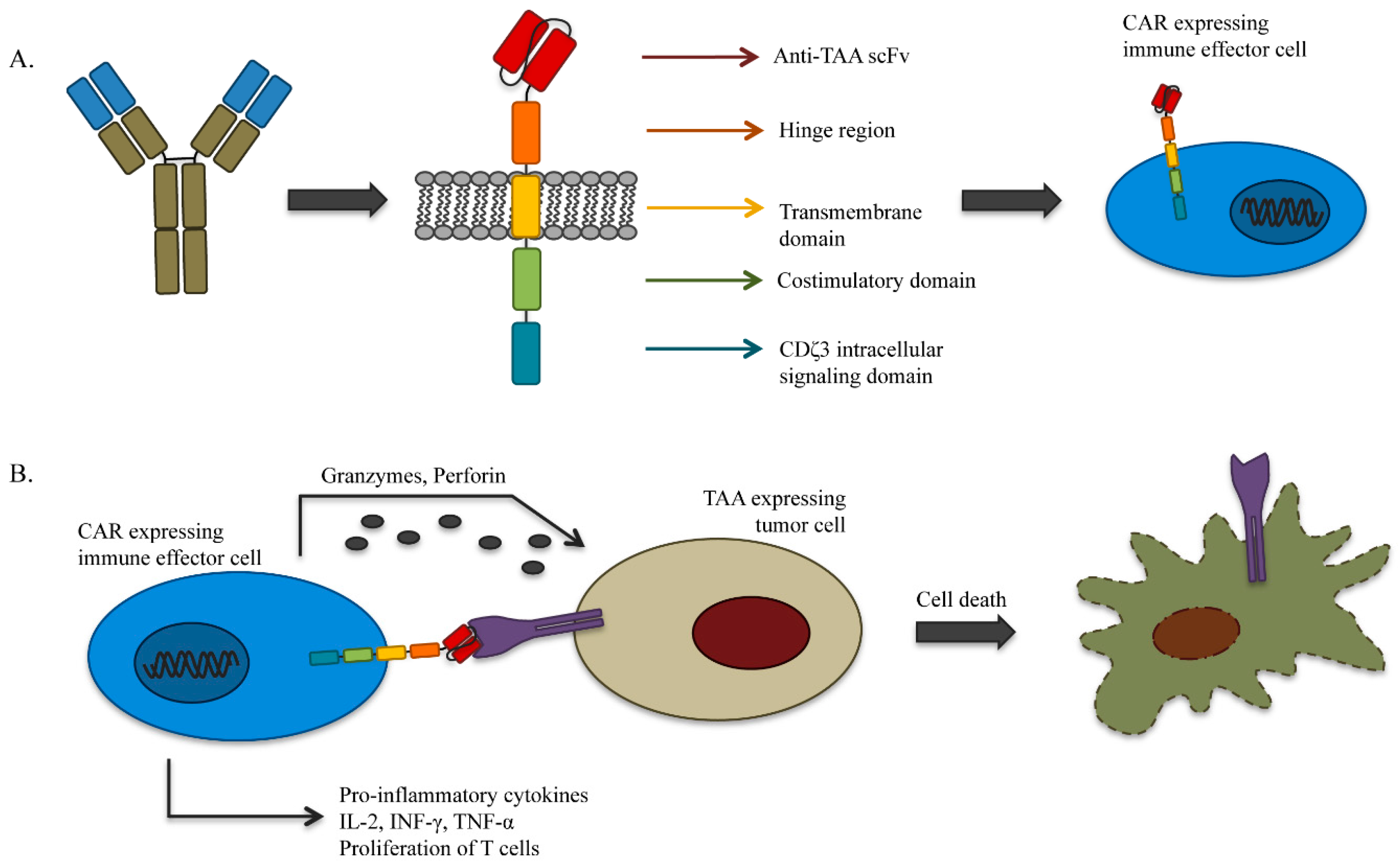
4.1. Genetically-Redirected Immune Cells
4.2. Dendritic Cell (DC)-Based Vaccines
5. Targets for Immunotherapy in MM
5.1. Targets on Myeloma Cells
5.1.1. SLAMF7, (CS1, CD319)
5.1.2. CD38
5.1.3. CD40
5.1.4. CD138 (Syndecan-1)
5.1.5. CD56 (NCAM1, Leu-19)
5.1.6. CD74
5.1.7. CD200 (MOX1, MRC, OX-2)
5.1.8. CD19
5.1.9. BCMA (TNFRSF17, CD269)
5.1.10. Cancer Testis Antigens (GAGE Family, LAGE, MAGE Family, NY-ESO-1, SSX Genes, etc.)
5.1.11. GRP78
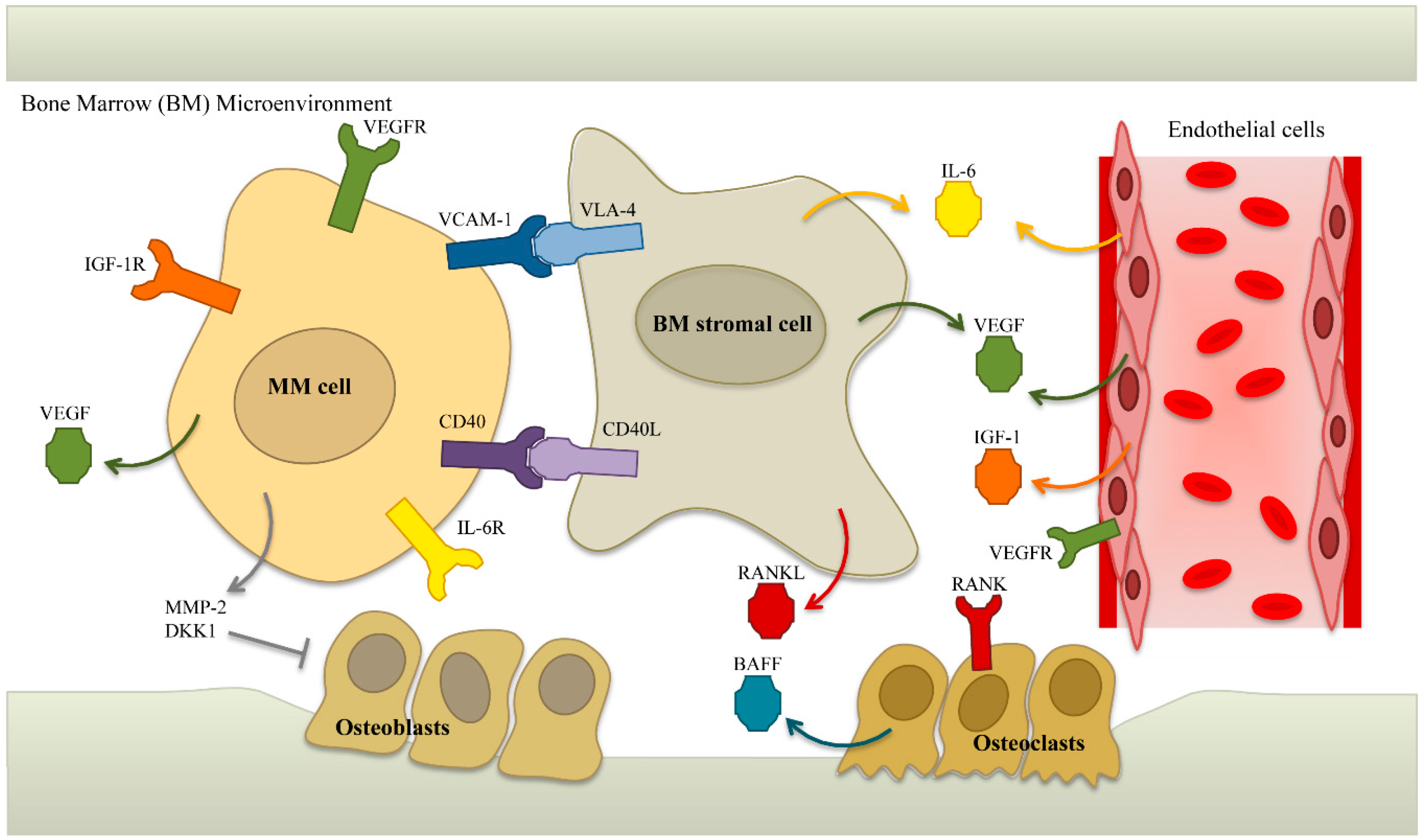
5.2. Targets in the Microenvironment
5.2.1. IL-6
5.2.2. PD-1/PD-L1
5.2.3. KIR
5.2.4. VEGF
5.2.5. BAFF/APRIL
6. Summary and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Palumbo, A.; Anderson, K. Multiple myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef] [PubMed]
- NCI. Surveillance, Epidemiology and End Results (Seer Database). Available online: http://seer.cancer.gov/statfacts/html/mulmy.html (accessed on 25 May 2016).
- Raab, M.S.; Podar, K.; Breitkreutz, I.; Richardson, P.G.; Anderson, K.C. Multiple myeloma. Lancet 2009, 374, 324–339. [Google Scholar] [CrossRef]
- Kuehl, W.M.; Bergsagel, P.L. Multiple myeloma: Evolving genetic events and host interactions. Nat. Rev. Cancer 2002, 2, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Avet-Loiseau, H.; Attal, M.; Moreau, P.; Charbonnel, C.; Garban, F.; Hulin, C.; Leyvraz, S.; Michallet, M.; Yakoub-Agha, I.; Garderet, L.; et al. Genetic abnormalities and survival in multiple myeloma: The experience of the intergroupe francophone du myelome. Blood 2007, 109, 3489–3495. [Google Scholar] [CrossRef] [PubMed]
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, R.; Blood, E.A.; Oken, M.M.; Kyle, R.A.; Dewald, G.W.; Bailey, R.J.; van Wier, S.A.; Henderson, K.J.; Hoyer, J.D.; Harrington, D.; et al. Myeloma and the t(11;14)(q13;q32); evidence for a biologically defined unique subset of patients. Blood 2002, 99, 3735–3741. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Facon, T.; Leleu, X.; Morineau, N.; Huyghe, P.; Harousseau, J.L.; Bataille, R.; Avet-Loiseau, H.; Intergroupe Francophone du, M. Recurrent 14q32 translocations determine the prognosis of multiple myeloma, especially in patients receiving intensive chemotherapy. Blood 2002, 100, 1579–1583. [Google Scholar] [CrossRef] [PubMed]
- Zhan, F.; Hardin, J.; Kordsmeier, B.; Bumm, K.; Zheng, M.; Tian, E.; Sanderson, R.; Yang, Y.; Wilson, C.; Zangari, M.; et al. Global gene expression profiling of multiple myeloma, monoclonal gammopathy of undetermined significance, and normal bone marrow plasma cells. Blood 2002, 99, 1745–1757. [Google Scholar] [CrossRef] [PubMed]
- Shou, Y.; Martelli, M.L.; Gabrea, A.; Qi, Y.; Brents, L.A.; Roschke, A.; Dewald, G.; Kirsch, I.R.; Bergsagel, P.L.; Kuehl, W.M. Diverse karyotypic abnormalities of the C-MYC locus associated with C-MYC dysregulation and tumor progression in multiple myeloma. Proc. Natl. Acad. Sci. USA 2000, 97, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Bezieau, S.; Devilder, M.C.; Avet-Loiseau, H.; Mellerin, M.P.; Puthier, D.; Pennarun, E.; Rapp, M.J.; Harousseau, J.L.; Moisan, J.P.; Bataille, R. High incidence of N and K-ras activating mutations in multiple myeloma and primary plasma cell leukemia at diagnosis. Hum. Mutat. 2001, 18, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Corradini, P.; Ladetto, M.; Voena, C.; Palumbo, A.; Inghirami, G.; Knowles, D.M.; Boccadoro, M.; Pileri, A. Mutational activation of N- and K-ras oncogenes in plasma cell dyscrasias. Blood 1993, 81, 2708–2713. [Google Scholar] [PubMed]
- Xiong, W.; Wu, X.; Starnes, S.; Johnson, S.K.; Haessler, J.; Wang, S.; Chen, L.; Barlogie, B.; Shaughnessy, J.D., Jr.; Zhan, F. An analysis of the clinical and biologic significance of TP53 loss and the identification of potential novel transcriptional targets of TP53 in multiple myeloma. Blood 2008, 112, 4235–4246. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, M.S.; Daggett, J.L.; Bender, T.P.; Kuehl, W.M.; Bergsagel, P.L.; Williams, M.E. Frequent inactivation of the cyclin-dependent kinase inhibitor p18 by homozygous deletion in multiple myeloma cell lines: Ectopic p18 expression inhibits growth and induces apoptosis. Leukemia 2002, 16, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Shivapurkar, N.; Reddy, J.; Shigematsu, H.; Miyajima, K.; Suzuki, M.; Toyooka, S.; Zochbauer-Muller, S.; Drach, J.; Parikh, G.; et al. DNA methylation profiles of lymphoid and hematopoietic malignancies. Clin. Cancer Res. 2004, 10, 2928–2935. [Google Scholar] [CrossRef] [PubMed]
- Roccaro, A.M.; Sacco, A.; Thompson, B.; Leleu, X.; Azab, A.K.; Azab, F.; Runnels, J.; Jia, X.; Ngo, H.T.; Melhem, M.R.; et al. MicroRNAs 15a and 16 regulate tumor proliferation in multiple myeloma. Blood 2009, 113, 6669–6680. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Mitsiades, C.; Tonon, G.; Richardson, P.G.; Anderson, K.C. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat. Rev. Cancer 2007, 7, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Urashima, M.; Chen, B.P.; Chen, S.; Pinkus, G.S.; Bronson, R.T.; Dedera, D.A.; Hoshi, Y.; Teoh, G.; Ogata, A.; Treon, S.P.; et al. The development of a model for the homing of multiple myeloma cells to human bone marrow. Blood 1997, 90, 754–765. [Google Scholar] [PubMed]
- Bellamy, W.T. Expression of vascular endothelial growth factor and its receptors in multiple myeloma and other hematopoietic malignancies. Semin. Oncol. 2001, 28, 551–559. [Google Scholar] [CrossRef]
- Bellamy, W.T.; Richter, L.; Frutiger, Y.; Grogan, T.M. Expression of vascular endothelial growth factor and its receptors in hematopoietic malignancies. Cancer Res. 1999, 59, 728–733. [Google Scholar] [PubMed]
- Chauhan, D.; Uchiyama, H.; Akbarali, Y.; Urashima, M.; Yamamoto, K.; Libermann, T.A.; Anderson, K.C. Multiple myeloma cell adhesion-induced interleukin-6 expression in bone marrow stromal cells involves activation of NF-kappa B. Blood 1996, 87, 1104–1112. [Google Scholar] [PubMed]
- Ge, N.L.; Rudikoff, S. Insulin-like growth factor I is a dual effector of multiple myeloma cell growth. Blood 2000, 96, 2856–2861. [Google Scholar] [PubMed]
- Lokhorst, H.M.; Lamme, T.; de Smet, M.; Klein, S.; de Weger, R.A.; van Oers, R.; Bloem, A.C. Primary tumor cells of myeloma patients induce interleukin-6 secretion in long-term bone marrow cultures. Blood 1994, 84, 2269–2277. [Google Scholar] [PubMed]
- Kline, M.; Donovan, K.; Wellik, L.; Lust, C.; Jin, W.; Moon-Tasson, L.; Xiong, Y.; Witzig, T.E.; Kumar, S.; Rajkumar, S.V.; et al. Cytokine and chemokine profiles in multiple myeloma; significance of stromal interaction and correlation of IL-8 production with disease progression. Leuk. Res. 2007, 31, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Moschetta, M.; Manier, S.; Glavey, S.; Gorgun, G.T.; Roccaro, A.M.; Anderson, K.C.; Ghobrial, I.M. Targeting the bone marrow microenvironment in multiple myeloma. Immunol. Rev. 2015, 263, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Vacca, A.; Ribatti, D.; Presta, M.; Minischetti, M.; Iurlaro, M.; Ria, R.; Albini, A.; Bussolino, F.; Dammacco, F. Bone marrow neovascularization, plasma cell angiogenic potential, and matrix metalloproteinase-2 secretion parallel progression of human multiple myeloma. Blood 1999, 93, 3064–3073. [Google Scholar] [PubMed]
- Dosani, T.; Carlsten, M.; Maric, I.; Landgren, O. The cellular immune system in myelomagenesis: NK cells and t cells in the development of myeloma [corrected] and their uses in immunotherapies. Blood Cancer J. 2015, 5, e306. [Google Scholar] [CrossRef] [PubMed]
- Dhodapkar, M.V.; Krasovsky, J.; Osman, K.; Geller, M.D. Vigorous premalignancy-specific effector T cell response in the bone marrow of patients with monoclonal gammopathy. J. Exp. Med. 2003, 198, 1753–1757. [Google Scholar] [CrossRef] [PubMed]
- Schutt, P.; Brandhorst, D.; Stellberg, W.; Poser, M.; Ebeling, P.; Muller, S.; Buttkereit, U.; Opalka, B.; Lindemann, M.; Grosse-Wilde, H.; et al. Immune parameters in multiple myeloma patients: Influence of treatment and correlation with opportunistic infections. Leuk. Lymphoma 2006, 47, 1570–1582. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.D.; Pope, B.; Murray, A.; Esdale, W.; Sze, D.M.; Gibson, J.; Ho, P.J.; Hart, D.; Joshua, D. Dendritic cells from patients with myeloma are numerically normal but functionally defective as they fail to up-regulate CD80 (B7–1) expression after HUCD40LT stimulation because of inhibition by transforming growth factor-beta1 and interleukin-10. Blood 2001, 98, 2992–2998. [Google Scholar] [CrossRef] [PubMed]
- Commes, T.; Klein, B.; Jourdan, M.; Clofent, G.; Houssiau, F.; Grenier, J.; Bataille, R. The defect in peripheral blood b-cell activation in patients with multiple myeloma is not due to a deficiency in the production of B-cell growth and differentiation factors. J. Clin. Immunol. 1989, 9, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Ratta, M.; Fagnoni, F.; Curti, A.; Vescovini, R.; Sansoni, P.; Oliviero, B.; Fogli, M.; Ferri, E.; Della Cuna, G.R.; Tura, S.; et al. Dendritic cells are functionally defective in multiple myeloma: The role of interleukin-6. Blood 2002, 100, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Attal, M.; Facon, T. Frontline therapy of multiple myeloma. Blood 2015, 125, 3076–3084. [Google Scholar] [CrossRef] [PubMed]
- Adams, J. The proteasome: A suitable antineoplastic target. Nat. Rev. Cancer 2004, 4, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, N.; Mitsiades, C.S.; Poulaki, V.; Chauhan, D.; Fanourakis, G.; Gu, X.; Bailey, C.; Joseph, M.; Libermann, T.A.; Treon, S.P.; et al. Molecular sequelae of proteasome inhibition in human multiple myeloma cells. Proc. Natl. Acad. Sci. USA 2002, 99, 14374–14379. [Google Scholar] [CrossRef] [PubMed]
- Quach, H.; Ritchie, D.; Stewart, A.K.; Neeson, P.; Harrison, S.; Smyth, M.J.; Prince, H.M. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia 2010, 24, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Davies, F.E.; Raje, N.; Hideshima, T.; Lentzsch, S.; Young, G.; Tai, Y.T.; Lin, B.; Podar, K.; Gupta, D.; Chauhan, D.; et al. Thalidomide and immunomodulatory derivatives augment natural killer cell cytotoxicity in multiple myeloma. Blood 2001, 98, 210–216. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, R.; Hideshima, T.; Catley, L.P.; Shringarpure, R.; Burger, R.; Mitsiades, N.; Mitsiades, C.; Cheema, P.; Chauhan, D.; Richardson, P.G.; et al. Immunomodulatory drug costimulates T cells via the B7-CD28 pathway. Blood 2004, 103, 1787–1790. [Google Scholar] [CrossRef] [PubMed]
- Hussein, M. Role of high-dose chemotherapy with hematopoietic stem-cell rescue for multiple myeloma. Leukemia 2004, 18, 893. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Callander, N.; Ganguly, S.; Gul, Z.; Hamadani, M.; Costa, L.; Sengsayadeth, S.; Abidi, M.; Hari, P.; Mohty, M.; et al. Hematopoietic stem cell transplantation for multiple myeloma: Guidelines from the american society for blood and marrow transplantation. Biol. Blood Marrow Transplant. 2015, 21, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Barlogie, B.; Tricot, G.; Anaissie, E.; Shaughnessy, J.; Rasmussen, E.; van Rhee, F.; Fassas, A.; Zangari, M.; Hollmig, K.; Pineda-Roman, M.; et al. Thalidomide and hematopoietic-cell transplantation for multiple myeloma. N. Engl. J. Med. 2006, 354, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Gahrton, G.; Tura, S.; Ljungman, P.; Belanger, C.; Brandt, L.; Cavo, M.; Facon, T.; Granena, A.; Gore, M.; Gratwohl, A.; et al. Allogeneic bone marrow transplantation in multiple myeloma. European group for bone marrow transplantation. N. Engl. J. Med. 1991, 325, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, M.; Kleber, M.; Udi, J.; Wasch, R.; Spencer, A.; Patriarca, F.; Knop, S.; Bruno, B.; Gramatzki, M.; Morabito, F.; et al. Consensus statement from european experts on the diagnosis, management, and treatment of multiple myeloma: From standard therapy to novel approaches. Leuk. Lymphoma 2010, 51, 1424–1443. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Rajkumar, S.V.; Dispenzieri, A.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Zeldenrust, S.R.; Dingli, D.; Russell, S.J.; Lust, J.A.; et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood 2008, 111, 2516–2520. [Google Scholar] [CrossRef] [PubMed]
- Khalil, D.N.; Smith, E.L.; Brentjens, R.J.; Wolchok, J.D. The future of cancer treatment: Immunomodulation, cars and combination immunotherapy. Nat. Rev. Clin. Oncol. 2016, 13, 273–290. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Lonial, S.; Jakubowiak, A.J.; Harousseau, J.L.; Anderson, K.C. Monoclonal antibodies in the treatment of multiple myeloma. Br. J. Haematol. 2011, 154, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.T.; Anderson, K.C. Antibody-based therapies in multiple myeloma. Bone Marrow Res. 2011, 2011, 924058. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Restifo, N.P.; Yang, J.C.; Morgan, R.A.; Dudley, M.E. Adoptive cell transfer: A clinical path to effective cancer immunotherapy. Nat. Rev. Cancer 2008, 8, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Restifo, N.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive immunotherapy for cancer: Harnessing the T cell response. Nat. Rev. Immunol. 2012, 12, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Noonan, K.A.; Huff, C.A.; Davis, J.; Lemas, M.V.; Fiorino, S.; Bitzan, J.; Ferguson, A.; Emerling, A.; Luznik, L.; Matsui, W.; et al. Adoptive transfer of activated marrow-infiltrating lymphocytes induces measurable antitumor immunity in the bone marrow in multiple myeloma. Sci. Transl. Med. 2015, 7, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Tricot, G.; Szmania, S.; Rosen, N.; Garg, T.K.; Malaviarachchi, P.A.; Moreno, A.; Dupont, B.; Hsu, K.C.; Baxter-Lowe, L.A.; et al. Infusion of haplo-identical killer immunoglobulin-like receptor ligand mismatched NK cells for relapsed myeloma in the setting of autologous stem cell transplantation. Br. J. Haematol. 2008, 143, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Szmania, S.; Lapteva, N.; Garg, T.; Greenway, A.; Lingo, J.; Nair, B.; Stone, K.; Woods, E.; Khan, J.; Stivers, J.; et al. Ex vivo-expanded natural killer cells demonstrate robust proliferation in vivo in high-risk relapsed multiple myeloma patients. J. Immunother. 2015, 38, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.L.L.; Kaur, I.; McCarty, J.; Yvon, E.; Shaim, H.; Muftuoglu, M.; Liu, E.; Sobieski, C.; Orlowski, R.Z.; Cooper, L.J.N.; et al. Infusion of ex vivo expanded allogeneic cord blood-derived natural killer cells in combination with autologous stem cell transplantation for multiple myeloma: Results of a phase I study. Blood 2015, 126, 1. [Google Scholar]
- Kerkar, S.P.; Sanchez-Perez, L.; Yang, S.; Borman, Z.A.; Muranski, P.; Ji, Y.; Chinnasamy, D.; Kaiser, A.D.; Hinrichs, C.S.; Klebanoff, C.A.; et al. Genetic engineering of murine CD8+ and CD4+ T cells for preclinical adoptive immunotherapy studies. J. Immunother. 2011, 34, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Bendle, G.M.; Linnemann, C.; Hooijkaas, A.I.; Bies, L.; de Witte, M.A.; Jorritsma, A.; Kaiser, A.D.; Pouw, N.; Debets, R.; Kieback, E.; et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat. Med. 2010, 16, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Dotti, G.; Gottschalk, S.; Savoldo, B.; Brenner, M.K. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol. Rev. 2014, 257, 107–126. [Google Scholar] [CrossRef] [PubMed]
- Neschadim, A.; Wang, J.C.; Sato, T.; Fowler, D.H.; Lavie, A.; Medin, J.A. Cell fate control gene therapy based on engineered variants of human deoxycytidine kinase. Mol. Ther. 2012, 20, 1002–1013. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Neschadim, A.; Konrad, M.; Fowler, D.H.; Lavie, A.; Medin, J.A. Engineered human TMPK/AZT as a novel enzyme/prodrug axis for suicide gene therapy. Mol. Ther. 2007, 15, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Scaife, M.; Pacienza, N.; Au, B.C.; Wang, J.C.; Devine, S.; Scheid, E.; Lee, C.J.; Lopez-Perez, O.; Neschadim, A.; Fowler, D.H.; et al. Engineered human tmpk fused with truncated cell-surface markers: Versatile cell-fate control safety cassettes. Gene Ther. 2013, 20, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Camino, V.; Harwood, S.L.; Alvarez-Mendez, A.; Alvarez-Vallina, L. Efficacy and toxicity management of CAR-T-cell immunotherapy: A matter of responsiveness control or tumour-specificity? Biochem. Soc. Trans. 2016, 44, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Glienke, W.; Esser, R.; Priesner, C.; Suerth, J.D.; Schambach, A.; Wels, W.S.; Grez, M.; Kloess, S.; Arseniev, L.; Koehl, U. Advantages and applications of car-expressing natural killer cells. Front. Pharmacol. 2015, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Ayed, A.O.; Chang, L.J.; Moreb, J.S. Immunotherapy for multiple myeloma: Current status and future directions. Crit. Rev. Oncol. Hematol. 2015, 96, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 2012, 12, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Titzer, S.; Christensen, O.; Manzke, O.; Tesch, H.; Wolf, J.; Emmerich, B.; Carsten, C.; Diehl, V.; Bohlen, H. Vaccination of multiple myeloma patients with idiotype-pulsed dendritic cells: Immunological and clinical aspects. Br. J. Haematol. 2000, 108, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Yi, Q.; Bergenbrant, S.; Osterborg, A.; Osby, E.; Ostman, R.; Bjorkholm, M.; Holm, G.; Lefvert, A.K. T-cell stimulation induced by idiotypes on monoclonal immunoglobulins in patients with monoclonal gammopathies. Scand. J. Immunol. 1993, 38, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Yi, Q.; Osterborg, A.; Bergenbrant, S.; Mellstedt, H.; Holm, G.; Lefvert, A.K. Idiotype-reactive T-cell subsets and tumor load in monoclonal gammopathies. Blood 1995, 86, 3043–3049. [Google Scholar] [PubMed]
- Lacy, M.Q.; Mandrekar, S.; Dispenzieri, A.; Hayman, S.; Kumar, S.; Buadi, F.; Dingli, D.; Litzow, M.; Wettstein, P.; Padley, D.; et al. Idiotype-pulsed antigen-presenting cells following autologous transplantation for multiple myeloma may be associated with prolonged survival. Am. J. Hematol. 2009, 84, 799–802. [Google Scholar] [CrossRef] [PubMed]
- Rosenblatt, J.; Vasir, B.; Uhl, L.; Blotta, S.; Macnamara, C.; Somaiya, P.; Wu, Z.; Joyce, R.; Levine, J.D.; Dombagoda, D.; et al. Vaccination with dendritic cell/tumor fusion cells results in cellular and humoral antitumor immune responses in patients with multiple myeloma. Blood 2011, 117, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Rosenblatt, J.; Avivi, I.; Vasir, B.; Uhl, L.; Munshi, N.C.; Katz, T.; Dey, B.R.; Somaiya, P.; Mills, H.; Campigotto, F.; et al. Vaccination with dendritic cell/tumor fusions following autologous stem cell transplant induces immunologic and clinical responses in multiple myeloma patients. Clin. Cancer Res. 2013, 19, 3640–3648. [Google Scholar] [CrossRef] [PubMed]
- Van de Donk, N.W.; Kamps, S.; Mutis, T.; Lokhorst, H.M. Monoclonal antibody-based therapy as a new treatment strategy in multiple myeloma. Leukemia 2012, 26, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Di Bernardo, A.; Macor, P.; Guarnotta, C.; Franco, G.; Florena, A.M.; Tedesco, F.; Tripodo, C. Humoral immunotherapy of multiple myeloma: Perspectives and perplexities. Expert Opin. Biol. Ther. 2010, 10, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Hsi, E.D.; Steinle, R.; Balasa, B.; Szmania, S.; Draksharapu, A.; Shum, B.P.; Huseni, M.; Powers, D.; Nanisetti, A.; Zhang, Y.; et al. CS1, a potential new therapeutic antibody target for the treatment of multiple myeloma. Clin. Cancer Res. 2008, 14, 2775–2784. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.T.; Dillon, M.; Song, W.; Leiba, M.; Li, X.F.; Burger, P.; Lee, A.I.; Podar, K.; Hideshima, T.; Rice, A.G.; et al. Anti-CS1 humanized monoclonal antibody HULUC63 inhibits myeloma cell adhesion and induces antibody-dependent cellular cytotoxicity in the bone marrow milieu. Blood 2008, 112, 1329–1337. [Google Scholar] [CrossRef] [PubMed]
- Van Rhee, F.; Szmania, S.M.; Dillon, M.; van Abbema, A.M.; Li, X.; Stone, M.K.; Garg, T.K.; Shi, J.; Moreno-Bost, A.M.; Yun, R.; et al. Combinatorial efficacy of anti-CS1 monoclonal antibody elotuzumab (HULUC63) and bortezomib against multiple myeloma. Mol. Cancer Ther. 2009, 8, 2616–2624. [Google Scholar] [CrossRef] [PubMed]
- Zonder, J.A.; Mohrbacher, A.F.; Singhal, S.; van Rhee, F.; Bensinger, W.I.; Ding, H.; Fry, J.; Afar, D.E.; Singhal, A.K. A phase 1, multicenter, open-label, dose escalation study of elotuzumab in patients with advanced multiple myeloma. Blood 2012, 120, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Dimopoulos, M.; Palumbo, A.; White, D.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; Moreau, P.; Mateos, M.V.; Magen, H.; et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N. Engl. J. Med. 2015, 373, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, J.E.; Deaglio, S.; Donati, D.; Beusan, I.S.; Corno, F.; Aranega, A.; Forni, M.; Falini, B.; Malavasi, F. Analysis of the distribution of human CD38 and of its ligand CD31 in normal tissues. J. Biol. Regul. Homeost. Agents 1998, 12, 81–91. [Google Scholar] [PubMed]
- Deaglio, S.; Mehta, K.; Malavasi, F. Human CD38: A (r)evolutionary story of enzymes and receptors. Leuk. Res. 2001, 25, 1–12. [Google Scholar] [CrossRef]
- Stevenson, F.K.; Bell, A.J.; Cusack, R.; Hamblin, T.J.; Slade, C.J.; Spellerberg, M.B.; Stevenson, G.T. Preliminary studies for an immunotherapeutic approach to the treatment of human myeloma using chimeric anti-CD38 antibody. Blood 1991, 77, 1071–1079. [Google Scholar] [PubMed]
- De Weers, M.; Tai, Y.T.; van der Veer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W.; et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J. Immunol. 2011, 186, 1840–1848. [Google Scholar] [CrossRef] [PubMed]
- Lokhorst, H.M.; Plesner, T.; Laubach, J.P.; Nahi, H.; Gimsing, P.; Hansson, M.; Minnema, M.C.; Lassen, U.; Krejcik, J.; Palumbo, A.; et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N. Engl. J. Med. 2015, 373, 1207–1219. [Google Scholar] [CrossRef] [PubMed]
- Deckert, J.; Wetzel, M.C.; Bartle, L.M.; Skaletskaya, A.; Goldmacher, V.S.; Vallee, F.; Zhou-Liu, Q.; Ferrari, P.; Pouzieux, S.; Lahoute, C.; et al. SAR650984, a novel humanized CD38-targeting antibody, demonstrates potent antitumor activity in models of multiple myeloma and other cd38+ hematologic malignancies. Clin. Cancer Res. 2014, 20, 4574–4583. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.G.; Strickland, S.A.; Glenn, M.; Zheng, W.; Daskalakis, N.; Mikhael, J.R. Sar650984, a cd38 monoclonal antibody in patients with selected CD38+ hematological malignancies—data from a dose-escalation phase I study. Blood 2013, 122, 284. [Google Scholar]
- Martin, T.G.; Baz, R.; Benson, D.M.; Lendvai, N.; Campana, F.; Charpentier, E.; Vij, R. A phase IB dose escalation trial of SAR650984 (anti-CD-38 mAb) in combination with lenalidomide and dexamethasone in relapsed/refractory multiple myeloma. Blood 2014, 124, 83. [Google Scholar]
- Tong, A.W.; Zhang, B.Q.; Mues, G.; Solano, M.; Hanson, T.; Stone, M.J. Anti-CD40 antibody binding modulates human multiple myeloma clonogenicity in vitro. Blood 1994, 84, 3026–3033. [Google Scholar] [PubMed]
- Westendorf, J.J.; Ahmann, G.J.; Armitage, R.J.; Spriggs, M.K.; Lust, J.A.; Greipp, P.R.; Katzmann, J.A.; Jelinek, D.F. CD40 expression in malignant plasma cells. Role in stimulation of autocrine IL-6 secretion by a human myeloma cell line. J. Immunol. 1994, 152, 117–128. [Google Scholar] [PubMed]
- Tai, Y.T.; Podar, K.; Gupta, D.; Lin, B.; Young, G.; Akiyama, M.; Anderson, K.C. CD40 activation induces p53-dependent vascular endothelial growth factor secretion in human multiple myeloma cells. Blood 2002, 99, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.T.; Podar, K.; Mitsiades, N.; Lin, B.; Mitsiades, C.; Gupta, D.; Akiyama, M.; Catley, L.; Hideshima, T.; Munshi, N.C.; et al. Cd40 induces human multiple myeloma cell migration via phosphatidylinositol 3-kinase/AKT/NF-kappa B signaling. Blood 2003, 101, 2762–2769. [Google Scholar] [CrossRef] [PubMed]
- Urashima, M.; Chauhan, D.; Hatziyanni, M.; Ogata, A.; Hollenbaugh, D.; Aruffo, A.; Anderson, K.C. CD40 ligand triggers interleukin-6 mediated B cell differentiation. Leuk. Res. 1996, 20, 507–515. [Google Scholar] [CrossRef]
- Tai, Y.T.; Catley, L.P.; Mitsiades, C.S.; Burger, R.; Podar, K.; Shringpaure, R.; Hideshima, T.; Chauhan, D.; Hamasaki, M.; Ishitsuka, K.; et al. Mechanisms by which SGN-40, a humanized anti-CD40 antibody, induces cytotoxicity in human multiple myeloma cells: Clinical implications. Cancer Res. 2004, 64, 2846–2852. [Google Scholar] [CrossRef] [PubMed]
- Hussein, M.; Berenson, J.R.; Niesvizky, R.; Munshi, N.; Matous, J.; Sobecks, R.; Harrop, K.; Drachman, J.G.; Whiting, N. A phase I multidose study of dacetuzumab (SGN-40; humanized anti-CD40 monoclonal antibody) in patients with multiple myeloma. Haematologica 2010, 95, 845–848. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.T.; Li, X.F.; Catley, L.; Coffey, R.; Breitkreutz, I.; Bae, J.; Song, W.; Podar, K.; Hideshima, T.; Chauhan, D.; et al. Immunomodulatory drug lenalidomide (CC-5013, IMID3) augments anti-CD40 SGN-40-induced cytotoxicity in human multiple myeloma: Clinical implications. Cancer Res. 2005, 65, 11712–11720. [Google Scholar] [CrossRef] [PubMed]
- Agura, E.N.R.; Matous, J.; Munshi, N.; Hussein, M.; Parameswaran, R.V.; Tarantolo, S.; Whiting, N.; Drachman, J.G.; Zonder, J.A. Dacetuzumab (SGN-40), lenalidomide, and weekly dexamethasone in relapsed or refractory multiple myeloma: Multiple responses observed in a phase 1b study. Blood 2009, 114, 2870. [Google Scholar]
- Tai, Y.T.; Li, X.; Tong, X.; Santos, D.; Otsuki, T.; Catley, L.; Tournilhac, O.; Podar, K.; Hideshima, T.; Schlossman, R.; et al. Human anti-CD40 antagonist antibody triggers significant antitumor activity against human multiple myeloma. Cancer Res. 2005, 65, 5898–5906. [Google Scholar] [CrossRef] [PubMed]
- Bensinger, W.; Maziarz, R.T.; Jagannath, S.; Spencer, A.; Durrant, S.; Becker, P.S.; Ewald, B.; Bilic, S.; Rediske, J.; Baeck, J.; et al. A phase 1 study of lucatumumab, a fully human anti-CD40 antagonist monoclonal antibody administered intravenously to patients with relapsed or refractory multiple myeloma. Br. J. Haematol. 2012, 159, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Byrd, J.C.; Kipps, T.J.; Flinn, I.W.; Cooper, M.; Odenike, O.; Bendiske, J.; Rediske, J.; Bilic, S.; Dey, J.; Baeck, J.; et al. Phase i study of the anti-cd40 humanized monoclonal antibody lucatumumab (HCD122) in relapsed chronic lymphocytic leukemia. Leuk. Lymphoma 2012, 53, 2136–2142. [Google Scholar] [CrossRef] [PubMed]
- Horton, H.M.; Bernett, M.J.; Peipp, M.; Pong, E.; Karki, S.; Chu, S.Y.; Richards, J.O.; Chen, H.; Repp, R.; Desjarlais, J.R.; et al. Fc-engineered anti-CD40 antibody enhances multiple effector functions and exhibits potent in vitro and in vivo antitumor activity against hematologic malignancies. Blood 2010, 116, 3004–3012. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, R.D.; Turnbull, J.E.; Gallagher, J.T.; Lander, A.D. Fine structure of heparan sulfate regulates syndecan-1 function and cell behavior. J. Biol. Chem. 1994, 269, 13100–13106. [Google Scholar] [PubMed]
- Wijdenes, J.; Vooijs, W.C.; Clement, C.; Post, J.; Morard, F.; Vita, N.; Laurent, P.; Sun, R.X.; Klein, B.; Dore, J.M. A plasmocyte selective monoclonal antibody (B-B4) recognizes syndecan-1. Br. J. Haematol. 1996, 94, 318–323. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, F.P.; Pinkus, J.L.; Pinkus, G.S. CD138 (syndecan-1), a plasma cell marker immunohistochemical profile in hematopoietic and nonhematopoietic neoplasms. Am. J. Clin. Pathol. 2004, 121, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Kambham, N.; Kong, C.; Longacre, T.A.; Natkunam, Y. Utility of syndecan-1 (CD138) expression in the diagnosis of undifferentiated malignant neoplasms: A tissue microarray study of 1754 cases. Appl. Immunohistochem. Mol. Morphol. 2005, 13, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Owens, R.; Tricot, G.; Wilson, C.S. Flow cytometric immunophenotypic analysis of 306 cases of multiple myeloma. Am. J. Clin. Pathol. 2004, 121, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Lamorte, S.; Ferrero, S.; Aschero, S.; Monitillo, L.; Bussolati, B.; Omede, P.; Ladetto, M.; Camussi, G. Syndecan-1 promotes the angiogenic phenotype of multiple myeloma endothelial cells. Leukemia 2012, 26, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Dhodapkar, M.V.; Kelly, T.; Theus, A.; Athota, A.B.; Barlogie, B.; Sanderson, R.D. Elevated levels of shed syndecan-1 correlate with tumour mass and decreased matrix metalloproteinase-9 activity in the serum of patients with multiple myeloma. Br. J. Haematol. 1997, 99, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yaccoby, S.; Liu, W.; Langford, J.K.; Pumphrey, C.Y.; Theus, A.; Epstein, J.; Sanderson, R.D. Soluble syndecan-1 promotes growth of myeloma tumors in vivo. Blood 2002, 100, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; MacLeod, V.; Dai, Y.; Khotskaya-Sample, Y.; Shriver, Z.; Venkataraman, G.; Sasisekharan, R.; Naggi, A.; Torri, G.; Casu, B.; et al. The syndecan-1 heparan sulfate proteoglycan is a viable target for myeloma therapy. Blood 2007, 110, 2041–2048. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Hideshima, T.; Fulciniti, M.; Lutz, R.J.; Yasui, H.; Okawa, Y.; Kiziltepe, T.; Vallet, S.; Pozzi, S.; Santo, L.; et al. The monoclonal antibody NBT062 conjugated to cytotoxic maytansinoids has selective cytotoxicity against cd138-positive multiple myeloma cells in vitro and in vivo. Clin. Cancer Res. 2009, 15, 4028–4037. [Google Scholar] [CrossRef] [PubMed]
- Heffner, L.T.; Jagannath, S.; Zimmerman, T.M.; Lee, K.P.; Rosenblatt, J.; Lonial, S.; Lutz, R.J.; Czeloth, N.; Osterroth, F.; Ruehle, M.; et al. BT062, an antibody-drug conjugate directed against CD138, given weekly for 3 weeks in each 4 week cycle: Safety and further evidence of clinical activity. Blood 2012, 120, 4042. [Google Scholar]
- Kelly, K.C.-K.A.; Heffner, L.; Somlo, G.; Siegel, D.S.; Zimmerman, T.; Karnad, A.; Munshi, N.; Jagannath, S.; Greenberg, A.; Lonial, S.; et al. Indatuximab ravtansine (BT062) in combination with lenalidomide and low-dose dexamethasone in patients with relapsed and/or refractory multiple myeloma: Clinical activity in patients already exposed to lenalidomide and bortezomib. Blood 2014, 124, 4736. [Google Scholar]
- Guo, B.; Chen, M.; Han, Q.; Hui, F.; Dai, H.; Zhang, W.; Zhang, Y.; Wang, Y.; Zhu, H.; Han, W. CD138-directed adoptive immunotherapy of chimeric antigen receptor (car)-modified T cells for multiple myeloma. J. Cell. Immunother. 2016, 2, 28–35. [Google Scholar] [CrossRef]
- Chaidos, A.; Barnes, C.P.; Cowan, G.; May, P.C.; Melo, V.; Hatjiharissi, E.; Papaioannou, M.; Harrington, H.; Doolittle, H.; Terpos, E.; et al. Clinical drug resistance linked to interconvertible phenotypic and functional states of tumor-propagating cells in multiple myeloma. Blood 2013, 121, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Goridis, C.; Brunet, J.F. Ncam: Structural diversity, function and regulation of expression. Semin. Cell. Biol. 1992, 3, 189–197. [Google Scholar] [CrossRef]
- Drach, J.; Gattringer, C.; Huber, H. Multiple myeloma with coexpression of myeloid and natural killer cell antigens. Blood 1990, 76, 265–266. [Google Scholar] [PubMed]
- Van Camp, B.; Durie, B.G.; Spier, C.; de Waele, M.; van Riet, I.; Vela, E.; Frutiger, Y.; Richter, L.; Grogan, T.M. Plasma cells in multiple myeloma express a natural killer cell-associated antigen: CD56 (NKH-1; LEU-19). Blood 1990, 76, 377–382. [Google Scholar] [PubMed]
- Jensen, M.; Berthold, F. Targeting the neural cell adhesion molecule in cancer. Cancer Lett. 2007, 258, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Tassone, P.; Gozzini, A.; Goldmacher, V.; Shammas, M.A.; Whiteman, K.R.; Carrasco, D.R.; Li, C.; Allam, C.K.; Venuta, S.; Anderson, K.C.; et al. In vitro and in vivo activity of the maytansinoid immunoconjugate hun901-N2′-deacetyl-N2′-(3-mercapto-1-oxopropyl)-maytansine against CD56+ multiple myeloma cells. Cancer Res. 2004, 64, 4629–4636. [Google Scholar] [CrossRef] [PubMed]
- Berdeja, J.G. Lorvotuzumab mertansine: Antibody-drug-conjugate for CD56+ multiple myeloma. Front. Biosci. 2014, 19, 163–170. [Google Scholar] [CrossRef]
- Stein, R.; Mattes, M.J.; Cardillo, T.M.; Hansen, H.J.; Chang, C.H.; Burton, J.; Govindan, S.; Goldenberg, D.M. CD74: A new candidate target for the immunotherapy of B-cell neoplasms. Clin. Cancer Res. 2007, 13, 5556s–5563s. [Google Scholar] [CrossRef] [PubMed]
- Claesson, L.; Larhammar, D.; Rask, L.; Peterson, P.A. Cdna clone for the human invariant gamma chain of class II histocompatibility antigens and its implications for the protein structure. Proc. Natl. Acad. Sci. USA 1983, 80, 7395–7399. [Google Scholar] [CrossRef] [PubMed]
- Burton, J.D.; Ely, S.; Reddy, P.K.; Stein, R.; Gold, D.V.; Cardillo, T.M.; Goldenberg, D.M. CD74 is expressed by multiple myeloma and is a promising target for therapy. Clin. Cancer Res. 2004, 10, 6606–6611. [Google Scholar] [CrossRef] [PubMed]
- Larson, D.F.; Horak, K. Macrophage migration inhibitory factor: Controller of systemic inflammation. Crit. Care 2006, 10, 138. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, J.L.; Niesvizky, R.; Stadtmauer, E.A.; Chanan-Khan, A.; Siegel, D.; Horne, H.; Wegener, W.A.; Goldenberg, D.M. Phase I, multicentre, dose-escalation trial of monotherapy with milatuzumab (humanized anti-CD74 monoclonal antibody) in relapsed or refractory multiple myeloma. Br. J. Haematol. 2013, 163, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Sapra, P.; Stein, R.; Pickett, J.; Qu, Z.; Govindan, S.V.; Cardillo, T.M.; Hansen, H.J.; Horak, I.D.; Griffiths, G.L.; Goldenberg, D.M. Anti-CD74 antibody-doxorubicin conjugate, IMMU-110, in a human multiple myeloma xenograft and in monkeys. Clin. Cancer Res. 2005, 11, 5257–5264. [Google Scholar] [CrossRef] [PubMed]
- Stein, R.; Smith, M.R.; Chen, S.; Zalath, M.; Goldenberg, D.M. Combining milatuzumab with bortezomib, doxorubicin, or dexamethasone improves responses in multiple myeloma cell lines. Clin. Cancer Res. 2009, 15, 2808–2817. [Google Scholar] [CrossRef] [PubMed]
- Barclay, A.N.; Wright, G.J.; Brooke, G.; Brown, M.H. Cd200 and membrane protein interactions in the control of myeloid cells. Trends Immunol. 2002, 23, 285–290. [Google Scholar] [CrossRef]
- Moreaux, J.; Hose, D.; Reme, T.; Jourdan, E.; Hundemer, M.; Legouffe, E.; Moine, P.; Bourin, P.; Moos, M.; Corre, J.; et al. CD200 is a new prognostic factor in multiple myeloma. Blood 2006, 108, 4194–4197. [Google Scholar] [CrossRef] [PubMed]
- Hoek, R.M.; Ruuls, S.R.; Murphy, C.A.; Wright, G.J.; Goddard, R.; Zurawski, S.M.; Blom, B.; Homola, M.E.; Streit, W.J.; Brown, M.H.; et al. Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science 2000, 290, 1768–1771. [Google Scholar] [CrossRef] [PubMed]
- McCaughan, G.W.; Clark, M.J.; Barclay, A.N. Characterization of the human homolog of the rat mrc OX-2 membrane glycoprotein. Immunogenetics 1987, 25, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Kretz-Rommel, A.; Qin, F.; Dakappagari, N.; Cofiell, R.; Faas, S.J.; Bowdish, K.S. Blockade of CD200 in the presence or absence of antibody effector function: Implications for anti-CD200 therapy. J. Immunol. 2008, 180, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wei, G.; Liu, D. CD19: A biomarker for b cell development, lymphoma diagnosis and therapy. Exp. Hematol. Oncol. 2012, 1, 36. [Google Scholar] [CrossRef] [PubMed]
- Ginaldi, L.; De Martinis, M.; Matutes, E.; Farahat, N.; Morilla, R.; Catovsky, D. Levels of expression of CD19 and CD20 in chronic B cell leukaemias. J. Clin. Pathol. 1998, 51, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Tembhare, P.R.; Yuan, C.M.; Venzon, D.; Braylan, R.; Korde, N.; Manasanch, E.; Zuchlinsky, D.; Calvo, K.; Kurlander, R.; Bhutani, M.; et al. Flow cytometric differentiation of abnormal and normal plasma cells in the bone marrow in patients with multiple myeloma and its precursor diseases. Leuk. Res. 2014, 38, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Garfall, A.L.; Maus, M.V.; Hwang, W.T.; Lacey, S.F.; Mahnke, Y.D.; Melenhorst, J.J.; Zheng, Z.; Vogl, D.T.; Cohen, A.D.; Weiss, B.M.; et al. Chimeric antigen receptor T cells against CD19 for multiple myeloma. N. Engl. J. Med. 2015, 373, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Hajek, R.; Okubote, S.A.; Svachova, H. Myeloma stem cell concepts, heterogeneity and plasticity of multiple myeloma. Br. J. Haematol. 2013, 163, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Garfall, A.L.; Maus, M.V.; Lacey, S.F.; Mahnke, Y.D.; Melenhorst, J.J.; Zheng, Z.; Vogl, D.T.; Cohen, A.D.; Weiss, B.M.; Dengel, K.; et al. Safety and efficacy of anti-CD19 chimeric antigen receptor (CAR)-modified autologous T cells (CTL019) in advanced multiple myeloma. J. Clin. Oncol. 2015, 33, 8517. [Google Scholar]
- Thompson, J.S.; Schneider, P.; Kalled, S.L.; Wang, L.; Lefevre, E.A.; Cachero, T.G.; MacKay, F.; Bixler, S.A.; Zafari, M.; Liu, Z.Y.; et al. Baff binds to the tumor necrosis factor receptor-like molecule B cell maturation antigen and is important for maintaining the peripheral B cell population. J. Exp. Med. 2000, 192, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Rennert, P.; Schneider, P.; Cachero, T.G.; Thompson, J.; Trabach, L.; Hertig, S.; Holler, N.; Qian, F.; Mullen, C.; Strauch, K.; et al. A soluble form of B cell maturation antigen, a receptor for the tumor necrosis factor family member april, inhibits tumor cell growth. J. Exp. Med. 2000, 192, 1677–1684. [Google Scholar] [CrossRef] [PubMed]
- Marsters, S.A.; Yan, M.; Pitti, R.M.; Haas, P.E.; Dixit, V.M.; Ashkenazi, A. Interaction of the tnf homologues BLYS and april with the TNF receptor homologues BCMA and TACI. Curr. Biol. 2000, 10, 785–788. [Google Scholar] [CrossRef]
- O’Connor, B.P.; Raman, V.S.; Erickson, L.D.; Cook, W.J.; Weaver, L.K.; Ahonen, C.; Lin, L.L.; Mantchev, G.T.; Bram, R.J.; Noelle, R.J. Bcma is essential for the survival of long-lived bone marrow plasma cells. J. Exp. Med. 2004, 199, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Novak, A.J.; Darce, J.R.; Arendt, B.K.; Harder, B.; Henderson, K.; Kindsvogel, W.; Gross, J.A.; Greipp, P.R.; Jelinek, D.F. Expression of BCMA, TACI, and BAFF-R in multiple myeloma: A mechanism for growth and survival. Blood 2004, 103, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Neri, P.; Kumar, S.; Fulciniti, M.T.; Vallet, S.; Chhetri, S.; Mukherjee, S.; Tai, Y.; Chauhan, D.; Tassone, P.; Venuta, S.; et al. Neutralizing B-cell activating factor antibody improves survival and inhibits osteoclastogenesis in a severe combined immunodeficient human multiple myeloma model. Clin. Cancer Res. 2007, 13, 5903–5909. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, R.; Alyea, E.P.; Chiaretti, S.; Wu, C.J.; Zorn, E.; Weller, E.; Wu, B.; Canning, C.; Schlossman, R.; Munshi, N.C.; et al. Graft-versus-tumor response in patients with multiple myeloma is associated with antibody response to BCMA, a PLASMA-cell membrane receptor. Blood 2005, 105, 3945–3950. [Google Scholar] [CrossRef] [PubMed]
- Moreaux, J.; Legouffe, E.; Jourdan, E.; Quittet, P.; Reme, T.; Lugagne, C.; Moine, P.; Rossi, J.F.; Klein, B.; Tarte, K. BAFF and APRIL protect myeloma cells from apoptosis induced by interleukin 6 deprivation and dexamethasone. Blood 2004, 103, 3148–3157. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, R.O.; Evbuomwan, M.O.; Pittaluga, S.; Rose, J.J.; Raffeld, M.; Yang, S.; Gress, R.E.; Hakim, F.T.; Kochenderfer, J.N. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin. Cancer Res. 2013, 19, 2048–2060. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.T.; Mayes, P.A.; Acharya, C.; Zhong, M.Y.; Cea, M.; Cagnetta, A.; Craigen, J.; Yates, J.; Gliddon, L.; Fieles, W.; et al. Novel anti-B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood 2014, 123, 3128–3138. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.A.; Shi, V.; Wang, M.; Stroncek, D.; Maric, I.; Brudno, J.N.; Stetler-Stevenson, M.; Rose, J.J.; Feldman, S.; Hansen, B.; et al. Remissions of multiple myeloma during a first-in-humans clinical trial of T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor. Blood 2015, 126, LBA-1. [Google Scholar]
- Susanne Hipp, P.D.; Wahl, J.; Blanset, D.; Thomas, O.; Rattel, B.; Adam, P.; Friedrich, M. BI 836909, a novel bispecific T cell engager for the treatment of multiple myeloma induces highly specific and efficacious lysis of multiple myeloma cells in vitro and shows anti-tumor activity in vivo. Blood 2015, 126, 2999. [Google Scholar]
- Van Rhee, F.; Szmania, S.M.; Zhan, F.; Gupta, S.K.; Pomtree, M.; Lin, P.; Batchu, R.B.; Moreno, A.; Spagnoli, G.; Shaughnessy, J.; et al. NY-ESO-1 is highly expressed in poor-prognosis multiple myeloma and induces spontaneous humoral and cellular immune responses. Blood 2005, 105, 3939–3944. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Germeau, C.; Vigneron, N.; Maernoudt, A.S.; Morel, S.; Boon, T.; Coulie, P.G.; van den Eynde, B.J. Two new tumor-specific antigenic peptides encoded by gene mage-C2 and presented to cytolytic T lymphocytes by HLA-A2. Int. J. Cancer 2004, 109, 698–702. [Google Scholar] [CrossRef] [PubMed]
- Atanackovic, D.; Luetkens, T.; Hildebrandt, Y.; Arfsten, J.; Bartels, K.; Horn, C.; Stahl, T.; Cao, Y.; Zander, A.R.; Bokemeyer, C.; et al. Longitudinal analysis and prognostic effect of cancer-testis antigen expression in multiple myeloma. Clin. Cancer Res. 2009, 15, 1343–1352. [Google Scholar] [CrossRef] [PubMed]
- Van Duin, M.; Broyl, A.; de Knegt, Y.; Goldschmidt, H.; Richardson, P.G.; Hop, W.C.; van der Holt, B.; Joseph-Pietras, D.; Mulligan, G.; Neuwirth, R.; et al. Cancer testis antigens in newly diagnosed and relapse multiple myeloma: Prognostic markers and potential targets for immunotherapy. Haematologica 2011, 96, 1662–1669. [Google Scholar] [CrossRef] [PubMed]
- Ghafouri-Fard, S.; Seifi-Alan, M.; Shamsi, R.; Esfandiary, A. Immunotherapy in multiple myeloma using cancer-testis antigens. Iran. J. Cancer Prev. 2015, 8, e3755. [Google Scholar] [CrossRef] [PubMed]
- Andrade, V.C.; Vettore, A.L.; Felix, R.S.; Almeida, M.S.; Carvalho, F.; Oliveira, J.S.; Chauffaille, M.L.; Andriolo, A.; Caballero, O.L.; Zago, M.A.; et al. Prognostic impact of cancer/testis antigen expression in advanced stage multiple myeloma patients. Cancer Immun. 2008, 8, 2. [Google Scholar] [PubMed]
- Condomines, M.; Hose, D.; Raynaud, P.; Hundemer, M.; De Vos, J.; Baudard, M.; Moehler, T.; Pantesco, V.; Moos, M.; Schved, J.F.; et al. Cancer/testis genes in multiple myeloma: Expression patterns and prognosis value determined by microarray analysis. J. Immunol. 2007, 178, 3307–3315. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, A.P.; Aqui, N.A.; Stadtmauer, E.A.; Vogl, D.T.; Xu, Y.Y.; Kalos, M.; Cai, L.; Fang, H.B.; Weiss, B.M.; Badros, A.; et al. Combination immunotherapy after ASCT for multiple myeloma using mage-A3/poly-ICLC immunizations followed by adoptive transfer of vaccine-primed and costimulated autologous T cells. Clin. Cancer Res. 2014, 20, 1355–1365. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, A.P.; Stadtmauer, E.A.; Binder-Scholl, G.K.; Goloubeva, O.; Vogl, D.T.; Lacey, S.F.; Badros, A.Z.; Garfall, A.; Weiss, B.; Finklestein, J.; et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med. 2015, 21, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. Glucose-regulated proteins in cancer: Molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 2014, 14, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Rasche, L.; Duell, J.; Morgner, C.; Chatterjee, M.; Hensel, F.; Rosenwald, A.; Einsele, H.; Topp, M.S.; Brandlein, S. The natural human igm antibody PAT-SM6 induces apoptosis in primary human multiple myeloma cells by targeting heat shock protein GRP78. PLoS ONE 2013, 8, e63414. [Google Scholar] [CrossRef] [PubMed]
- Rasche, L.; Duell, J.; Castro, I.C.; Dubljevic, V.; Chatterjee, M.; Knop, S.; Hensel, F.; Rosenwald, A.; Einsele, H.; Topp, M.S.; et al. GRP78-directed immunotherapy in relapsed or refractory multiple myeloma—Results from a phase 1 trial with the monoclonal immunoglobulin m antibody PAT-SM6. Haematologica 2015, 100, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.; Untergasser, G.; Zenzmaier, C.; Sarg, B.; Gastl, G.; Gunsilius, E.; Steurer, M. GRP-78 secreted by tumor cells blocks the antiangiogenic activity of bortezomib. Blood 2009, 114, 3960–3967. [Google Scholar] [CrossRef] [PubMed]
- Abdel Malek, M.A.; Jagannathan, S.; Malek, E.; Sayed, D.M.; Elgammal, S.A.; Abd El-Azeem, H.G.; Thabet, N.M.; Driscoll, J.J. Molecular chaperone GRP78 enhances aggresome delivery to autophagosomes to promote drug resistance in multiple myeloma. Oncotarget 2015, 6, 3098–3110. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.H.; Piao, S.F.; Dey, S.; McAfee, Q.; Karakousis, G.; Villanueva, J.; Hart, L.S.; Levi, S.; Hu, J.; Zhang, G.; et al. Targeting ER stress-induced autophagy overcomes braf inhibitor resistance in melanoma. J. Clin. Investig. 2014, 124, 1406–1417. [Google Scholar] [CrossRef] [PubMed]
- Rauschert, N.; Brandlein, S.; Holzinger, E.; Hensel, F.; Muller-Hermelink, H.K.; Vollmers, H.P. A new tumor-specific variant of GRP78 as target for antibody-based therapy. Lab. Investig. 2008, 88, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Hensel, F.; Eckstein, M.; Rosenwald, A.; Brandlein, S. Early development of PAT-SM6 for the treatment of melanoma. Melanoma Res. 2013, 23, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Burger, R. Impact of interleukin-6 in hematological malignancies. Transfus. Med. Hemother. 2013, 40, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Van Oers, M.H.; van Zaanen, H.C.; Lokhorst, H.M. Interleukin-6, a new target for therapy in multiple myeloma? Ann. Hematol. 1993, 66, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Hunsucker, S.A.; Magarotto, V.; Kuhn, D.J.; Kornblau, S.M.; Wang, M.; Weber, D.M.; Thomas, S.K.; Shah, J.J.; Voorhees, P.M.; Xie, H.; et al. Blockade of interleukin-6 signalling with siltuximab enhances melphalan cytotoxicity in preclinical models of multiple myeloma. Br. J. Haematol. 2011, 152, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Voorhees, P.M.; Chen, Q.; Small, G.W.; Kuhn, D.J.; Hunsucker, S.A.; Nemeth, J.A.; Orlowski, R.Z. Targeted inhibition of interleukin-6 with cnto 328 sensitizes pre-clinical models of multiple myeloma to dexamethasone-mediated cell death. Br. J. Haematol. 2009, 145, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Kurzrock, R.; Voorhees, P.M.; Casper, C.; Furman, R.R.; Fayad, L.; Lonial, S.; Borghaei, H.; Jagannath, S.; Sokol, L.; Usmani, S.Z.; et al. A phase I, open-label study of siltuximab, an anti-IL-6 monoclonal antibody, in patients with B-cell non-hodgkin lymphoma, multiple myeloma, or castleman disease. Clin. Cancer Res. 2013, 19, 3659–3670. [Google Scholar] [CrossRef] [PubMed]
- Voorhees, P.M.; Manges, R.F.; Sonneveld, P.; Jagannath, S.; Somlo, G.; Krishnan, A.; Lentzsch, S.; Frank, R.C.; Zweegman, S.; Wijermans, P.W.; et al. A phase 2 multicentre study of siltuximab, an anti-interleukin-6 monoclonal antibody, in patients with relapsed or refractory multiple myeloma. Br. J. Haematol. 2013, 161, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, R.Z.; Gercheva, L.; Williams, C.; Sutherland, H.; Robak, T.; Masszi, T.; Goranova-Marinova, V.; Dimopoulos, M.A.; Cavenagh, J.D.; Spicka, I.; et al. A phase 2, randomized, double-blind, placebo-controlled study of siltuximab (anti-IL-6 mab) and bortezomib versus bortezomib alone in patients with relapsed or refractory multiple myeloma. Am. J. Hematol. 2015, 90, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. Pd-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubata, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Atanackovic, D.; Luetkens, T.; Kroger, N. Coinhibitory molecule PD-1 as a potential target for the immunotherapy of multiple myeloma. Leukemia 2014, 28, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.M., Jr.; Bakan, C.E.; Mishra, A.; Hofmeister, C.C.; Efebera, Y.; Becknell, B.; Baiocchi, R.A.; Zhang, J.; Yu, J.; Smith, M.K.; et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: A therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood 2010, 116, 2286–2294. [Google Scholar] [CrossRef] [PubMed]
- Tamura, H.; Ishibashi, M.; Yamashita, T.; Tanosaki, S.; Okuyama, N.; Kondo, A.; Hyodo, H.; Shinya, E.; Takahashi, H.; Dong, H.; et al. Marrow stromal cells induce B7-H1 expression on myeloma cells, generating aggressive characteristics in multiple myeloma. Leukemia 2013, 27, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-l1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [PubMed]
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.; Halwani, A.; Gutierrez, M.; Millenson, M.; Cohen, A.; Schuster, S.; Lebovic, D.; et al. Preliminary results of a phase I study of nivolumab (BMS-936558) in patients with relapsed or refractory lymphoid malignancies. Blood 2014, 124, 291. [Google Scholar]
- Badros, A.K.M.; Ma, N.; Rapoport, A.; Lederer, E.; Philip, S.; Lesho, P.D.C.; Hardy, N.; Yared, J.; Goloubeva, O.; Singh, Z. A phase II study of anti PD-1 antibody pembrolizumab, pomalidomide anddexamethasone in patients with relapsed/refractory multiple myeloma RRMM. Blood 2015, 126, 506. [Google Scholar]
- Berger, R.; Rotem-Yehudar, R.; Slama, G.; Landes, S.; Kneller, A.; Leiba, M.; Koren-Michowitz, M.; Shimoni, A.; Nagler, A. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin. Cancer Res. 2008, 14, 3044–3051. [Google Scholar] [CrossRef] [PubMed]
- Ferrini, S.; Cambiaggi, A.; Meazza, R.; Sforzini, S.; Marciano, S.; Mingari, M.C.; Moretta, L. T cell clones expressing the natural killer cell-related p58 receptor molecule display heterogeneity in phenotypic properties and p58 function. Eur. J. Immunol. 1994, 24, 2294–2298. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.H.; Gumperz, J.E.; Parham, P.; Lanier, L.L. Superantigen-dependent, cell-mediated cytotoxicity inhibited by MHC class I receptors on T lymphocytes. Science 1995, 268, 403–405. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.S.; Purdy, A.K. Structure/function of human killer cell immunoglobulin-like receptors: Lessons from polymorphisms, evolution, crystal structures and mutations. Immunology 2011, 132, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.S.; Hasegawa, J. Natural killer cell biology: An update and future directions. J. Allergy Clin. Immunol. 2013, 132, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.M., Jr.; Bakan, C.E.; Zhang, S.; Collins, S.M.; Liang, J.; Srivastava, S.; Hofmeister, C.C.; Efebera, Y.; Andre, P.; Romagne, F.; et al. IPH2101, a novel anti-inhibitory KIR antibody, and lenalidomide combine to enhance the natural killer cell versus multiple myeloma effect. Blood 2011, 118, 6387–6391. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.M., Jr.; Hofmeister, C.C.; Padmanabhan, S.; Suvannasankha, A.; Jagannath, S.; Abonour, R.; Bakan, C.; Andre, P.; Efebera, Y.; Tiollier, J.; et al. A phase 1 trial of the anti-KIR antibody IPH2101 in patients with relapsed/refractory multiple myeloma. Blood 2012, 120, 4324–4333. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.M., Jr.; Cohen, A.D.; Jagannath, S.; Munshi, N.C.; Spitzer, G.; Hofmeister, C.C.; Efebera, Y.A.; Andre, P.; Zerbib, R.; Caligiuri, M.A. A phase I trial of the anti-KIR antibody iph2101 and lenalidomide in patients with relapsed/refractory multiple myeloma. Clin. Cancer Res. 2015, 21, 4055–4061. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Witzig, T.E.; Timm, M.; Haug, J.; Wellik, L.; Fonseca, R.; Greipp, P.R.; Rajkumar, S.V. Expression of vegf and its receptors by myeloma cells. Leukemia 2003, 17, 2025–2031. [Google Scholar] [CrossRef] [PubMed]
- Grothey, A.; Galanis, E. Targeting angiogenesis: Progress with anti-VEGF treatment with large molecules. Nat. Rev. Clin. Oncol. 2009, 6, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Callander, N.S.; Markovina, S.; Juckett, M.B. The addition of bevacizumab (B) to lenalidomide and low dose dexamethasone does not significantly increase response in relapsed or refractory multiple myeloma. Blood 2009, 114, 22. [Google Scholar]
- Somlo, G.; Lashkari, A.; Bellamy, W.; Zimmerman, T.M.; Tuscano, J.M.; O'Donnell, M.R.; Mohrbacher, A.F.; Forman, S.J.; Frankel, P.; Chen, H.X.; et al. Phase II randomized trial of bevacizumab versus bevacizumab and thalidomide for relapsed/refractory multiple myeloma: A california cancer consortium trial. Br. J. Haematol. 2011, 154, 533–535. [Google Scholar] [CrossRef] [PubMed]
- White, D.; Kassim, A.; Bhaskar, B.; Yi, J.; Wamstad, K.; Paton, V.E. Results from amber, a randomized phase 2 study of bevacizumab and bortezomib versus bortezomib in relapsed or refractory multiple myeloma. Cancer 2013, 119, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Hengeveld, P.J.; Kersten, M.J. B-cell activating factor in the pathophysiology of multiple myeloma: A target for therapy? Blood Cancer J. 2015, 5, e282. [Google Scholar] [CrossRef] [PubMed]
- Bolkun, L.; Lemancewicz, D.; Jablonska, E.; Kulczynska, A.; Bolkun-Skornicka, U.; Kloczko, J.; Dzieciol, J. Baff and april as TNF superfamily molecules and angiogenesis parallel progression of human multiple myeloma. Ann. Hematol. 2014, 93, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Fragioudaki, M.; Boula, A.; Tsirakis, G.; Psarakis, F.; Spanoudakis, M.; Papadakis, I.S.; Pappa, C.A.; Alexandrakis, M.G. B cell-activating factor: Its clinical significance in multiple myeloma patients. Ann. Hematol. 2012, 91, 1413–1418. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.T.; Li, X.F.; Breitkreutz, I.; Song, W.; Neri, P.; Catley, L.; Podar, K.; Hideshima, T.; Chauhan, D.; Raje, N.; et al. Role of B-cell-activating factor in adhesion and growth of human multiple myeloma cells in the bone marrow microenvironment. Cancer Res. 2006, 66, 6675–6682. [Google Scholar] [CrossRef] [PubMed]
- Manetta, J.; Bina, H.; Ryan, P.; Fox, N.; Witcher, D.R.; Kikly, K. Generation and characterization of tabalumab, a human monoclonal antibody that neutralizes both soluble and membrane-bound B-cell activating factor. J. Inflamm. Res. 2014, 7, 121–131. [Google Scholar] [PubMed]
- Raje, N.; Faber, E.A., Jr.; Richardson, P.G.; Schiller, G.; Hohl, R.; Cohen, A.; Forero, A.; Carpenter, S.P.; Cronier, D.; Kaiser, C.; et al. Phase 1 study of tabalumab, a human anti-BAFF antibody and bortezomib in patients with previously-treated multiple myeloma. Blood 2012, 120, 447. [Google Scholar]
- Abe, M.; Kido, S.; Hiasa, M.; Nakano, A.; Oda, A.; Amou, H.; Matsumoto, T. Baff and april as osteoclast-derived survival factors for myeloma cells: A rationale for TACI-FC treatment in patients with multiple myeloma. Leukemia 2006, 20, 1313–1315. [Google Scholar] [CrossRef] [PubMed]
- Yaccoby, S.; Pennisi, A.; Li, X.; Dillon, S.R.; Zhan, F.; Barlogie, B.; Shaughnessy, J.D., Jr. Atacicept (TACI-IG) inhibits growth of TACI(high) primary myeloma cells in SCID-HU mice and in coculture with osteoclasts. Leukemia 2008, 22, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Rossi, J.F.; Moreaux, J.; Hose, D.; Requirand, G.; Rose, M.; Rouille, V.; Nestorov, I.; Mordenti, G.; Goldschmidt, H.; Ythier, A.; et al. Atacicept in relapsed/refractory multiple myeloma or active waldenstrom’s macroglobulinemia: A phase I study. Br. J. Cancer 2009, 101, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Malek, E.; de Lima, M.; Letterio, J.J.; Kim, B.G.; Finke, J.H.; Driscoll, J.J.; Giralt, S.A. Myeloid-derived suppressor cells: The green light for myeloma immune escape. Blood Rev. 2016. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Hujaily, E.M.; Oldham, R.A.A.; Hari, P.; Medin, J.A. Development of Novel Immunotherapies for Multiple Myeloma. Int. J. Mol. Sci. 2016, 17, 1506. https://doi.org/10.3390/ijms17091506
Al-Hujaily EM, Oldham RAA, Hari P, Medin JA. Development of Novel Immunotherapies for Multiple Myeloma. International Journal of Molecular Sciences. 2016; 17(9):1506. https://doi.org/10.3390/ijms17091506
Chicago/Turabian StyleAl-Hujaily, Ensaf M., Robyn A. A. Oldham, Parameswaran Hari, and Jeffrey A. Medin. 2016. "Development of Novel Immunotherapies for Multiple Myeloma" International Journal of Molecular Sciences 17, no. 9: 1506. https://doi.org/10.3390/ijms17091506
APA StyleAl-Hujaily, E. M., Oldham, R. A. A., Hari, P., & Medin, J. A. (2016). Development of Novel Immunotherapies for Multiple Myeloma. International Journal of Molecular Sciences, 17(9), 1506. https://doi.org/10.3390/ijms17091506