
DNA Interaction Studies of Selected Polyamine Conjugates

 , and
, and 
Abstract
:
1. Introduction
2. Results
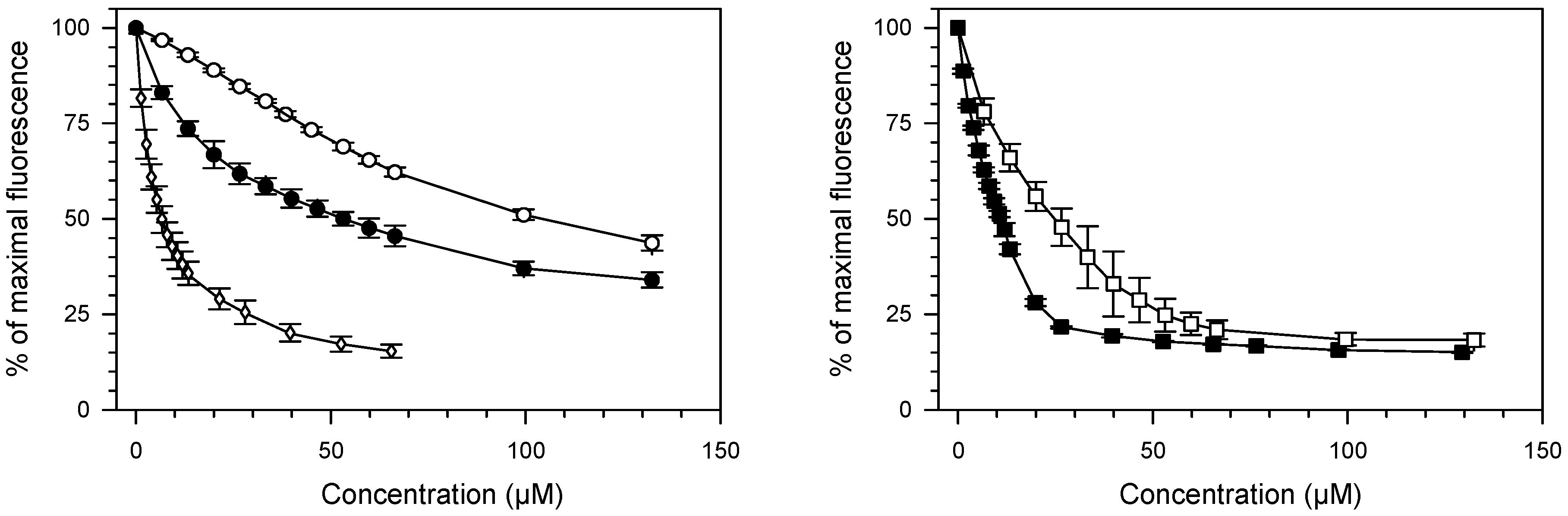
2.1. Ethidium Bromide Displacement Assay
2.2. Thermal Melting Studies
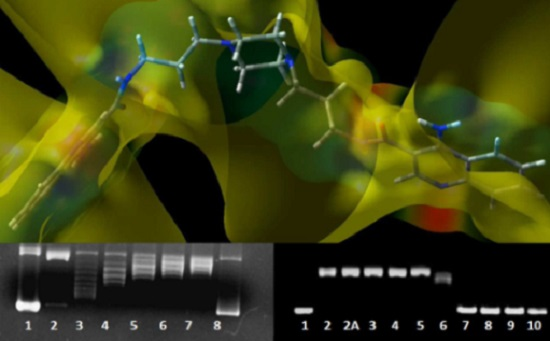
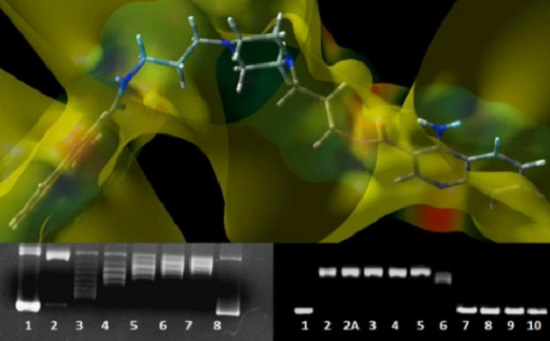
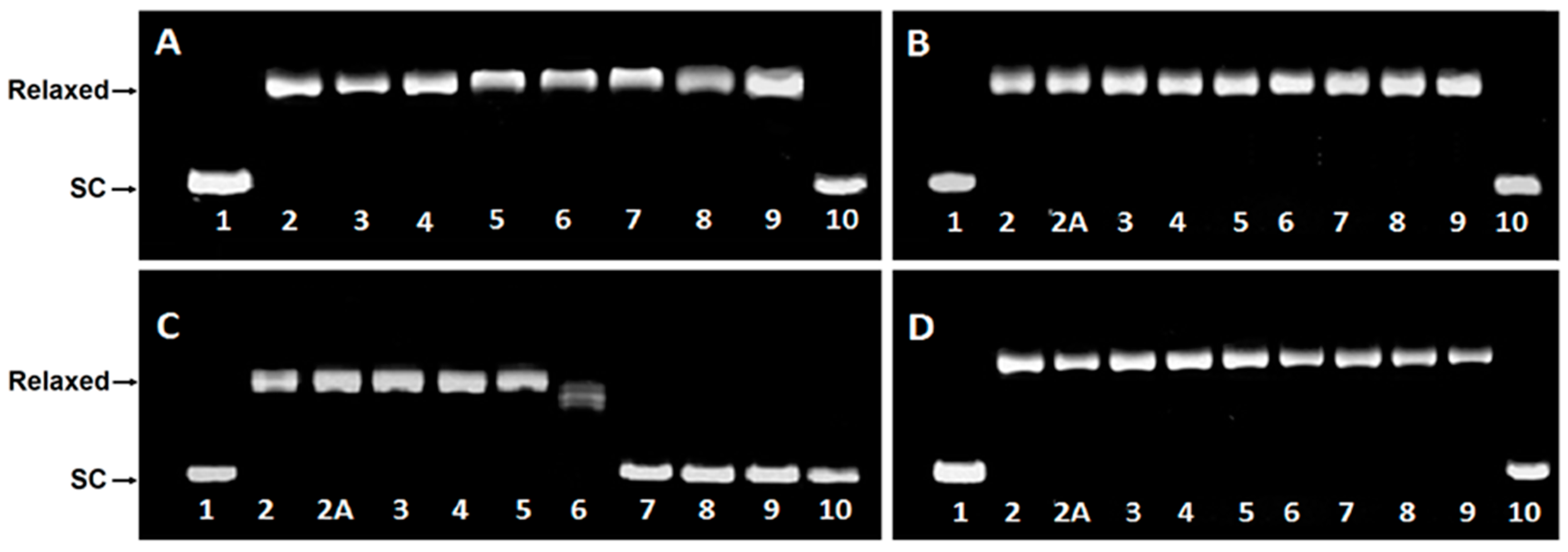
2.3. DNA Unwinding Assay
2.4. Topoisomerase I Activity Assay
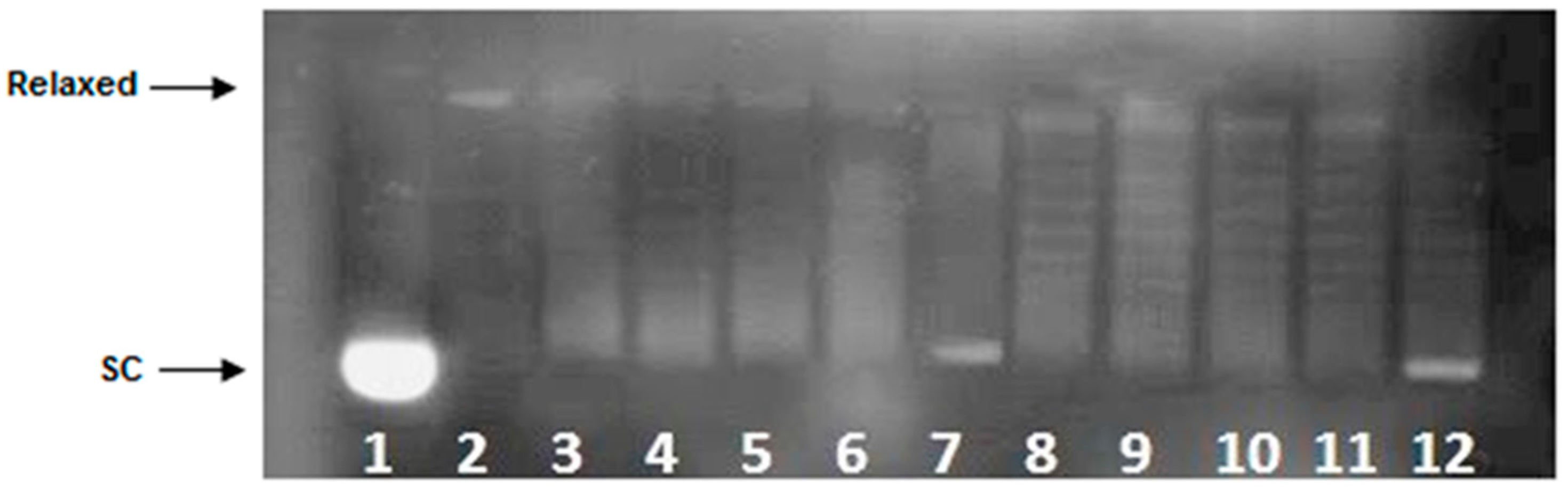
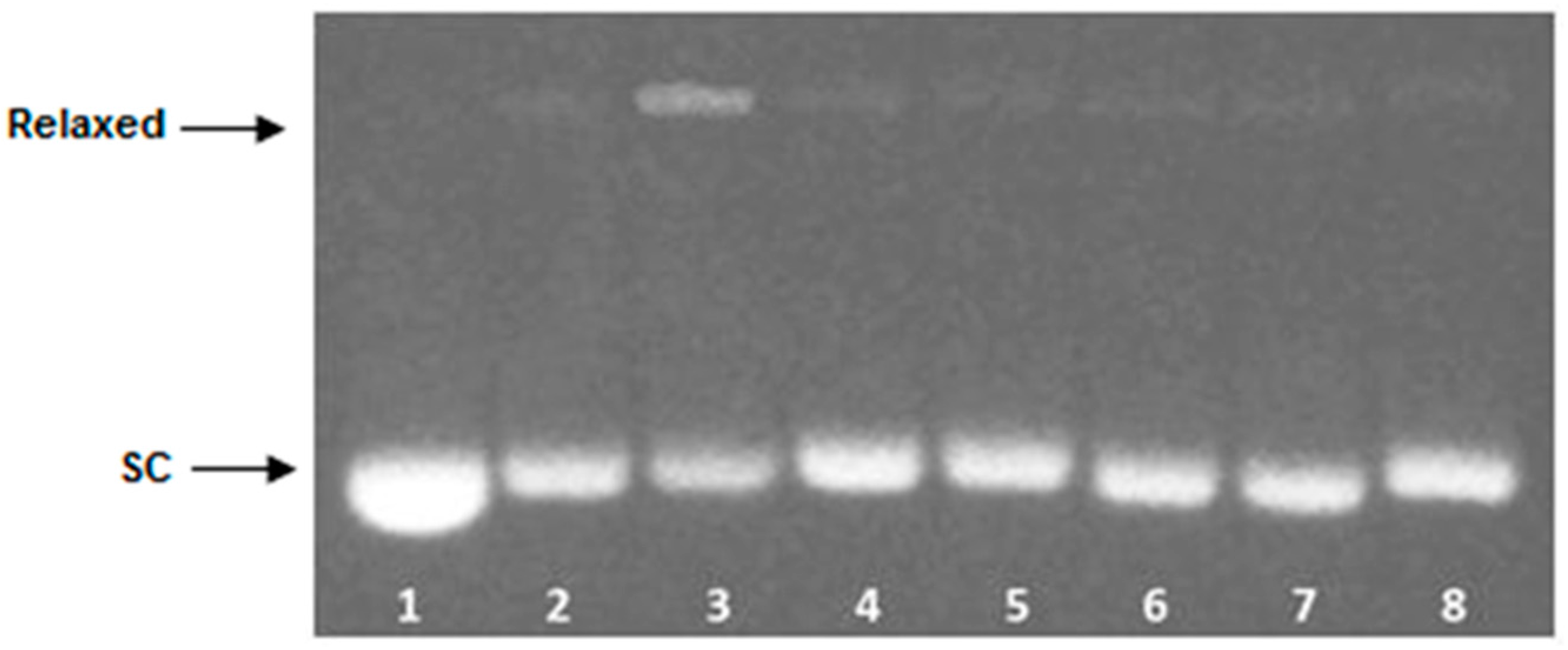
2.5. Topoisomerase II DNA Cleavage Assay
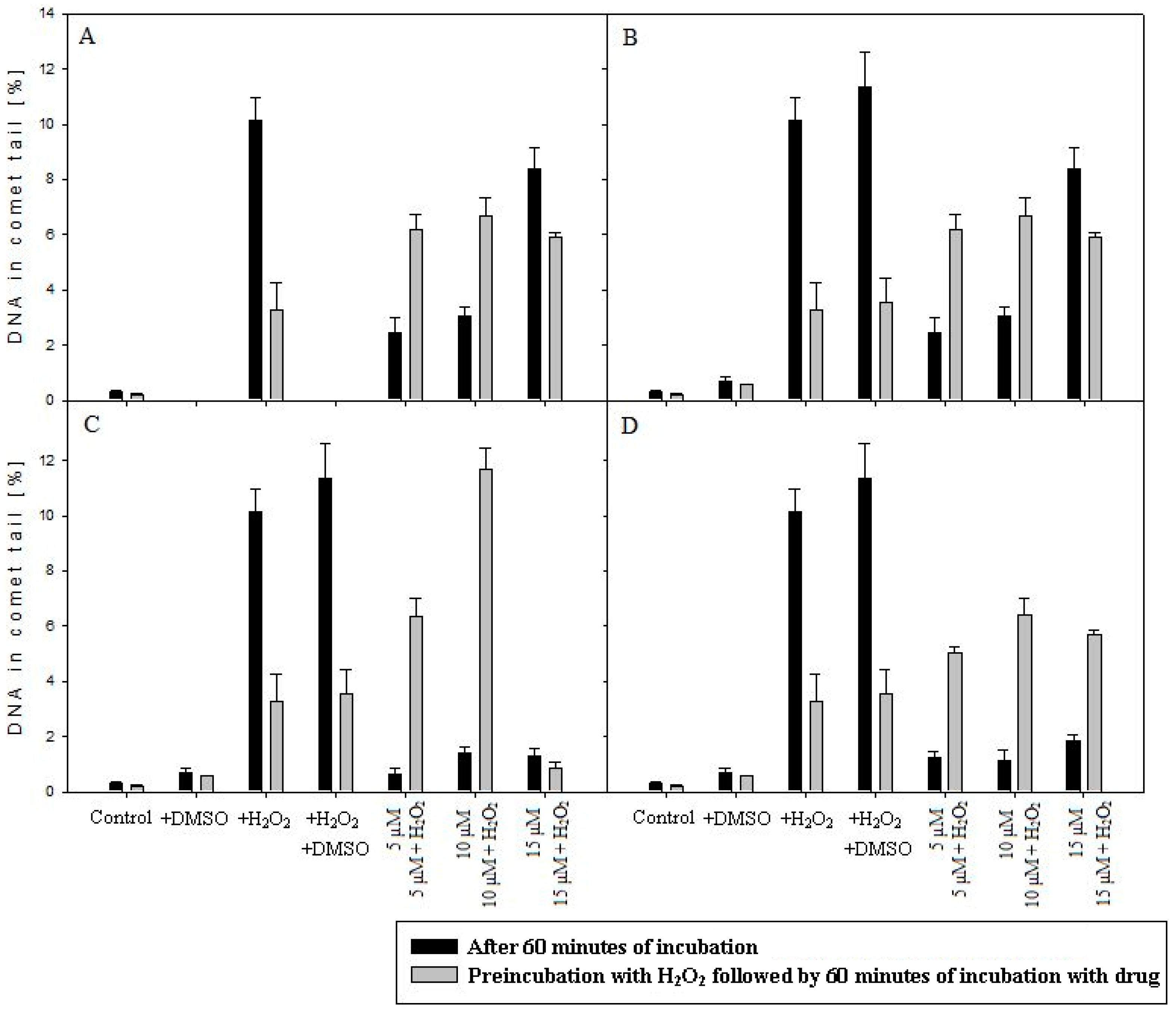
2.6. Comet Assay
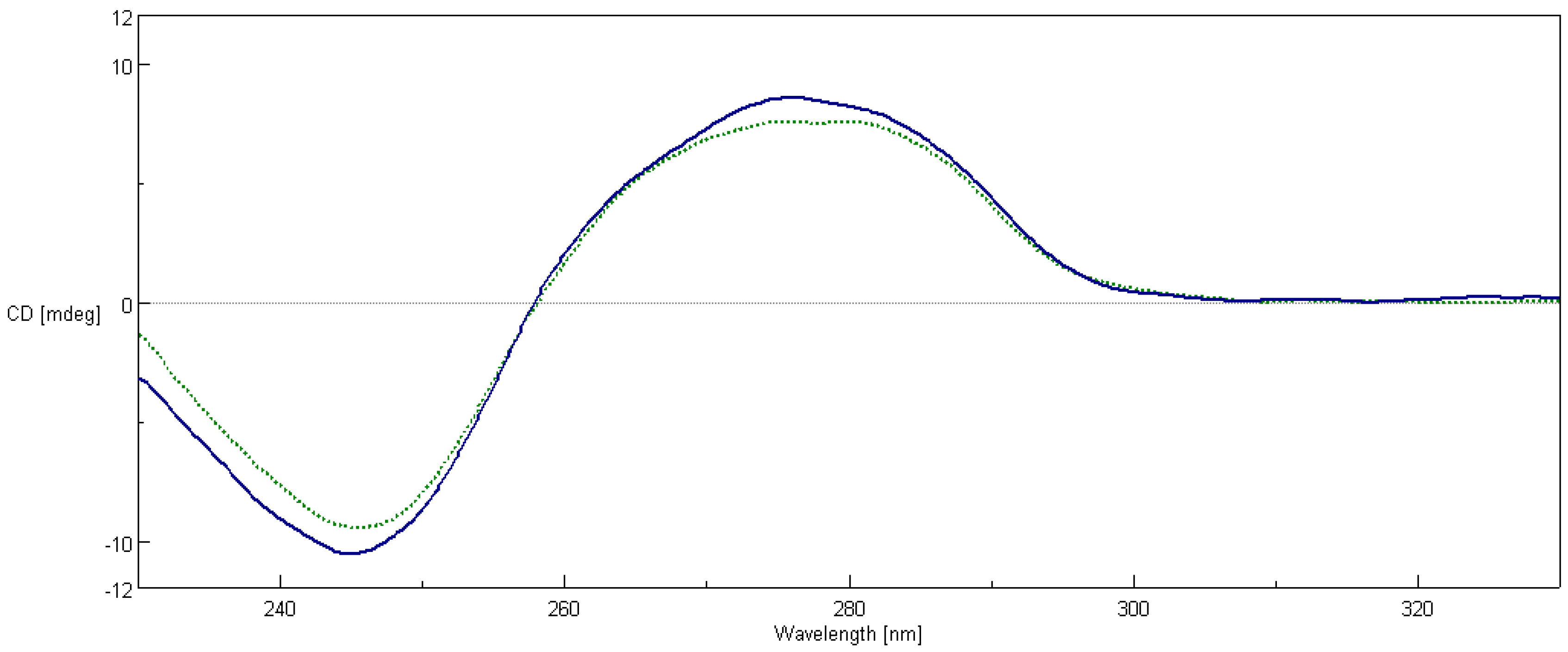
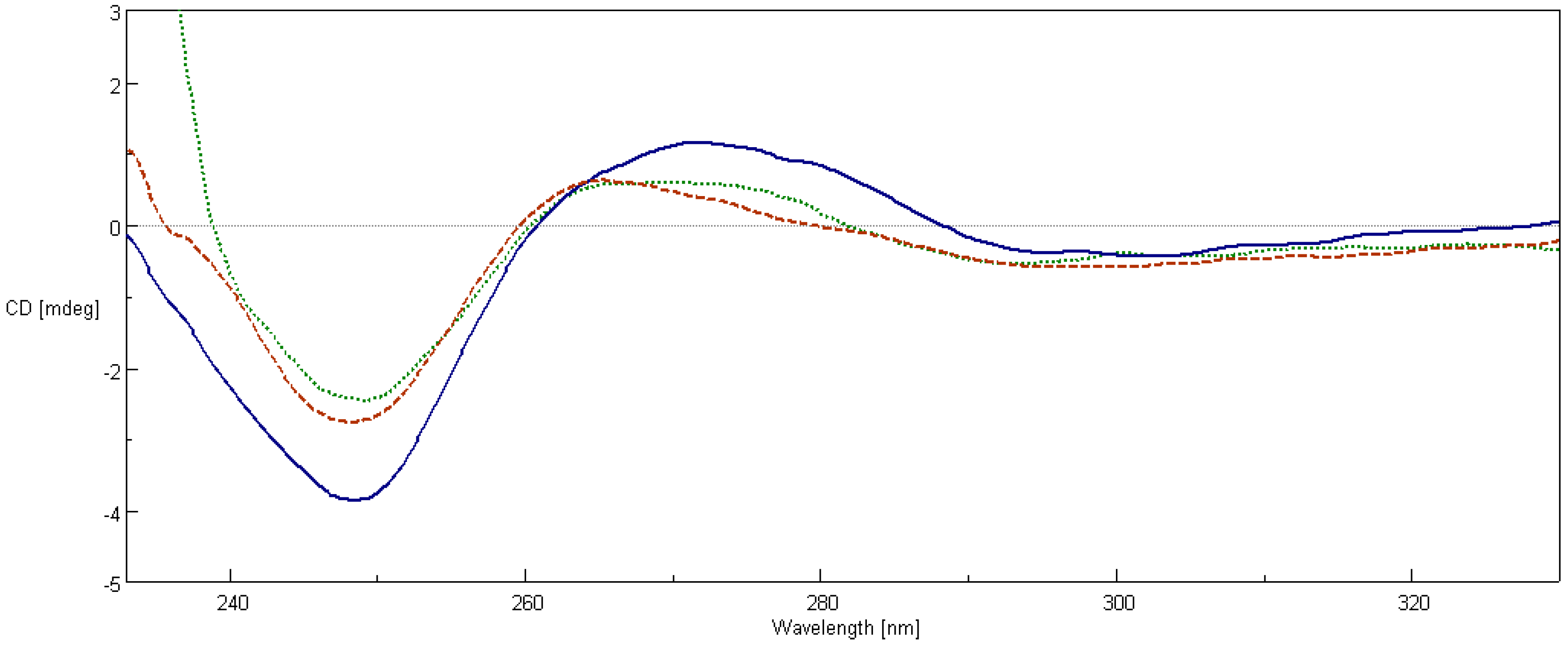
2.7. Circular Dichroism Spectroscopy
3. Discussion
4. Materials and Methods
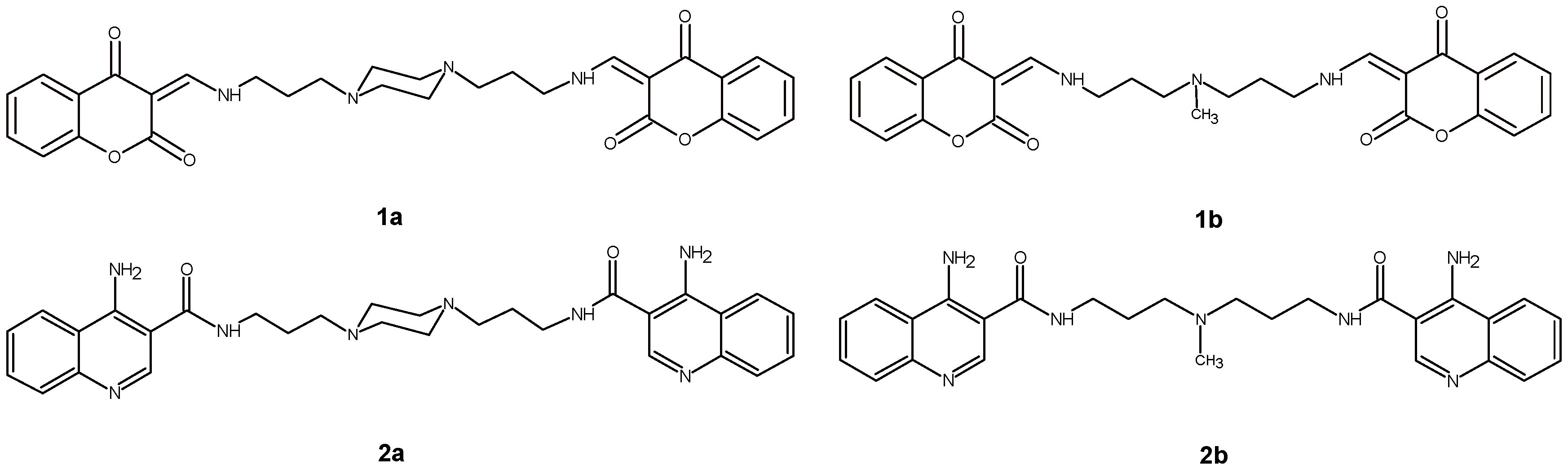
4.1. Examined Compounds
4.2. Ethidium Bromide Displacement Assay
4.3. Thermal Melting Studies
- 1: 5′-AAATTAATATGTATTGTATATAAATTATT-3′
- 2: 3′-TTTAATTATACATAACATATATTTAATAA-5′
4.4. DNA Unwinding Assay
4.4.1. Strains and Media
4.4.2. Bacterial Culture and Plasmid Isolation
4.5. Topoisomerase I Activity Assay
4.6. Topoisomerase II DNA Cleavage Assay
4.7. Comet Assay
4.8. Circular Dichroism Spectroscopy
4.9. Computational Methodology
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 9AA | 9-aminoacridine |
| B3LYP | Becke-Lee-Yang_Parr hybrid method |
| BSA | Bovine Serum Albumin |
| C50 | the concentration of added compound required to reduce the fluorescence of the DNA/EtBr complex to 50% |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DFT | density functional theory |
| DMSO | dimethyl sulfoxide |
| dsDNA | double stranded Deoxyribonucleic acid |
| DTT | Dithiothreitol |
| ct-DNA | Calf thymus DNA |
| EDTA | Ethylenediaminetetraacetic acid (as disodium salt) |
| EtBr | Ethidium Bromide |
| GGA | generalized gradient approximation |
| Kapp | apparent binding constant of a compound |
| NBO | natural bond order |
| NPA | natural population analyses |
| Tm | melting temperature |
| Topo I | topoisomerase I |
| Tris | Tris(hydroxymethyl)aminomethane |
| TBS | Tris-buffered saline |
References
- Cheung-Ong, K.; Giaever, G.; Nislow, C. DNA-damaging agents in cancer chemotherapy: Serendipity and chemical biology. Chem. Biol. 2013, 20, 648–659. [Google Scholar] [CrossRef] [PubMed]
- Hurley, L.H. DNA and its associated processes as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Brana, M.F.; Cacho, M.; Gradillas, A.; de Pascual-Teresa, B.; Ramos, A. Intercalators as anticancer drugs. Curr. Pharm. Des. 2001, 7, 1745–1780. [Google Scholar] [CrossRef] [PubMed]
- Markovits, J.; Pommier, Y.; Mattern, M.R.; Esnault, C.; Roques, B.P.; Le Pecq, J.B.; Kohn, K.W. Effects of the bifunctional antitumor intercalator ditercalinium on DNA in mouse leukemia L1210 cells and DNA topoisomerase II. Cancer Res. 1986, 46, 5821–5826. [Google Scholar] [PubMed]
- Zhang, G.S.; Fang, L.Y.; Zhu, L.Z.; Sun, D.X.; Wang, P.G. Syntheses and biological activity of bisdaunorubicins. Bioorg. Med. Chem. 2006, 14, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Chaires, J.B.; Leng, F.F.; Przewloka, T.; Fokt, I.; Ling, Y.H.; PerezSoler, R.; Priebe, W. Structure-based design of a new bisintercalating anthracycline antibiotic. J. Med. Chem. 1997, 40, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Martinez, R.; Chacon-Garcia, L. The search of DNA-intercalators as antitumoral drugs: What worked and what did not work. Curr. Med. Chem. 2005, 12, 127–151. [Google Scholar] [CrossRef] [PubMed]
- Romerdahl, C.A.; Brana, M.F. Bis-Naphthalimides. In Cancer Therapeutics: Experimental and Clinical Agents; Teicher, B.A., Ed.; Humana Press: New York, NY, USA, 1997; Chapter 7; pp. 215–226. [Google Scholar]
- Le Pecq, J.B.; Le Bret, M.; Barbet, J.; Roques, B. DNA polyintercalating drugs: DNA binding of diacridine derivatives. Proc. Natl. Acad. Sci. USA 1975, 72, 2915–2919. [Google Scholar] [CrossRef] [PubMed]
- Gaugain, B.; Barbet, J.; Capelle, N.; Roques, B.P.; Le Pecq, J.B.; Lebret, M. DNA bifunctional intercalators. 2. Fluorescence properties and DNA binding interaction of an ethidium homodimer and an acridine ethidium heterodimer. Biochemistry 1978, 17, 5078–5088. [Google Scholar] [CrossRef] [PubMed]
- Cholody, W.M.; Hernandez, L.; Hassner, L.; Scudiero, D.A.; Djurickovic, D.B.; Michejda, C.J. Bisimidazoacridones and related-compounds—New antineoplastic agents with high selectivity against colon tumors. J. Med. Chem. 1995, 38, 3043–3052. [Google Scholar] [CrossRef] [PubMed]
- Stanczak, A.; Szumilak, M. Bisinterkalatory jako potencjalne leki przeciwnowotworowe. Wiad. Chem. 2009, 63, 847–875. [Google Scholar]
- Thomas, T.; Thomas, T.J. Polyamines in cell growth and cell death: Molecular mechanisms and therapeutic applications. Cell. Mol. Life Sci. 2001, 58, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Bloomfield, V.A.; Benevides, J.M.; Thomas, G.J. Structural basis of polyamine-DNA recognition: Spermidine and spermine interactions with genomic B-DNAs of different GC content probed by Raman spectroscopy. Nucleic Acids Res. 2000, 28, 3379–3385. [Google Scholar] [CrossRef] [PubMed]
- N’Soukpoe-Kossi, C.N.; Ouameur, A.A.; Thomas, T.; Shirahata, A.; Thomas, T.J.; Tajmir-Riahi, H.A. DNA interaction with antitumor polyamine analogues: A comparison with biogenic polyamines. Biomacromolecules 2008, 9, 2712–2718. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.J.; Wallace, H.M. The polyamine transport system as a target for anticancer drug development. Amino Acids 2010, 38, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Vijayanathan, V.; Thomas, T.; Shirahata, A.; Thomas, T.J. DNA condensation by polyamines: A laser light scattering study of structural effects. Biochemistry 2001, 40, 13644–13651. [Google Scholar] [CrossRef] [PubMed]
- Vijayanathan, V.; Thomas, T.; Antony, T.; Shirahata, A.; Thomas, T.J. Formation of DNA nanoparticles in the presence of novel polyamine analogues: A laser light scattering and atomic force microscopic study. Nucleic Acids Res. 2004, 32, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.J.; Tajmir-Riahi, H.A.; Thomas, T. Polyamine-DNA interactions and development of gene delivery vehicles. Amino Acids 2016. [Google Scholar] [CrossRef] [PubMed]
- Todd, B.A.; Parsegian, V.A.; Shirahata, A.; Thomas, T.J.; Rau, D.C. Attractive forces between cation condensed DNA double helices. Biophys. J. 2008, 94, 4775–4782. [Google Scholar] [CrossRef] [PubMed]
- Vijayanathan, V.; Thomas, T.; Thomas, T.J. DNA nanoparticles and development of DNA delivery vehicles for gene therapy. Biochemistry 2002, 41, 14085–14094. [Google Scholar] [CrossRef] [PubMed]
- Vijayanathan, V.; Lyall, J.; Thomas, T.; Shirahata, A.; Thomas, T.J. Ionic, structural, and temperature effects on dna nanoparticles formed by natural and synthetic polyamines. Biomacromolecules 2005, 6, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Vijayanathan, V.; Agostinelli, E.; Thomas, T.; Thomas, T.J. Innovative approaches to the use of polyamines for DNA nanoparticle preparation for gene therapy. Amino Acids 2014, 46, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Agostinelli, E.; Vianello, F.; Magliulo, G.; Thomas, T.; Thomas, T.J. Nanoparticle strategies for cancer therapeutics: Nucleic acids, polyamines, bovine serum amine oxidase and iron oxide nanoparticles (review). Int. J. Oncol. 2015, 46, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Cullis, P.M.; Green, R.E.; Merson-Davies, L.; Travis, N. Probing the mechanism of transport and compartmentalisation of polyamines in mammalian cells. Chem. Biol. 1999, 6, 717–729. [Google Scholar] [CrossRef]
- Wallace, H.M.; Fraser, A.V.; Hughes, A. A perspective of polyamine metabolism. Biochem. J. 2003, 376, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wallace, H.M.; Niiranen, K. Polyamine analogues—An update. Amino Acids 2007, 33, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Christensen, U.B.; Wamberg, M.; El-Essawy, F.A.G.; Ismail, A.E.H.; Nielsen, C.B.; Filichev, V.V.; Jessen, C.H.; Petersen, M.; Pedersen, E.B. Intercalating nucleic acids: The influence of linker length and intercalator type on their duplex stabilities. Nucleoside Nucleotides Nucleic 2004, 23, 207–225. [Google Scholar] [CrossRef]
- Ferguson, L.R.; Denny, W.A. Genotoxicity of non-covalent interactions: DNA intercalators. Mutat. Res. 2007, 623, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Szumilak, M.; Lewgowd, W.; Stanczak, A. In silico ADME studies of polyamine conjugates as potential anticancer drugs. Acta Pol. Pharm. 2016, 73, 717–723. [Google Scholar]
- Szulawska-Mroczek, A.; Szumilak, M.; Szczesio, M.; Olczak, A.; Nazarski, R.B.; Lewgowd, W.; Czyz, M.; Stanczak, A. Synthesis and biological evaluation of new bischromone derivatives with antiproliferative activity. Arch. Pharm. 2013, 346, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Szumilak, M.; Szulawska-Mroczek, A.; Koprowska, K.; Stasiak, M.; Lewgowd, W.; Stanczak, A.; Czyz, M. Synthesis and in vitro biological evaluation of new polyamine conjugates as potential anticancer drugs. Eur. J. Med. Chem. 2010, 45, 5744–5751. [Google Scholar] [CrossRef] [PubMed]
- Szumilak, M.; Galdyszynska, M.; Dominska, K.; Stanczak, A.; Piastowska-Ciesielska, A. Antitumor activity of polyamine conjugates in human prostate and breast cancer. Acta Biochim. Pol. 2016, in press. [Google Scholar]
- Palchaudhuri, R.; Hergenrother, P.J. DNA as a target for anticancer compounds: Methods to determine the mode of binding and the mechanism of action. Curr. Opin. Biotechnol. 2007, 18, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Cain, B.F.; Baguley, B.C.; Denny, W.A. Potential antitumor agents. 28. Deoxyribonucleic-acid polyintercalating agents. J. Med. Chem. 1978, 21, 658–668. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, T.C. Optical absorbance and fluorescence techniques for measuring DNA-drug interactions. In Drug-DNA Interaction Protocols; Fox, K.R., Ed.; Humana Press: Totowa, NY, USA, 1997; Volume 90, pp. 195–218. [Google Scholar]
- Guedin, A.; Lacroix, L.; Mergny, J.-L. Thermal melting studies of ligand DNA interactions. In Drug-DNA Interaction Protocols, 2nd ed.; Fox, K.R., Ed.; Humana Press: New York, NY, USA, 2010; Volume 613, pp. 25–35. [Google Scholar]
- Gentry, A.C.; Juul, S.; Veigaard, C.; Knudsen, B.R.; Osheroff, N. The geometry of DNA supercoils modulates the DNA cleavage activity of human topoisomerase I. Nucleic Acids Res. 2011, 39, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Sappal, D.S.; McClendon, A.K.; Fleming, J.A.; Thoroddsen, V.; Connolly, K.; Reimer, C.; Blackman, R.K.; Bulawa, C.E.; Osheroff, N.; Charlton, P. Biological characterization of MLN944: A potent DNA binding agent. Mol. Cancer Ther. 2004, 3, 47–58. [Google Scholar] [PubMed]
- Lodish, H.; Berk, A.; Xipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. The Role of topoisomerases in DNA replication. In Molecular Cell Biology, 4th ed.W. H. Freeman: New York, NY, USA, 2000; Chapter 12.3. Available online: http://www.ncbi.nlm.nih.gov/books/NBK21703/ (accessed on 1 July 2016). [Google Scholar]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Burden, D.A.; Kingma, P.S.; Froelich-Ammon, S.J.; Bjornsti, M.A.; Patchan, M.W.; Thompson, R.B.; Osheroff, N. Topoisomerase II etoposide interactions direct the formation of drug-induced enzyme-DNA cleavage complexes. J. Biol. Chem. 1996, 271, 29238–29244. [Google Scholar] [PubMed]
- Tabatabaee, M.; Bordbar, M.; Ghassemzadeh, M.; Tahriri, M.; Tahrir, M.; Lighvan, Z.M.; Neumüller, B. Two new neutral copper(II) complexes with dipicolinic acid and 3-amino-1H-1,2,4-triazole formed under different reaction conditions: Synthesis, characterization, molecular structures and DNA-binding studies. Eur. J. Med. Chem. 2003, 70, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Maheswari, P.U.; Palaniandavar, M. DNA binding and cleavage properties of certain tetrammine ruthenium(II) complexes of modified 1,10-phenanthrolines—Effect of hydrogen-bonding on DNA-binding affinity. J. Inorg. Biochem. 2004, 98, 219–230. [Google Scholar] [CrossRef]
- Norden, B.; Tjerneld, F. Structure of methylene blue-DNA complexes studied by linear and circular dichroism spectroscopy. Biopolymers 1982, 21, 1713–1734. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zheng, K.C.; Chen, Y.; Li, Y.Z.; Lin, L.J.; Li, H.; Zhang, P.X.; Ji, L.N. Effects of ligand planarity on the interaction of polypyridyl Ru(II) complexes with DNA. Dalton Trans. 2003, 11, 2260–2268. [Google Scholar] [CrossRef]
- Tidd, D.M.; Broughton, C.M.; Clark, R.E. Cpg oligodeoxynucleotide 5mer-induced apoptosis in MOLT-4 leukemia cells does not require caspase 3 or new protein synthesis. Nucleic Acids Res. 2003, 31, 2297–2304. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.P.; McCoy, M.T.; Tice, R.R.; Schneider, E.L. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp. Cell Res. 1988, 175, 184–191. [Google Scholar] [CrossRef]
- Kibbe, W.A. OligoCalc: An online oligonucleotide properties calculator. Nucleic Acids Res. 2007, 27, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. 3. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Wiberg, K.B.; Radom, L.; Schleyer, P.V.R.; Pople, J.A. Ab initio molecular orbital theory. J. Comput. Chem. 1986, 7, 379–379. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.W.; Schlegel, H.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, T.; Kudin, K.N.; Burant, J.C. Gaussian 03, Revision C. 02; Gaussian, Inc.: Wallingford, CT, USA, 2008. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | * C50 ± SD | † Kapp × 106 |
|---|---|---|
| (µM) | (M−1) | |
| 1a | 52.90 ± 3.18 | 0.23 |
| 1b | 104.67 ± 5.01 | 0.11 |
| 2a | 2.81 ± 0.53 | 4.26 |
| 2b | 11.10 ± 0.36 | 1.08 |
| 9AA | 6.62 ± 0.93 | 1.81 |
| Additive | Oligonucleotide | Melting Temperature, Tm (°C) |
|---|---|---|
| None (negative control) | dsDNA | 64.97 ± 0.20 |
| 1a | dsDNA | 64.02 ± 0.00 |
| 1b | dsDNA | 64.07 ± 0.14 |
| 2a | dsDNA | 70.7 ± 0.71 |
| 2b | dsDNA | 64.5 ± 0.71 |
| 9AA (positive control) | dsDNA | 78.00 ± 0.00 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szumilak, M.; Merecz, A.; Strek, M.; Stanczak, A.; Inglot, T.W.; Karwowski, B.T. DNA Interaction Studies of Selected Polyamine Conjugates. Int. J. Mol. Sci. 2016, 17, 1560. https://doi.org/10.3390/ijms17091560
Szumilak M, Merecz A, Strek M, Stanczak A, Inglot TW, Karwowski BT. DNA Interaction Studies of Selected Polyamine Conjugates. International Journal of Molecular Sciences. 2016; 17(9):1560. https://doi.org/10.3390/ijms17091560
Chicago/Turabian StyleSzumilak, Marta, Anna Merecz, Malgorzata Strek, Andrzej Stanczak, Tadeusz W. Inglot, and Boleslaw T. Karwowski. 2016. "DNA Interaction Studies of Selected Polyamine Conjugates" International Journal of Molecular Sciences 17, no. 9: 1560. https://doi.org/10.3390/ijms17091560
APA StyleSzumilak, M., Merecz, A., Strek, M., Stanczak, A., Inglot, T. W., & Karwowski, B. T. (2016). DNA Interaction Studies of Selected Polyamine Conjugates. International Journal of Molecular Sciences, 17(9), 1560. https://doi.org/10.3390/ijms17091560