
Pathophysiology and the Monitoring Methods for Cardiac Arrest Associated Brain Injury

 , ,
, ,
Abstract
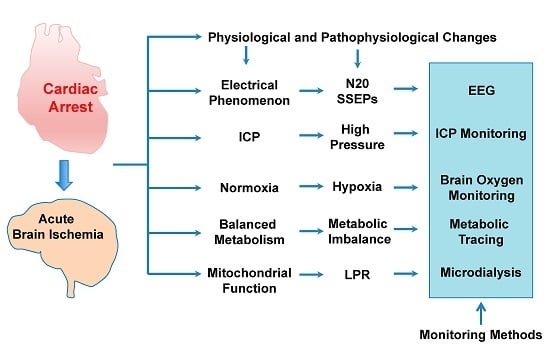
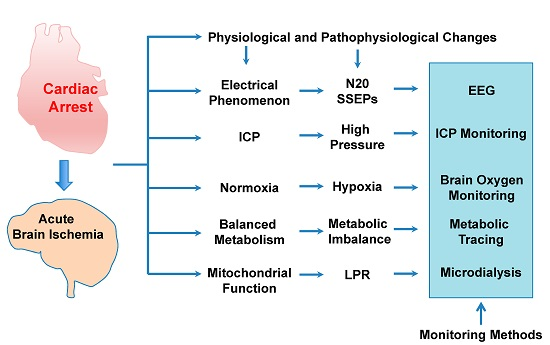
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
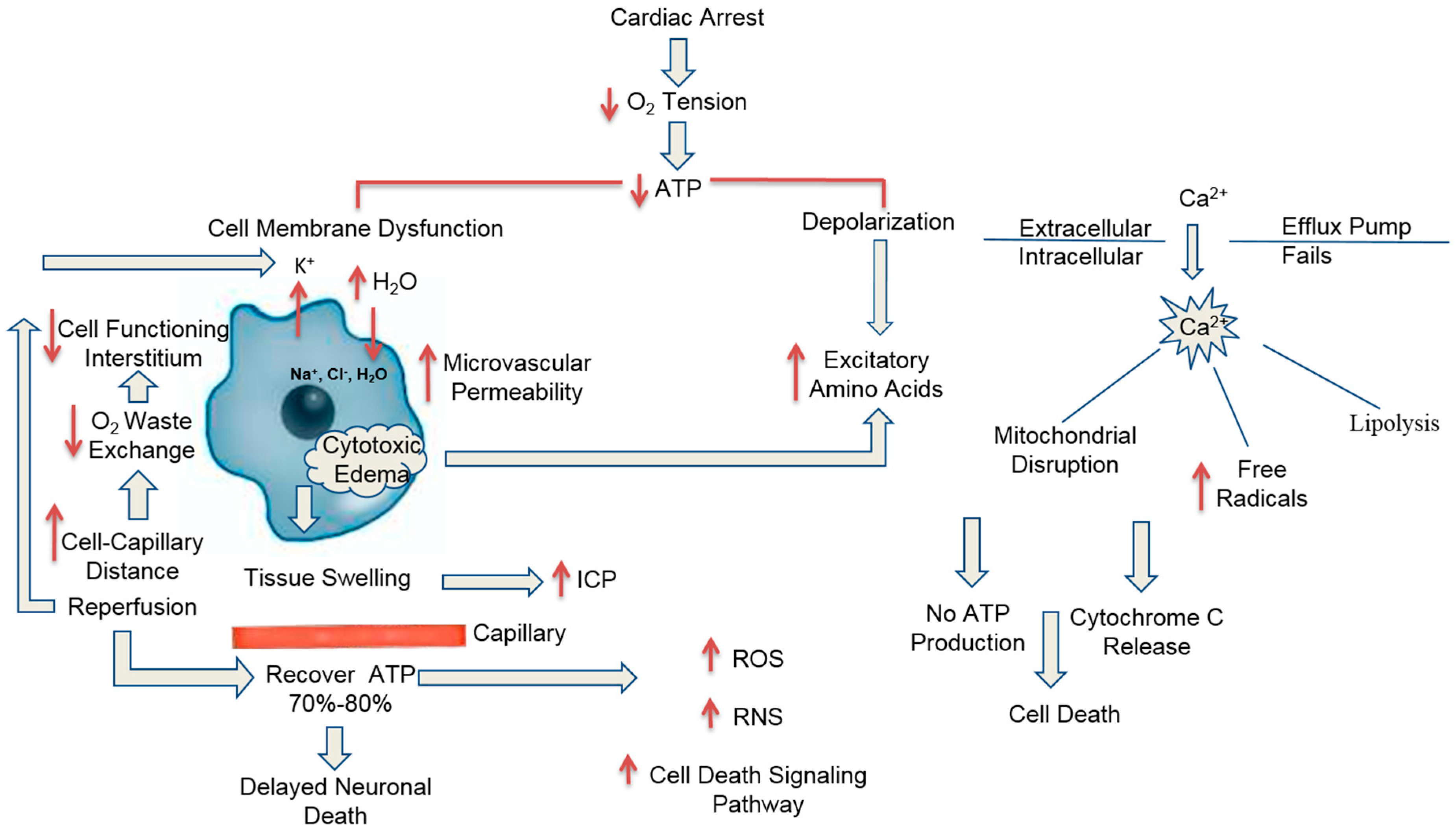
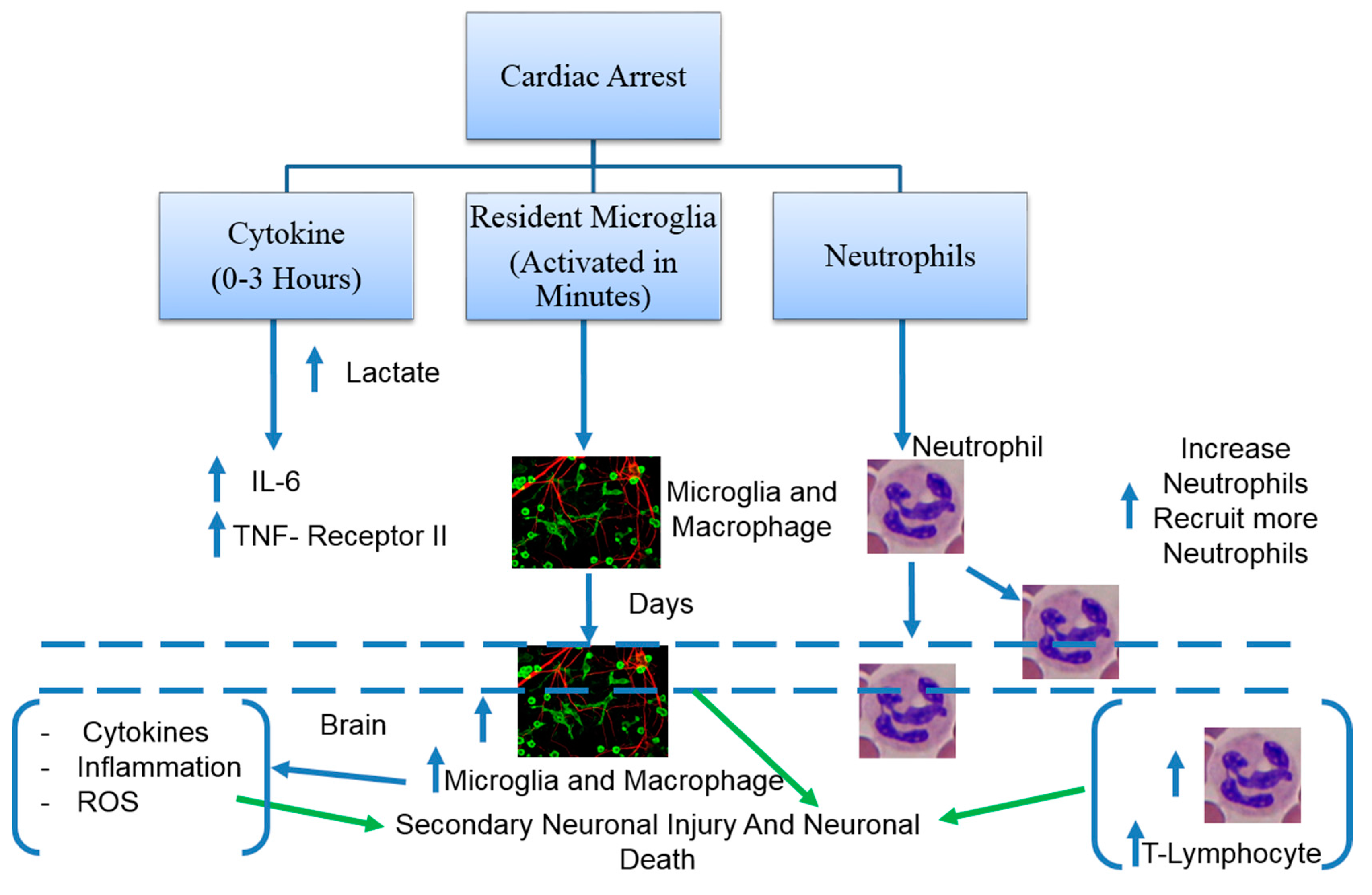
3. Pathophysiology
4. Electrophysiologic Monitoring of Brain Injury after Cardiac Arrest
5. Intracranial Pressure Monitoring and Cerebral Autoregulation
6. Brain Oxygen Monitoring
7. Metabolic Imaging Modalities for Cardiac Arrest Brain Injury
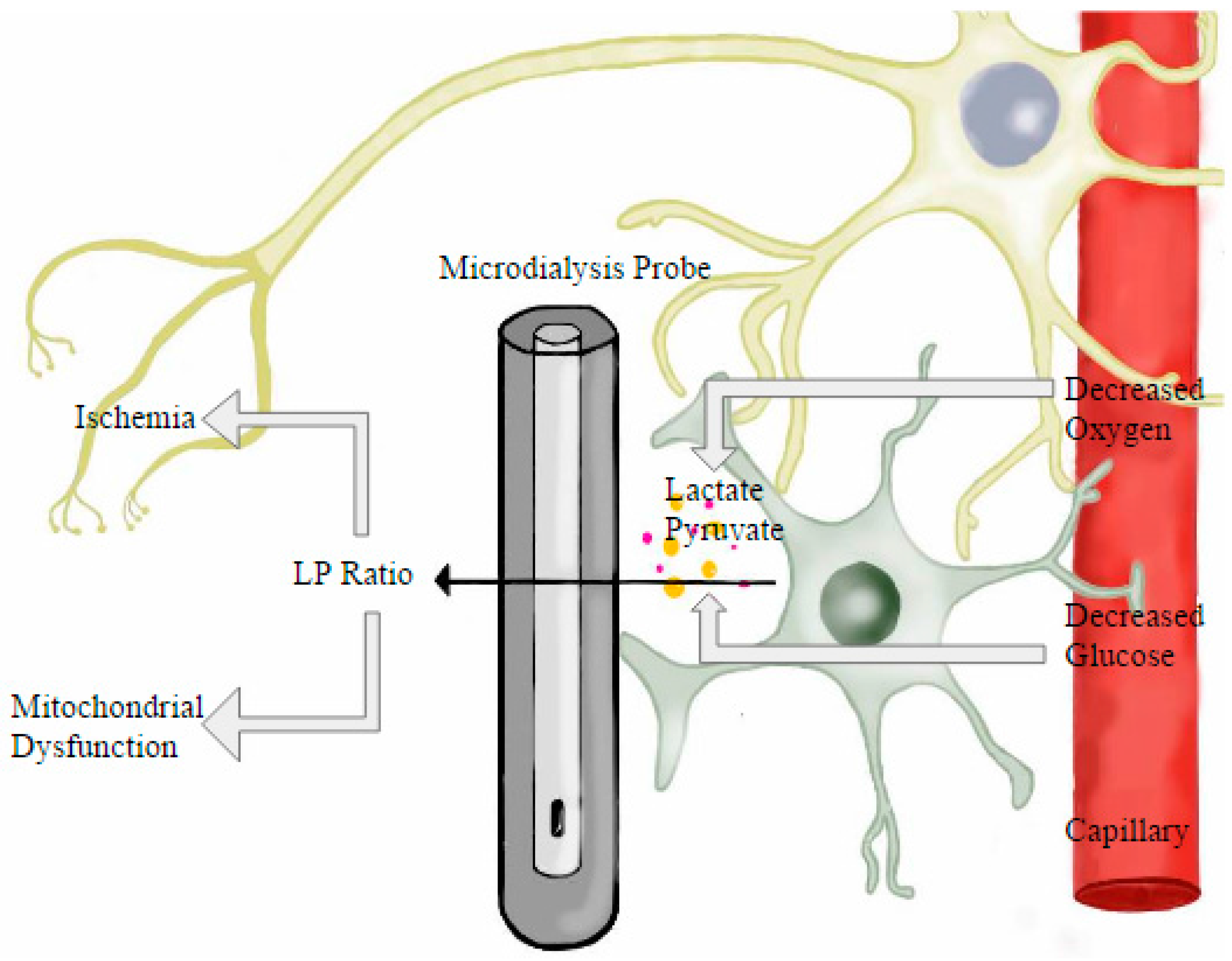
8. Microdialysis
9. Conclusions
Conflicts of Interest
References
- Berdowski, J.; Berg, R.A.; Tijssen, J.G.; Koster, R.W. Global incidences of out-of-hospital cardiac arrest and survival rates: Systematic review of 67 prospective studies. Resuscitation 2010, 81, 1479–1487. [Google Scholar] [CrossRef] [PubMed]
- Fugate, J.E.; Brinjikji, W.; Mandrekar, J.N.; Cloft, H.J.; White, R.D.; Wijdicks, E.F.; Rabinstein, A.A. Post-cardiac arrest mortality is declining: A study of the US national inpatient sample 2001 to 2009. Circulation 2012, 126, 546–550. [Google Scholar] [CrossRef]
- Rossetti, A.O.; Rabinstein, A.A.; Oddo, M. Neurological prognostication of outcome in patients in coma after cardiac arrest. Lancet Neurol. 2016, 15, 597–609. [Google Scholar] [CrossRef]
- Horowitz, R.E.; Naritoku, W.Y. The autopsy as a performance measure and teaching tool. Hum. Pathol. 2007, 38, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Scordi-Bello, I.A.; Kalb, T.H.; Lento, P.A. Clinical setting and extent of premortem evaluation do not predict autopsy discrepancy rates. Mod. Pathol. 2010, 23, 1225–1230. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Kass, D.A. Heart failure with preserved ejection fraction: Mechanisms, clinical features, and therapies. Circ. Res. 2014, 115, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Hossmann, K.A.; Grosse Ophoff, B. Recovery of monkey brain after prolonged ischemia. I. Electrophysiology and brain electrolytes. J. Cereb. Blood Flow Metab. 1986, 6, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Cavus, E.; Bein, B.; Dorges, V.; Stadlbauer, K.H.; Wenzel, V.; Steinfath, M.; Hanss, R.; Scholz, J. Brain tissue oxygen pressure and cerebral metabolism in an animal model of cardiac arrest and cardiopulmonary resuscitation. Resuscitation 2006, 71, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Eleff, S.M.; Maruki, Y.; Monsein, L.H.; Traystman, R.J.; Bryan, R.N.; Koehler, R.C. Sodium, ATP, and intracellular pH transients during reversible complete ischemia of dog cerebrum. Stroke 1991, 22, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Nishijima, M.K.; Koehler, R.C.; Hurn, P.D.; Eleff, S.M.; Norris, S.; Jacobus, W.E.; Traystman, R.J. Postischemic recovery rate of cerebral ATP, phosphocreatine, pH, and evoked potentials. Am. J. Physiol. 1989, 257, H1860–H1870. [Google Scholar] [PubMed]
- Rincon, F.; Mayer, S.A. Therapeutic hypothermia for brain injury after cardiac arrest. Semin. Neurol. 2006, 26, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.J. Effect of anoxia on ion distribution in the brain. Physiol. Rev. 1985, 65, 101–148. [Google Scholar] [PubMed]
- Siesjo, B.K.; Katsura, K.; Zhao, Q.; Folbergrova, J.; Pahlmark, K.; Siesjo, P.; Smith, M.L. Mechanisms of secondary brain damage in global and focal ischemia: A speculative synthesis. J. Neurotrauma 1995, 12, 943–956. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Ibarra, F.P.; Varon, J.; Lopez-Meza, E.G. Therapeutic hypothermia: Critical review of the molecular mechanisms of action. Front. Neurol. 2011, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Katsura, K.; Rodriguez de Turco, E.B.; Folbergrova, J.; Bazan, N.G.; Siesjo, B.K. Coupling among energy failure, loss of ion homeostasis, and phospholipase A2 and C activation during ischemia. J. Neurochem. 1993, 61, 1677–1684. [Google Scholar] [CrossRef] [PubMed]
- Piantadosi, C.A.; Zhang, J. Mitochondrial generation of reactive oxygen species after brain ischemia in the rat. Stroke 1996, 27, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Kluck, R.M.; Bossy-Wetzel, E.; Green, D.R.; Newmeyer, D.D. The release of cytochrome c from mitochondria: A primary site for Bcl-2 regulation of apoptosis. Science 1997, 275, 1132–1136. [Google Scholar] [CrossRef] [PubMed]
- Broughton, B.R.; Reutens, D.C.; Sobey, C.G. Apoptotic mechanisms after cerebral ischemia. Stroke 2009, 40, e331–e339. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Vazquez, E.J.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Production of reactive oxygen species by mitochondria: Central role of complex III. J. Biol. Chem. 2003, 278, 36027–36031. [Google Scholar] [CrossRef] [PubMed]
- Lipton, P. Ischemic cell death in brain neurons. Physiol. Rev. 1999, 79, 1431–1568. [Google Scholar] [PubMed]
- Sugawara, T.; Fujimura, M.; Noshita, N.; Kim, G.W.; Saito, A.; Hayashi, T.; Narasimhan, P.; Maier, C.M.; Chan, P.H. Neuronal death/survival signaling pathways in cerebral ischemia. NeuroRx 2004, 1, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Zhang, S.; Arnold, T.C.; Alexander, J.S.; Huang, J.; Carden, D.L.; Conrad, S.A. Mild hypothermia induced before cardiac arrest reduces brain edema formation in rats. Acad. Emerg. Med. 2002, 9, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Gidday, J.M.; Park, T.S.; Gonzales, E.R.; Beetsch, J.W. CD18-dependent leukocyte adherence and vascular injury in pig cerebral circulation after ischemia. Am. J. Physiol. 1997, 272, H2622–H2629. [Google Scholar] [PubMed]
- Pluta, R.; Lossinsky, A.S.; Wisniewski, H.M.; Mossakowski, M.J. Early blood-brain barrier changes in the rat following transient complete cerebral ischemia induced by cardiac arrest. Brain Res. 1994, 633, 41–52. [Google Scholar] [CrossRef]
- Cristia, C.; Ho, M.L.; Levy, S.; Andersen, L.W.; Perman, S.M.; Giberson, T.; Salciccioli, J.D.; Saindon, B.Z.; Cocchi, M.N.; Donnino, M.W. The association between a quantitative computed tomography (CT) measurement of cerebral edema and outcomes in post-cardiac arrest—A validation study. Resuscitation 2014, 85, 1348–1353. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, M.; Okuchi, K.; Sakaki, T.; Hiramatsu, K.; Miyamoto, S.; Iwasaki, S. Specific changes in human brain following reperfusion after cardiac arrest. Stroke 1994, 25, 2091–2095. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, Y.; Kemmotsu, O.; Kitami, K.; Matsubara, I.; Tedo, I. Acute brain swelling after out-of-hospital cardiac arrest: Pathogenesis and outcome. Crit. Care Med. 1993, 21, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Dolinak, D.; Smith, C.; Graham, D.I. Global hypoxia per se is an unusual cause of axonal injury. Acta Neuropathol. 2000, 100, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, S.; Amiry-Moghaddam, M.; Ottersen, O.P.; Bhardwaj, A. Conivaptan, a Selective Arginine Vasopressin V1a and V2 Receptor Antagonist Attenuates Global Cerebral Edema Following Experimental Cardiac Arrest via Perivascular Pool of Aquaporin-4. Neurocrit. Care 2016, 24, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Kalimo, H.; Garcia, J.H.; Kamijyo, Y.; Tanaka, J.; Trump, B.F. The ultrastructure of “brain death”. II. Electron microscopy of feline cortex after complete ischemia. Virchows Arch. B Cell Pathol. 1977, 25, 207–220. [Google Scholar] [PubMed]
- Kirino, T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982, 239, 57–69. [Google Scholar] [CrossRef]
- Pulsinelli, W.A.; Brierley, J.B.; Plum, F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann. Neurol. 1982, 11, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, A.; Takeyama, N.; Nakatani, T.; Tanaka, T. Mitochondrial permeability transition and cytochrome c release in ischemia-reperfusion injury of the rat liver. J. Surg. Res. 2003, 111, 240–247. [Google Scholar] [CrossRef]
- Takeyama, N.; Miki, S.; Hirakawa, A.; Tanaka, T. Role of the mitochondrial permeability transition and cytochrome c release in hydrogen peroxide-induced apoptosis. Exp. Cell Res. 2002, 274, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Kuida, K.; Haydar, T.F.; Kuan, C.Y.; Gu, Y.; Taya, C.; Karasuyama, H.; Su, M.S.; Rakic, P.; Flavell, R.A. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell 1998, 94, 325–337. [Google Scholar] [CrossRef]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef]
- Yoshida, H.; Kong, Y.Y.; Yoshida, R.; Elia, A.J.; Hakem, A.; Hakem, R.; Penninger, J.M.; Mak, T.W. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell 1998, 94, 739–750. [Google Scholar] [CrossRef]
- Zou, H.; Henzel, W.J.; Liu, X.; Lutschg, A.; Wang, X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 1997, 90, 405–413. [Google Scholar] [CrossRef]
- Slee, E.A.; Harte, M.T.; Kluck, R.M.; Wolf, B.B.; Casiano, C.A.; Newmeyer, D.D.; Wang, H.G.; Reed, J.C.; Nicholson, D.W.; Alnemri, E.S.; et al. Ordering the cytochrome c-initiated caspase cascade: Hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J. Cell Biol. 1999, 144, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.; Du, C.; Wu, J.W.; Kyin, S.; Wang, X.; Shi, Y. Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature 2000, 406, 855–862. [Google Scholar] [PubMed]
- Miller, L.K. An exegesis of IAPs: Salvation and surprises from BIR motifs. Trends Cell Biol. 1999, 9, 323–328. [Google Scholar] [CrossRef]
- Namura, S.; Zhu, J.; Fink, K.; Endres, M.; Srinivasan, A.; Tomaselli, K.J.; Yuan, J.; Moskowitz, M.A. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J. Neurosci. 1998, 18, 3659–3668. [Google Scholar] [PubMed]
- Adrie, C.; Adib-Conquy, M.; Laurent, I.; Monchi, M.; Vinsonneau, C.; Fitting, C.; Fraisse, F.; Dinh-Xuan, A.T.; Carli, P.; Spaulding, C.; et al. Successful cardiopulmonary resuscitation after cardiac arrest as a “sepsis-like” syndrome. Circulation 2002, 106, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Carter, J.; Traystman, R.J.; Wagner, D.H.; Herson, P.S. Pro-inflammatory T-lymphocytes rapidly infiltrate into the brain and contribute to neuronal injury following cardiac arrest and cardiopulmonary resuscitation. J. Neuroimmunol. 2014, 274, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Larmann, J.; Schmidt, C.; Gammelin, H.; van Aken, H.K.; Frenzel, T.; Lanckohr, C.; Lox, M.; Boese, N.; Jurk, K.; Theilmeier, G. Intercellular adhesion molecule-1 inhibition attenuates neurologic and hepatic damage after resuscitation in mice. Anesthesiology 2005, 103, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Zhao, H.; Wang, J.; Zhang, L.; Liu, A.; Chen, Y. Inflammatory mechanisms involved in brain injury following cardiac arrest and cardiopulmonary resuscitation. Biomed. Rep. 2016, 5, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Preston-Hurlburt, P.; Janeway, C.A., Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 1997, 388, 394–397. [Google Scholar] [PubMed]
- Asmussen, A.; Fink, K.; Busch, H.J.; Helbing, T.; Bourgeois, N.; Bode, C.; Grundmann, S. Inflammasome and toll-like receptor signaling in human monocytes after successful cardiopulmonary resuscitation. Crit. Care 2016, 20, 170. [Google Scholar] [CrossRef] [PubMed]
- Aloisi, F. Immune function of microglia. Glia 2001, 36, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Yenari, M.A.; Kauppinen, T.M.; Swanson, R.A. Microglial activation in stroke: Therapeutic targets. Neurotherapeutics 2010, 7, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.; Xiong, W.; Jia, X. Electrophysiological Monitoring of Brain Injury and Recovery after Cardiac Arrest. Int. J. Mol. Sci. 2015, 16, 25999–26018. [Google Scholar] [CrossRef] [PubMed]
- Claassen, J.; Vespa, P. Participants in the International Multi-disciplinary Consensus Conference on Multimodality, M. Electrophysiologic monitoring in acute brain injury. Neurocrit. Care 2014, 21 (Suppl. 2), S129–S147. [Google Scholar] [CrossRef] [PubMed]
- Leary, M.; Fried, D.A.; Gaieski, D.F.; Merchant, R.M.; Fuchs, B.D.; Kolansky, D.M.; Edelson, D.P.; Abella, B.S. Neurologic prognostication and bispectral index monitoring after resuscitation from cardiac arrest. Resuscitation 2010, 81, 1133–1137. [Google Scholar] [CrossRef] [PubMed]
- Selig, C.; Riegger, C.; Dirks, B.; Pawlik, M.; Seyfried, T.; Klingler, W. Bispectral index (BIS) and suppression ratio (SR) as an early predictor of unfavourable neurological outcome after cardiac arrest. Resuscitation 2014, 85, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Borges, M.A.; Botos, H.J.; Bastos, R.F.; Godoy, M.F.; Marchi, N.S. Emergency EEG: Study of survival. Arq. Neuropsiquiatr. 2010, 68, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Su, Y.; Hussain, M.; Chen, W.; Ye, H.; Gao, D.; Tian, F. Poor outcome prediction by burst suppression ratio in adults with post-anoxic coma without hypothermia. Neurol. Res. 2014, 36, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, G.L.; Rasmussen, S.B.; Gyllenborg, J.; Benedek, K.; Lauritzen, M. Prognostic value of periodic electroencephalographic discharges for neurological patients with profound disturbances of consciousness. Clin. Neurophysiol. 2013, 124, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Soholm, H.; Kjaer, T.W.; Kjaergaard, J.; Cronberg, T.; Bro-Jeppesen, J.; Lippert, F.K.; Kober, L.; Wanscher, M.; Hassager, C. Prognostic value of electroencephalography (EEG) after out-of-hospital cardiac arrest in successfully resuscitated patients used in daily clinical practice. Resuscitation 2014, 85, 1580–1585. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Su, Y.Y.; Haupt, W.F.; Zhao, J.W.; Xiao, S.Y.; Li, H.L.; Pang, Y.; Yang, Q.L. Application of electrophysiologic techniques in poor outcome prediction among patients with severe focal and diffuse ischemic brain injury. J. Clin. Neurophysiol. 2011, 28, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, V.; Oddo, M.; Rossetti, A.O. Stimulus-induced rhythmic, periodic or ictal discharges (SIRPIDs) in comatose survivors of cardiac arrest: Characteristics and prognostic value. Clin. Neurophysiol. 2013, 124, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Juan, E.; Novy, J.; Suys, T.; Oddo, M.; Rossetti, A.O. Clinical evolution after a non-reactive hypothermic EEG following cardiac arrest. Neurocrit. Care 2015, 22, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Sandroni, C.; Cavallaro, F.; Callaway, C.W.; D’Arrigo, S.; Sanna, T.; Kuiper, M.A.; Biancone, M.; Della Marca, G.; Farcomeni, A.; Nolan, J.P. Predictors of poor neurological outcome in adult comatose survivors of cardiac arrest: A systematic review and meta-analysis. Part 2: Patients treated with therapeutic hypothermia. Resuscitation 2013, 84, 1324–1338. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.; Jiang, G.; Li, Z.; Wang, X. Prognostic value of electroencephalography (EEG) for brain injury after cardiopulmonary resuscitation. Neurol. Sci. 2016, 37, 843–849. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, A.O.; Urbano, L.A.; Delodder, F.; Kaplan, P.W.; Oddo, M. Prognostic value of continuous EEG monitoring during therapeutic hypothermia after cardiac arrest. Crit. Care 2010, 14, R173. [Google Scholar] [CrossRef] [PubMed]
- Wijdicks, E.F.; Hijdra, A.; Young, G.B.; Bassetti, C.L.; Wiebe, S. Practice parameter: Prediction of outcome in comatose survivors after cardiopulmonary resuscitation (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2006, 67, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Leithner, C.; Ploner, C.J.; Hasper, D.; Storm, C. Does hypothermia influence the predictive value of bilateral absent N20 after cardiac arrest? Neurology 2010, 74, 965–969. [Google Scholar] [CrossRef] [PubMed]
- Forgacs, P.B.; Fridman, E.A.; Goldfine, A.M.; Schiff, N.D. Isolation Syndrome after Cardiac Arrest and Therapeutic Hypothermia. Front. Neurosci. 2016, 10, 259. [Google Scholar] [CrossRef] [PubMed]
- Borjigin, J.; Lee, U.; Liu, T.; Pal, D.; Huff, S.; Klarr, D.; Sloboda, J.; Hernandez, J.; Wang, M.M.; Mashour, G.A. Surge of neurophysiological coherence and connectivity in the dying brain. Proc. Natl. Acad. Sci. USA 2013, 110, 14432–14437. [Google Scholar] [CrossRef] [PubMed]
- Young, G.B.; Doig, G.S. Continuous EEG monitoring in comatose intensive care patients: Epileptiform activity in etiologically distinct groups. Neurocrit. Care 2005, 2, 5–10. [Google Scholar] [CrossRef]
- Rittenberger, J.C.; Popescu, A.; Brenner, R.P.; Guyette, F.X.; Callaway, C.W. Frequency and timing of nonconvulsive status epilepticus in comatose post-cardiac arrest subjects treated with hypothermia. Neurocrit. Care 2012, 16, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Karapetkova, M.; Koenig, M.A.; Jia, X. Early prognostication markers in cardiac arrest patients treated with hypothermia. Eur. J. Neurol. 2016, 23, 476–848. [Google Scholar] [CrossRef] [PubMed]
- Sakabe, T.; Tateishi, A.; Miyauchi, Y.; Maekawa, T.; Matsumoto, M.; Tsutsui, T.; Takeshita, H. Intracranial pressure following cardiopulmonary resuscitation. Intensive Care Med. 1987, 13, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Badri, S.; Chen, J.; Barber, J.; Temkin, N.R.; Dikmen, S.S.; Chesnut, R.M.; Deem, S.; Yanez, N.D.; Treggiari, M.M. Mortality and long-term functional outcome associated with intracranial pressure after traumatic brain injury. Intensive Care Med. 2012, 38, 1800–1809. [Google Scholar] [CrossRef] [PubMed]
- Cocchi, M.N.; Lucas, J.M.; Salciccioli, J.; Carney, E.; Herman, S.; Zimetbaum, P.; Donnino, M.W. The role of cranial computed tomography in the immediate post-cardiac arrest period. Intern. Emerg. Med. 2010, 5, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Narayan, R.K.; Kishore, P.R.; Becker, D.P.; Ward, J.D.; Enas, G.G.; Greenberg, R.P.; Domingues Da Silva, A.; Lipper, M.H.; Choi, S.C.; Mayhall, C.G.; et al. Intracranial pressure: To monitor or not to monitor? A review of our experience with severe head injury. J. Neurosurg. 1982, 56, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Kasotakis, G.; Michailidou, M.; Bramos, A.; Chang, Y.; Velmahos, G.; Alam, H.; King, D.; de Moya, M.A. Intraparenchymal vs extracranial ventricular drain intracranial pressure monitors in traumatic brain injury: Less is more? J. Am. Coll. Surg. 2012, 214, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Cardim, D.; Schmidt, B.; Robba, C.; Donnelly, J.; Puppo, C.; Czosnyka, M.; Smielewski, P. Transcranial Doppler Monitoring of Intracranial Pressure Plateau Waves. Neurocrit. Care 2016, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Raffiz, M.; Abdullah, J.M. Optic nerve sheath diameter measurement: A means of detecting raised ICP in adult traumatic and non-traumatic neurosurgical patients. Am. J. Emerg. Med. 2017, 35, 150–153. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.W.; Gombart, Z.J.; Rogers, S.; Gardiner, S.K.; Cecil, S.; Bullock, R.M. Pupillary reactivity as an early indicator of increased intracranial pressure: The introduction of the Neurological Pupil index. Surg. Neurol. Int. 2011, 2, 82. [Google Scholar] [CrossRef] [PubMed]
- Sundgreen, C.; Larsen, F.S.; Herzog, T.M.; Knudsen, G.M.; Boesgaard, S.; Aldershvile, J. Autoregulation of cerebral blood flow in patients resuscitated from cardiac arrest. Stroke 2001, 32, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Mirski, A.M.; Denchev, I.D.; Schnitzer, S.M.; Hanley, F.D. Comparison between hypertonic saline and mannitol in the reduction of elevated intracranial pressure in a rodent model of acute cerebral injury. J. Neurosurg. Anesthesiol. 2000, 12, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Hiploylee, C.; Colbourne, F. Intracranial pressure measured in freely moving rats for days after intracerebral hemorrhage. Exp. Neurol. 2014, 255, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Uldall, M.; Juhler, M.; Skjolding, A.D.; Kruuse, C.; Jansen-Olesen, I.; Jensen, R. A novel method for long-term monitoring of intracranial pressure in rats. J. Neurosci. Methods 2014, 227, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Limbrick, D.D., Jr.; Lake, S.; Talcott, M.; Alexander, B.; Wight, S.; Willie, J.T.; Richard, W.D.; Genin, G.M.; Leuthardt, E.C. The baric probe: A novel long-term implantable intracranial pressure monitor with ultrasound-based interrogation. J. Neurosurg. Pediatr. 2012, 10, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Parnia, S.; Yang, J.; Nguyen, R.; Ahn, A.; Zhu, J.; Inigo-Santiago, L.; Nasir, A.; Golder, K.; Ravishankar, S.; Bartlett, P.; et al. Cerebral Oximetry During Cardiac Arrest: A Multicenter Study of Neurologic Outcomes and Survival. Crit. Care Med. 2016, 44, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Genbrugge, C.; Dens, J.; Meex, I.; Boer, W.; Eertmans, W.; Sabbe, M.; Jans, F.; de Deyne, C. Regional Cerebral Oximetry During Cardiopulmonary Resuscitation: Useful or Useless? J. Emerg. Med. 2016, 50, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Citerio, G.; Piper, I.; Chambers, I.R.; Galli, D.; Enblad, P.; Kiening, K.; Ragauskas, A.; Sahuquillo, J.; Gregson, B.; Brain, I.T.G. Multicenter clinical assessment of the Raumedic Neurovent-P intracranial pressure sensor: A report by the BrainIT group. Neurosurgery 2008, 63, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Kilgannon, J.H.; Roberts, B.W.; Jones, A.E.; Mittal, N.; Cohen, E.; Mitchell, J.; Chansky, M.E.; Trzeciak, S. Arterial blood pressure and neurologic outcome after resuscitation from cardiac arrest. Crit. Care Med. 2014, 42, 2083–2091. [Google Scholar] [CrossRef] [PubMed]
- Ameloot, K.; Genbrugge, C.; Meex, I.; Eertmans, W.; Jans, F.; de Deyne, C.; Dens, J.; Mullens, W.; Ferdinande, B.; Dupont, M. Is venous congestion associated with reduced cerebral oxygenation and worse neurological outcome after cardiac arrest? Crit. Care 2016, 20, 146. [Google Scholar] [CrossRef] [PubMed]
- Murkin, J.M.; Arango, M. Near-infrared spectroscopy as an index of brain and tissue oxygenation. Br. J. Anaesth. 2009, 103 (Suppl. 1), i3–i13. [Google Scholar] [CrossRef]
- Scheeren, T.W.; Schober, P.; Schwarte, L.A. Monitoring tissue oxygenation by near infrared spectroscopy (NIRS): Background and current applications. J. Clin. Monit. Comput. 2012, 26, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Schewe, J.C.; Thudium, M.O.; Kappler, J.; Steinhagen, F.; Eichhorn, L.; Erdfelder, F.; Heister, U.; Ellerkmann, R. Monitoring of cerebral oxygen saturation during resuscitation in out-of-hospital cardiac arrest: A feasibility study in a physician staffed emergency medical system. Scand. J. Trauma Resusc. Emerg. Med. 2014, 22, 58. [Google Scholar] [CrossRef] [PubMed]
- Asim, K.; Gokhan, E.; Ozlem, B.; Ozcan, Y.; Deniz, O.; Kamil, K.; Murat, Z.; Aydin, C.; Selman, Y. Near infrared spectrophotometry (cerebral oximetry) in predicting the return of spontaneous circulation in out-of-hospital cardiac arrest. Am. J. Emerg. Med. 2014, 32, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.W.; Trammell, A.R.; Austin, H.; Barbour, K.; Onuorah, E.; House, D.; Miller, H.L.; Tutt, C.; Combs, D.; Phillips, R.; et al. Cerebral Oximetry as a Real-Time Monitoring Tool to Assess Quality of In-Hospital Cardiopulmonary Resuscitation and Post Cardiac Arrest Care. J. Am. Heart Assoc. 2015, 4, e001859. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Settecase, F.; Xiao, J.; Yu, Z.; Flexman, A.M.; Higashida, R.T. Initial clinical experience with near-infrared spectroscopy in assessing cerebral tissue oxygen saturation in cerebral vasospasm before and after intra-arterial verapamil injection. J. Clin. Neurosci. 2016, 26, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Neumar, R.W.; Nolan, J.P.; Adrie, C.; Aibiki, M.; Berg, R.A.; Bottiger, B.W.; Callaway, C.; Clark, R.S.; Geocadin, R.G.; Jauch, E.C.; et al. Post-cardiac arrest syndrome: Epidemiology, pathophysiology, treatment, and prognostication. A consensus statement from the International Liaison Committee on Resuscitation (American Heart Association, Australian and New Zealand Council on Resuscitation, European Resuscitation Council, Heart and Stroke Foundation of Canada, InterAmerican Heart Foundation, Resuscitation Council of Asia, and the Resuscitation Council of Southern Africa); the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; and the Stroke Council. Circulation 2008, 118, 2452–2483. [Google Scholar] [PubMed]
- Chalkias, A.; Xanthos, T. Post-cardiac arrest brain injury: Pathophysiology and treatment. J. Neurol. Sci. 2012, 315, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, C.S.; Wu, C.J.; Yang, J.; Hang, C.C. Comparison of Cerebral Metabolism between Pig Ventricular Fibrillation and Asphyxial Cardiac Arrest Models. Chin. Med. J. 2015, 128, 1643–1648. [Google Scholar] [PubMed]
- Schaafsma, A.; de Jong, B.M.; Bams, J.L.; Haaxma-Reiche, H.; Pruim, J.; Zijlstra, J.G. Cerebral perfusion and metabolism in resuscitated patients with severe post-hypoxic encephalopathy. J. Neurol. Sci. 2003, 210, 423–430. [Google Scholar] [CrossRef]
- Hahn, D.K.; Geocadin, R.G.; Greer, D.M. Quality of evidence in studies evaluating neuroimaging for neurologic prognostication in adult patients resuscitated from cardiac arrest. Resuscitation 2014, 85, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Krep, H.; Bottiger, B.W.; Bock, C.; Kerskens, C.M.; Radermacher, B.; Fischer, M.; Hoehn, M.; Hossmann, K.A. Time course of circulatory and metabolic recovery of cat brain after cardiac arrest assessed by perfusion-and diffusion-weighted imaging and MR-spectroscopy. Resuscitation 2003, 58, 337–348. [Google Scholar] [CrossRef]
- Tang, Z.R.; Li, C.S.; Zhao, H.; Gong, P.; Zhang, M.Y.; Su, Z.Y.; Wang, S. Effects of hypothermia on brain injury assessed by magnetic resonance imaging after cardiopulmonary resuscitation in a porcine model of cardiac arrest. Am. J. Emerg. Med. 2013, 31, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Li, C.; Han, Y.; Yin, X.; Guo, M. Evaluation of cerebral metabolism by 1H-magnetic resonance spectroscopy for 4 °C saline-induced therapeutic hypothermia in pig model of cardiac arrest. Am. J. Emerg. Med. 2011, 29, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.W.; Chow, A.M.; Chan, K.C.; Yang, J.; Wu, E.X. Magnetic resonance spectroscopy of the brain under mild hypothermia indicates changes in neuroprotection-related metabolites. Neurosci. Lett. 2010, 475, 150–155. [Google Scholar] [CrossRef]
- Chefer, V.I.; Thompson, A.C.; Zpata, A.; Shippenberg, T.S. Overview of Brain Microdialysis. Curr. Protoc. Neurosci. 2010. [Google Scholar] [CrossRef]
- Uchino, H.; Ogihara, Y.; Fukui, H.; Chijiiwa, M.; Sekine, S.; Hara, N.; Elmér, E. Brain injury following cardiac arrest: Pathophysiology for neurocritical care. J. Intensive Care 2016, 4, 31. [Google Scholar] [CrossRef]
- Roh, D.; Park, S. Brain Multimodality Monitoring: Updated Perspectives. Curr. Neurol. Neurosci. Rep. 2016, 16, 56. [Google Scholar] [CrossRef] [PubMed]
- Reis, C.; Wang, Y.; Akyol, O.; Ho, W.M.; Ii, R.A.; Stier, G.; Martin, R.; Zhang, J.H. What’s New in Traumatic Brain Injury: Update on Tracking, Monitoring and Treatment. Int. J. Mol. Sci. 2015, 16, 11903–11965. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Valverde, T.; Sánchez-Guerrero, A.; Vidal-Jorge, M.; Torné, R.; Castro, L.; Gandara, D.; Munar, F.; Poca, M.A.; Sahuquillo, J. Characterization of the Ionic Profile of the Extracellular Space of the Injured and Ischemic Brain: A Microdialysis Study. J. Neurotrauma 2016, 34, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, P.; Claassen, J.; Schmidt, J.M.; Helbok, R.; Hanafy, K.A.; Presciutti, M.; Lantigua, H.; Connolly, E.S.; Lee, K.; Badjatia, N.; et al. Reduced Brain/Serum Glucose Ratios Predict Cerebral Metabolic Distress and Mortality After Severe Brain Injury. Neurocrit. Care 2013, 19, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Young, B.; Kalanuria, A.; Kumar, M.; Burke, K.; Balu, R.; Amendolia, O.; McNulty, K.; Marion, B.; Beckmann, B.; Ciocco, L.; et al. Cerebral Microdialysis. Crit. Care Nurs. Clin. N. Am. 2016, 28, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.H.; Schalén, W.; Stāl, N.; Toft, P.; Reinstrup, P.; Nordström, C.H. Bedside diagnosis of mitochondrial dysfunction after malignant middle cerebral artery infarction. Neurocrit. Care 2014, 21, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Nordström, C.H.; Nielsen, T.H.; Schalén, W.; Reinstrup, P.; Ungerstedt, U. Biochemical indications of cerebral ischemia and mitochondrial dysfunction in severe brain trauma analysed with regard to type of lesion. Acta Neurochir. 2016, 158, 1231–1240. [Google Scholar] [CrossRef]
- Jacobsen, A.; Nielsen, T.H.; Nilsson, O.; Schalén, W.; Nordström, C.H. Bedside diagnosis of mitochondrial dysfunction in aneurysmal subarachnoid hemorrhage. Acta Neurol. Scand. 2014, 130, 156–163. [Google Scholar] [CrossRef]
- Hosmann, A.; Schober, A.; Gruber, A.; Sterz, F.; Testori, C.; Warenits, A.; Weihs, W.; Högler, S.; Scherer, T.; Janata, A. Cerebral and Peripheral Merabolsim to Predict Successful Reperfusion After Cardiac Arrest in Rats—A Microdialysis study. Neurocrit. Care 2015, 24, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Schober, A.; Warenits, A.M.; Testori, C.; Weihs, W.; Hosmann, A.; Högler, S.; Sterz, F.; Janata, A.; Scherer, T.; Magnet, I.A.; et al. Microdialysis assessment of cerebral perfusion during cardiac arrest, extracorporeal life support and cardiopulmonary resuscitation in rats—A pilot trial. PLoS ONE 2016, 11, e0155303. [Google Scholar] [CrossRef] [PubMed]
- Nordström, C.H. Cerebral energy metabolism and microdialysis in neurocritical care. Child's Nerv. Syst. 2010, 26, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Stover, J.F. Actual evidence for neuromonitoring-guided intensive care following severe trauamtic brain injury. Swiss Med. Wkly. 2011, 141, w13245. [Google Scholar] [PubMed]
- Makarenko, S.; Griesdale, D.E.; Gooderham, P.; Sekhon, M.S. Multimodal neuromonitoring for traumatic brain injury: A shift towards individualized therapy. J. Clin. Neurosci. 2016, 26, 8–13. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reis, C.; Akyol, O.; Araujo, C.; Huang, L.; Enkhjargal, B.; Malaguit, J.; Gospodarev, V.; Zhang, J.H. Pathophysiology and the Monitoring Methods for Cardiac Arrest Associated Brain Injury. Int. J. Mol. Sci. 2017, 18, 129. https://doi.org/10.3390/ijms18010129
Reis C, Akyol O, Araujo C, Huang L, Enkhjargal B, Malaguit J, Gospodarev V, Zhang JH. Pathophysiology and the Monitoring Methods for Cardiac Arrest Associated Brain Injury. International Journal of Molecular Sciences. 2017; 18(1):129. https://doi.org/10.3390/ijms18010129
Chicago/Turabian StyleReis, Cesar, Onat Akyol, Camila Araujo, Lei Huang, Budbazar Enkhjargal, Jay Malaguit, Vadim Gospodarev, and John H. Zhang. 2017. "Pathophysiology and the Monitoring Methods for Cardiac Arrest Associated Brain Injury" International Journal of Molecular Sciences 18, no. 1: 129. https://doi.org/10.3390/ijms18010129
APA StyleReis, C., Akyol, O., Araujo, C., Huang, L., Enkhjargal, B., Malaguit, J., Gospodarev, V., & Zhang, J. H. (2017). Pathophysiology and the Monitoring Methods for Cardiac Arrest Associated Brain Injury. International Journal of Molecular Sciences, 18(1), 129. https://doi.org/10.3390/ijms18010129