
Treating the Synapse in Major Psychiatric Disorders: The Role of Postsynaptic Density Network in Dopamine-Glutamate Interplay and Psychopharmacologic Drugs Molecular Actions




 ,
, 

 ,
, 
Abstract
:1. Introduction
2. Involvement of PSD Molecules in Psychiatric Drugs Mechanisms of Actions
2.1. Antipsychotic Drugs
2.2. Mood-Stabilizing Drugs
2.3. Antidepressant Drugs
3. Novel Putative Therapeutic Strategies Based on PSD Molecules Modulation
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Sheng, M.; Hoogenraad, C.C. The postsynaptic architecture of excitatory synapses: A more quantitative view. Annu. Rev. Biochem. 2007, 76, 823–847. [Google Scholar] [CrossRef] [PubMed]
- De Bartolomeis, A.; Sarappa, C.; Magara, S.; Iasevoli, F. Targeting glutamate system for novel antipsychotic approaches: Relevance for residual psychotic symptoms and treatment resistant schizophrenia. Eur. J. Pharmacol. 2012, 682, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Tronson, N.C.; Radulovic, J. Modulation of behavior by scaffolding proteins of the post-synaptic density. Neurobiol. Learn. Mem. 2013, 105, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Iasevoli, F.; Tomasetti, C.; de Bartolomeis, A. Scaffolding proteins of the post-synaptic density contribute to synaptic plasticity by regulating receptor localization and distribution: Relevance for neuropsychiatric diseases. Neurochem. Res. 2013, 38, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.O.; Husi, H.; Yu, L.; Brandon, J.M.; Anderson, C.N.; Blackstock, W.P.; Choudhary, J.S.; Grant, S.G. Molecular characterization and comparison of the components and multiprotein complexes in the postsynaptic proteome. J. Neurochem. 2006, 97, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Boeckers, T.M. The postsynaptic density. Cell Tissue Res. 2006, 326, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.G. A frontier in the understanding of synaptic plasticity: Solving the structure of the postsynaptic density. Bioessays 2012, 34, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Emes, R.D.; Pocklington, A.J.; Anderson, C.N.; Bayes, A.; Collins, M.O.; Vickers, C.A.; Croning, M.D.; Malik, B.R.; Choudhary, J.S.; Armstrong, J.D.; et al. Evolutionary expansion and anatomical specialization of synapse proteome complexity. Nat. Neurosci. 2008, 11, 799–806. [Google Scholar] [CrossRef] [PubMed]
- De Bartolomeis, A.; Buonaguro, E.F.; Iasevoli, F.; Tomasetti, C. The emerging role of dopamine-glutamate interaction and of the postsynaptic density in bipolar disorder pathophysiology: Implications for treatment. J. Psychopharmacol. 2014, 28, 505–526. [Google Scholar] [CrossRef] [PubMed]
- Kneussel, M. Postsynaptic scaffold proteins at non-synaptic sites. The role of postsynaptic scaffold proteins in motor-protein-receptor complexes. EMBO Rep. 2005, 6, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Trinidad, J.C.; Thalhammer, A.; Specht, C.G.; Lynn, A.J.; Baker, P.R.; Schoepfer, R.; Burlingame, A.L. Quantitative analysis of synaptic phosphorylation and protein expression. Mol. Cell. Proteom. 2008, 7, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Wibrand, K.; Panja, D.; Tiron, A.; Ofte, M.L.; Skaftnesmo, K.O.; Lee, C.S.; Pena, J.T.; Tuschl, T.; Bramham, C.R. Differential regulation of mature and precursor microRNA expression by NMDA and metabotropic glutamate receptor activation during LTP in the adult dentate gyrus in vivo. Eur. J. Neurosci. 2010, 31, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Lugli, G.; Larson, J.; Demars, M.P.; Smalheiser, N.R. Primary microRNA precursor transcripts are localized at post-synaptic densities in adult mouse forebrain. J. Neurochem. 2012, 123, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Vessey, J.P.; Karra, D. More than just synaptic building blocks: Scaffolding proteins of the post-synaptic density regulate dendritic patterning. J. Neurochem. 2007, 102, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Bayes, A.; van de Lagemaat, L.N.; Collins, M.O.; Croning, M.D.R.; Whittle, I.R.; Choudhary, J.S.; Grant, S.G.N. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat. Neurosci. 2011, 14, 19–21. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.F. PSD-95-like membrane associated guanylate kinases (PSD-MAGUKs) and synaptic plasticity. Curr. Opin. Neurobiol. 2011, 21, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Sheng, M. PDZ domain proteins of synapses. Nat. Rev. Neurosci. 2004, 5, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.P.; Xu, T.X.; Hallett, P.J.; Watanabe, M.; Grant, S.G.N.; Isacson, O.; Yao, W.D. PSD-95 Uncouples Dopamine-Glutamate Interaction in the D-1/PSD-95/NMDA Receptor Complex. J. Neurosci. 2009, 29, 2948–2960. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lewis, S.M.; Kuhlman, B.; Lee, A.L. Supertertiary Structure of the MAGUK Core from PSD-95. Structure 2013, 21, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi-Yamaguchi, Y.; Furuichi, T. The Homer family proteins. Genome Biol. 2007, 8, 206. [Google Scholar] [CrossRef] [PubMed]
- Kammermeier, P. Regulation of mGlur signaling by endogenous homer proteins. Neuropharmacology 2008, 55, 604. [Google Scholar]
- Ango, F.; Prezeau, L.; Muller, T.; Tu, J.C.; Xiao, B.; Worley, P.F.; Pin, J.P.; Bockaert, J.; Fagni, L. Agonist-independent activation of metabotropic glutamate receptors by the intracellular protein Homer. Nature 2001, 411, 962–965. [Google Scholar] [CrossRef] [PubMed]
- De Bartolomeis, A.; Tomasetti, C. Calcium-dependent networks in dopamine-glutamate interaction: The role of postsynaptic scaffolding proteins. Mol. Neurobiol. 2012, 46, 275–296. [Google Scholar] [CrossRef] [PubMed]
- Boeckers, T.M.; Bockmann, J.; Kreutz, M.R.; Gundelfinger, E.D. ProSAP/Shank proteins—A family of higher order organizing molecules of the postsynaptic density with an emerging role in human neurological disease. J. Neurochem. 2002, 81, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.K.; Tang, C.Y.; Verpelli, C.; Narayanan, R.; Stearns, M.H.; Xu, R.M.; Li, H.L.; Sala, C.; Hayashi, Y. The Postsynaptic Density Proteins Homer and Shank Form a Polymeric Network Structure. Cell 2009, 137, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Dosemeci, A.; Makusky, A.J.; Jankowska-Stephens, E.; Yang, X.; Slotta, D.J.; Markey, S.P. Composition of the synaptic PSD-95 complex. Mol. Cell. Proteom. 2007, 6, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.J.; Xue, S.; Pei, L.; Vukusic, B.; Chery, N.; Wang, Y.; Wang, Y.T.; Niznik, H.B.; Yu, X.M.; Liu, F. Dual regulation of NMDA receptor functions by direct protein-protein interactions with the dopamine D1 receptor. Cell 2002, 111, 219–230. [Google Scholar] [CrossRef]
- Lee, F.J.; Liu, F. Direct interactions between NMDA and D1 receptors: A tale of tails. Biochem. Soc. Trans. 2004, 32, 1032–1036. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.H.; Yang, S.; Shi, W.X.; Jin, G.Z.; Zhen, X.C. Requirement of PSD-95 for dopamine D1 receptor modulating glutamate NR1a/NR2B receptor function. Acta Pharmacol. Sin. 2007, 28, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Sokoloff, P.; Le Foll, B. The dopamine D3 receptor, a quarter century later. Eur. J. Neurosci. 2016, 45, 2–19. [Google Scholar] [CrossRef] [PubMed]
- Guillin, O.; Diaz, J.; Carroll, P.; Griffon, N.; Schwartz, J.C.; Sokoloff, P. BDNF controls dopamine D3 receptor expression and triggers behavioural sensitization. Nature 2001, 411, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Inta, D.; Cameron, H.A.; Gass, P. New neurons in the adult striatum: From rodents to humans. Trends Neurosci. 2015, 38, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Diaz, J.; Pilon, C.; Le Foll, B.; Gros, C.; Triller, A.; Schwartz, J.C.; Sokoloff, P. Dopamine D3 receptors expressed by all mesencephalic dopamine neurons. J. Neurosci. 2000, 20, 8677–8684. [Google Scholar]
- Bernard, V.; Bolam, J.P. Subcellular and subsynaptic distribution of the NR1 subunit of the NMDA receptor in the neostriatum and globus pallidus of the rat: Co-Localization at synapses with the GluR2/3 subunit of the AMPA receptor. Eur. J. Neurosci. 1998, 10, 3721–3736. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Y.; Mao, L.M.; Zhang, G.C.; Papasian, C.J.; Fibuch, E.E.; Lan, H.X.; Zhou, H.F.; Xu, M.; Wang, J.Q. Activity-dependent modulation of limbic dopamine D3 receptors by CaMKII. Neuron 2009, 61, 425–438. [Google Scholar] [CrossRef] [PubMed]
- De Bartolomeis, A.; Marmo, F.; Buonaguro, E.F.; Rossi, R.; Tomasetti, C.; Iasevoli, F. Imaging brain gene expression profiles by antipsychotics: Region-specific action of amisulpride on postsynaptic density transcripts compared to haloperidol. Eur. Neuropsychopharmacol. 2013, 23, 1516–1529. [Google Scholar] [CrossRef] [PubMed]
- De Bartolomeis, A.; Marmo, F.; Buonaguro, E.F.; Latte, G.; Tomasetti, C.; Iasevoli, F. Switching antipsychotics: Imaging the differential effect on the topography of postsynaptic density transcripts in antipsychotic-naive vs. antipsychotic-exposed rats. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2016, 70, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Grabrucker, A.M. A Role for Synaptic Zinc in ProSAP/Shank PSD Scaffold Malformation in Autism Spectrum Disorders. Dev. Neurobiol. 2014, 74, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Grabrucker, S.; Jannetti, L.; Eckert, M.; Gaub, S.; Chhabra, R.; Pfaender, S.; Mangus, K.; Reddy, P.P.; Rankovic, V.; Schmeisser, M.J.; et al. Zinc deficiency dysregulates the synaptic ProSAP/Shank scaffold and might contribute to autism spectrum disorders. Brain 2014, 137, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Grabrucker, S.; Proepper, C.; Mangus, K.; Eckert, M.; Chhabra, R.; Schmeisser, M.J.; Boeckers, T.M.; Grabrucker, A.M. The PSD protein ProSAP2/Shank3 displays synapto-nuclear shuttling which is deregulated in a schizophrenia-associated mutation. Exp. Neurol. 2014, 253, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Grant, S.G.N. Synaptopathies: Diseases of the synaptome. Curr.Opin. Neurobiol. 2012, 22, 522–529. [Google Scholar] [CrossRef] [PubMed]
- De Bartolomeis, A.; Iasevoli, F.; Marmo, F.; Buonaguro, E.F.; Eramo, A.; Rossi, R.; Avvisati, L.; Latte, G.; Tomasetti, C. Progressive recruitment of cortical and striatal regions by inducible postsynaptic density transcripts after increasing doses of antipsychotics with different receptor profiles: Insights for psychosis treatment. Eur. Neuropsychopharmacol. 2015, 25, 566–582. [Google Scholar] [CrossRef] [PubMed]
- Iasevoli, F.; Tomasetti, C.; Marmo, F.; Bravi, D.; Arnt, J.; de Bartolomeis, A. Divergent acute and chronic modulation of glutamatergic postsynaptic density genes expression by the antipsychotics haloperidol and sertindole. Psychopharmacology 2010, 212, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Peykov, S.; Berkel, S.; Schoen, M.; Weiss, K.; Degenhardt, F.; Strohmaier, J.; Weiss, B.; Proepper, C.; Schratt, G.; Nothen, M.M.; et al. Identification and functional characterization of rare SHANK2 variants in schizophrenia. Mol. Psychiatry 2015, 20, 1489–1498. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.C.; Wu, J.; Ward, M.D.; Yang, S.G.; Chuang, Y.A.; Xiao, M.F.; Li, R.J.; Leahy, D.J.; Worley, P.F. Structural basis of arc binding to synaptic proteins: Implications for cognitive disease. Neuron 2015, 86, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.R.; Kimura, H.; Wang, C.Y.; Ishizuka, K.; Kushima, I.; Arioka, Y.; Yoshimi, A.; Nakamura, Y.; Shiino, T.; Oya-Ito, T.; et al. Resequencing and Association Analysis of Six PSD-95-Related Genes as Possible Susceptibility Genes for Schizophrenia and Autism Spectrum Disorders. Sci. Rep. 2016, 6, 27491. [Google Scholar] [CrossRef] [PubMed]
- Leblond, C.S.; Nava, C.; Polge, A.; Gauthier, J.; Huguet, G.; Lumbroso, S.; Giuliano, F.; Stordeur, C.; Depienne, C.; Mouzaf, K.; et al. Meta-analysis of SHANK mutations in autism spectrum disorders: A gradient of severity in cognitive impairments. PLoS Genet. 2014, 10, e1004580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matosin, N.; Green, M.J.; Andrews, J.L.; Newell, K.A.; Fernandez-Enright, F. Possibility of a sex-specific role for a genetic variant in FRMPD4 in schizophrenia, but not cognitive function. Neuroreport 2016, 27, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.; Trent, S.; Thomas, K.L.; O'Donovan, M.C.; Owen, M.J. Genetic risk for schizophrenia: Convergence on synaptic pathways involved in plasticity. Biol. Psychiatry 2015, 77, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Karolewicz, B.; Szebeni, K.; Gilmore, T.; Maciag, D.; Stockmeier, C.A.; Ordway, G.A. Elevated levels of NR2A and PSD-95 in the lateral amygdala in depression. Int. J. Neuropsychopharmacol. 2009, 12, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Krueger-Burg, D.; Winkler, D.; Mitkovski, M.; Daher, F.; Ronnenberg, A.; Schluter, O.M.; Dere, E.; Ehrenreich, H. The socioBox: A novel paradigm to assess complex social recognition in male mice. Front. Behav. Neurosci. 2016, 10, 151. [Google Scholar] [CrossRef] [PubMed]
- De Bartolomeis, A.; Errico, F.; Aceto, G.; Tomasetti, C.; Usiello, A.; Iasevoli, F. d-aspartate dysregulation in Ddo-/- mice modulates phencyclidine-induced gene expression changes of postsynaptic density molecules in cortex and striatum. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2015, 62, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.P.; Saur, T.; Duke, A.N.; Grant, S.G.N.; Platt, D.M.; Rowlett, J.K.; Isacson, O.; Yao, W.D. Motor impairments, striatal degeneration, and altered dopamine-glutamate interplay in mice lacking PSD-95. J. Neurogenet. 2014, 28, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.H.; Ehlers, M.D. Modeling Autism by SHANK Gene Mutations in Mice. Neuron 2013, 78, 8–27. [Google Scholar] [CrossRef]
- Manago, F.; Mereu, M.; Mastwal, S.; Mastrogiacomo, R.; Scheggia, D.; Emanuele, M.; de Luca, M.A.; Weinberger, D.R.; Wang, K.H.; Papaleo, F. Genetic disruption of Arc/Arg3.1 in mice causes alterations in dopamine and neurobehavioral phenotypes related to schizophrenia. Cell Rep. 2016, 16, 2116–2128. [Google Scholar] [CrossRef] [PubMed]
- Fossati, G.; Morini, R.; Corradini, I.; Antonucci, F.; Trepte, P.; Edry, E.; Sharma, V.; Papale, A.; Pozzi, D.; Defilippi, P.; et al. Reduced SNAP-25 increases PSD-95 mobility and impairs spine morphogenesis. Cell Death Differ. 2015, 22, 1425–1436. [Google Scholar] [CrossRef] [PubMed]
- Meshul, C.K.; Tan, S.E. Haloperidol-induced morphological alterations are associated with changes in calcium/calmodulin kinase II activity and glutamate immunoreactivity. Synapse 1994, 18, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Meshul, C.K.; Stallbaumer, R.K.; Taylor, B.; Janowsky, A. Haloperidol-induced morphological changes in striatum are associated with glutamate synapses. Brain Res. 1994, 648, 181–195. [Google Scholar] [CrossRef]
- Vincent, S.L.; McSparren, J.; Wang, R.Y.; Benes, F.M. Evidence for ultrastructural changes in cortical axodendritic synapses following long-term treatment with haloperidol or clozapine. Neuropsychopharmacology 1991, 5, 147–155. [Google Scholar] [PubMed]
- Polese, D.; de Serpis, A.A.; Ambesi-Impiombato, A.; Muscettola, G.; de Bartolomeis, A. Homer 1a gene expression modulation by antipsychotic drugs: Involvement of the glutamate metabotropic system and effects of D-cycloserine. Neuropsychopharmacology 2002, 27, 906–913. [Google Scholar] [CrossRef]
- De Bartolomeis, A.; Aloj, L.; Ambesi-Impiombato, A.; Bravi, D.; Caraco, C.; Muscettola, G.; Barone, P. Acute administration of antipsychotics modulates Homer striatal gene expression differentially. Brain Res. Mol. Brain Res. 2002, 98, 124–129. [Google Scholar] [CrossRef]
- Tomasetti, C.; Dellaversano, C.; Iasevoli, F.; de Bartolomeis, A. Homer splice variants modulation within cortico-subcortical regions by dopamine D2 antagonists, a partial agonist, and an indirect agonist: Implication for glutamatergic postsynaptic density in antipsychotics action. Neuroscience 2007, 150, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, F.; Frasca, A.; Racagni, G.; Riva, M.A. Dynamic regulation of glutamatergic postsynaptic activity in rat prefrontal cortex by repeated administration of antipsychotic drugs. Mol. Pharmacol. 2008, 73, 1484–1490. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.I.; Yadav, P.N.; Yao, W.D.; Arbuckle, M.I.; Grant, S.G.; Caron, M.G.; Roth, B.L. PSD-95 is essential for hallucinogen and atypical antipsychotic drug actions at serotonin receptors. J. Neurosci. 2009, 29, 7124–7136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bartolomeis, A.; Latte, G.; Tomasetti, C.; Iasevoli, F. Glutamatergic postsynaptic density protein dysfunctions in synaptic plasticity and dendritic spines morphology: Relevance to schizophrenia and other behavioral disorders pathophysiology, and implications for novel therapeutic approaches. Mol. Neurobiol. 2014, 49, 484–511. [Google Scholar] [CrossRef] [PubMed]
- De Bartolomeis, A.; Iasevoli, F.; Tomasetti, C.; Buonaguro, E.F. MicroRNAs in Schizophrenia: Implications for synaptic plasticity and dopamine-glutamate interaction at the postsynaptic density. New avenues for antipsychotic treatment under a theranostic perspective. Mol. Neurobiol. 2015, 52, 1771–1790. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, R.F.; Schloesser, R.J.; Gould, T.D.; Manji, H.K. Mood stabilizers target cellular plasticity and resilience cascades: Implications for the development of novel therapeutics. Mol. Neurobiol. 2005, 32, 173–202. [Google Scholar] [CrossRef]
- Du, J.; Quiroz, J.; Yuan, P.; Zarate, C.; Manji, H.K. Bipolar disorder: Involvement of signaling cascades and AMPA receptor trafficking at synapses. Neuron Glia Biol. 2004, 1, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhang, G.Y. Lithium reduced N-methyl-d-aspartate receptor subunit 2A tyrosine phosphorylation and its interactions with Src and Fyn mediated by PSD-95 in rat hippocampus following cerebral ischemia. Neurosci. Lett. 2003, 348, 185–189. [Google Scholar] [CrossRef]
- Kim, H.J.; Thayer, S.A. Lithium increases synapse formation between hippocampal neurons by depleting phosphoinositides. Mol. Pharmacol. 2009, 75, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- De Bartolomeis, A.; Tomasetti, C.; Cicale, M.; Yuan, P.X.; Manji, H.K. Chronic treatment with lithium or valproate modulates the expression of Homer1b/c and its related genes Shank and Inositol 1,4,5-trisphosphate receptor. Eur. Neuropsychopharmacol. 2012, 22, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Dell'Aversano, C.; Iasevoli, F.; Marmo, F.; de Bartolomeis, A. The acute and chronic effects of combined antipsychotic-mood stabilizing treatment on the expression of cortical and striatal postsynaptic density genes. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, J.M. Converging evidence for regulation of dopamine neurotransmission by lithium: An editorial highlight for ‘chronic lithium treatment rectifies maladaptive dopamine release in the nucleus accumbens’. J. Neurochem. 2016, 139, 520–522. [Google Scholar] [CrossRef] [PubMed]
- De Bartolomeis, A.; Avvisati, L.; Iasevoli, F.; Tomasetti, C. Intracellular pathways of antipsychotic combined therapies: Implication for psychiatric disorders treatment. Eur. J. Pharmacol. 2013, 718, 502–523. [Google Scholar] [CrossRef] [PubMed]
- Quiroz, J.A.; Singh, J.; Gould, T.D.; Denicoff, K.D.; Zarate, C.A.; Manji, H.K. Emerging experimental therapeutics for bipolar disorder: Clues from the molecular pathophysiology. Mol. Psychiatry 2004, 9, 756–776. [Google Scholar] [CrossRef] [PubMed]
- Gould, T.D.; Quiroz, J.A.; Singh, J.; Zarate, C.A.; Manji, H.K. Emerging experimental therapeutics for bipolar disorder: Insights from the molecular and cellular actions of current mood stabilizers. Mol. Psychiatry 2004, 9, 734–755. [Google Scholar] [CrossRef] [PubMed]
- Qiao, H.; Li, M.X.; Xu, C.; Chen, H.B.; An, S.C.; Ma, X.M. Dendritic Spines in Depression: What We Learned from Animal Models. Neural Plast. 2016, 2016, 8056370. [Google Scholar] [CrossRef] [PubMed]
- Dean, B.; Gibbons, A.S.; Boer, S.; Uezato, A.; Meador-Woodruff, J.; Scarr, E.; McCullumsmith, R.E. Changes in cortical N-methyl-d-aspartate receptors and post-synaptic density protein 95 in schizophrenia, mood disorders and suicide. Aust. N. Z. J. Psychiatry 2016, 50, 275–283. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, O.F.; Wu, X.; Castren, E. Chronic fluoxetine treatment increases expression of synaptic proteins in the hippocampus of the ovariectomized rat: Role of BDNF signalling. Psychoneuroendocrinology 2009, 34, 367–381. [Google Scholar] [CrossRef] [PubMed]
- Ampuero, E.; Rubio, F.J.; Falcon, R.; Sandoval, M.; Diaz-Veliz, G.; Gonzalez, R.E.; Earle, N.; Dagnino-Subiabre, A.; Aboitiz, F.; Orrego, F.; et al. Chronic fluoxetine treatment induces structural plasticity and selective changes in glutamate receptor subunits in the rat cerebral cortex. Neuroscience 2010, 169, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Stan, T.L.; Sousa, V.C.; Zhang, X.; Ono, M.; Svenningsson, P. Lurasidone and fluoxetine reduce novelty-induced hypophagia and NMDA receptor subunit and PSD-95 expression in mouse brain. Eur. Neuropsychopharmacol. 2015, 25, 1714–1722. [Google Scholar] [CrossRef] [PubMed]
- Luoni, A.; Macchi, F.; Papp, M.; Molteni, R.; Riva, M.A. Lurasidone exerts antidepressant properties in the chronic mild stress model through the regulation of synaptic and neuroplastic mechanisms in the rat prefrontal cortex. Int. J. Neuropsychopharmacol. 2014, 18. [Google Scholar] [CrossRef] [PubMed]
- Luoni, A.; Berry, A.; Calabrese, F.; Capoccia, S.; Bellisario, V.; Gass, P.; Cirulli, F.; Riva, M.A. Delayed BDNF alterations in the prefrontal cortex of rats exposed to prenatal stress: Preventive effect of lurasidone treatment during adolescence. Eur. Neuropsychopharmacol. 2014, 24, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Dell'aversano, C.; Tomasetti, C.; Iasevoli, F.; de Bartolomeis, A. Antipsychotic and antidepressant co-treatment: Effects on transcripts of inducible postsynaptic density genes possibly implicated in behavioural disorders. Brain Res. Bull. 2009, 79, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Englisch, S.; Inta, D.; Eer, A.; Zink, M. Bupropion for depression in schizophrenia. Clin. Neuropharmacol. 2010, 33, 257–259. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.X.; Wang, J.; Xie, Z.M.; Xu, N.; Zhang, G.F.; Jia, M.; Zhou, Z.Q.; Hashimoto, K.; Yang, J.J. Regulation of glutamate transporter 1 via BDNF-TrkB signaling plays a role in the anti-apoptotic and antidepressant effects of ketamine in chronic unpredictable stress model of depression. Psychopharmacology 2016, 233, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.J.; Fuchikami, M.; Dwyer, J.M.; Lepack, A.E.; Duman, R.S.; Aghajanian, G.K. GSK-3 inhibition potentiates the synaptogenic and antidepressant-like effects of subthreshold doses of ketamine. Neuropsychopharmacology 2013, 38, 2268–2277. [Google Scholar] [CrossRef] [PubMed]
- Zanos, P.; Moaddel, R.; Morris, P.J.; Georgiou, P.; Fischell, J.; Elmer, G.I.; Alkondon, M.; Yuan, P.; Pribut, H.J.; Singh, N.S.; et al. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature 2016, 533, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Wiedholz, L.M.; Owens, W.A.; Horton, R.E.; Feyder, M.; Karlsson, R.M.; Hefner, K.; Sprengel, R.; Celikel, T.; Daws, L.C.; Holmes, A. Mice lacking the AMPA GluR1 receptor exhibit striatal hyperdopaminergia and “schizophrenia-related” behaviors. Mol. Psychiatry 2008, 13, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Chourbaji, S.; Vogt, M.A.; Fumagalli, F.; Sohr, R.; Frasca, A.; Brandwein, C.; Hortnagl, H.; Riva, M.A.; Sprengel, R.; Gass, P. AMPA receptor subunit 1 (GluR-A) knockout mice model the glutamate hypothesis of depression. FASEB J. 2008, 22, 3129–3134. [Google Scholar] [CrossRef] [PubMed]
- Inta, D.; Vogt, M.A.; Elkin, H.; Weber, T.; Lima-Ojeda, J.M.; Schneider, M.; Luoni, A.; Riva, M.A.; Gertz, K.; Hellmann-Regen, J.; et al. Phenotype of mice with inducible ablation of GluA1 AMPA receptors during late adolescence: Relevance for mental disorders. Hippocampus 2014, 24, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Vogt, M.A.; Elkin, H.; Pfeiffer, N.; Sprengel, R.; Gass, P.; Inta, D. Impact of adolescent GluA1 AMPA receptor ablation in forebrain excitatory neurons on behavioural correlates of mood disorders. Eur. Arch. Psychiatry Clin. Neurosci. 2014, 264, 625–629. [Google Scholar] [CrossRef] [PubMed]
- Nosyreva, E.; Autry, A.E.; Kavalali, E.T.; Monteggia, L.M. Age dependence of the rapid antidepressant and synaptic effects of acute NMDA receptor blockade. Front. Mol. Neurosci. 2014, 7, 94. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Sanchez, L.; Campa, L.; Auberson, Y.P.; Adell, A. The role of GluN2A and GluN2B subunits on the effects of NMDA receptor antagonists in modeling schizophrenia and treating refractory depression. Neuropsychopharmacology 2014, 39, 2673–2680. [Google Scholar] [CrossRef] [PubMed]
- Kiselycznyk, C.; Jury, N.J.; Halladay, L.R.; Nakazawa, K.; Mishina, M.; Sprengel, R.; Grant, S.G.; Svenningsson, P.; Holmes, A. NMDA receptor subunits and associated signaling molecules mediating antidepressant-related effects of NMDA-GluN2B antagonism. Behav. Brain Res. 2015, 287, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Egbujo, C.N.; Sinclair, D.; Borgmann-Winter, K.E.; Arnold, S.E.; Turetsky, B.I.; Hahn, C.G. Molecular evidence for decreased synaptic efficacy in the postmortem olfactory bulb of individuals with schizophrenia. Schizophr. Res. 2015, 168, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Grabrucker, A.M.; Ruozi, B.; Belletti, D.; Pederzoli, F.; Forni, F.; Vandelli, M.A.; Tosi, G. Nanoparticle transport across the blood brain barrier. Tissue Barriers 2016, 4, e1153568. [Google Scholar] [CrossRef] [PubMed]
- Tabansky, I.; Messina, M.D.; Bangeranye, C.; Goldstein, J.; Blitz-Shabbir, K.M.; Machado, S.; Jeganathan, V.; Wright, P.; Najjar, S.; Cao, Y.H.; et al. Advancing drug delivery systems for the treatment of multiple sclerosis. Immunol. Res. 2015, 63, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Ali, J.; Ali, M.; Baboota, S.; Sahni, J.K.; Ramassamy, C.; Dao, L.; Bhavna. Potential of nanoparticulate drug delivery systems by intranasal administration. Curr. Pharm. Des. 2010, 16, 1644–1653. [Google Scholar] [CrossRef] [PubMed]
- Ferres-Coy, A.; Galofre, M.; Pilar-Cuellar, F.; Vidal, R.; Paz, V.; Ruiz-Bronchal, E.; Campa, L.; Pazos, A.; Caso, J.R.; Leza, J.C.; et al. Therapeutic antidepressant potential of a conjugated siRNA silencing the serotonin transporter after intranasal administration. Mol. Psychiatry 2016, 21, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, A.; Constantine-Paton, M. Postsynaptic localization of PSD-95 is regulated by all three pathways downstream of TrkB signaling. Front. Synaptic Neurosci. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Willard, S.; Koochekpour, S. Glutamate, glutamate receptors, and downstream signaling pathways. Int. J. Biol. Sci. 2013, 9, 948–959. [Google Scholar] [CrossRef] [PubMed]
- Serchov, T.; Clement, H.W.; Schwarz, M.K.; Iasevoli, F.; Tosh, D.K.; Idzko, M.; Jacobson, K.A.; de Bartolomeis, A.; Normann, C.; Biber, K.; et al. Increased signaling via adenosine A(1) Receptors, sleep deprivation, imipramine, and ketamine inhibit depressive-like behavior via induction of Homer1a. Neuron 2015, 87, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Buonaguro, E.F.; Tomasetti, C.; Chiodini, P.; Marmo, F.; Latte, G.; Rossi, R.; Avvisati, L.; Iasevoli, F.; de Bartolomeis, A. Postsynaptic density protein transcripts are differentially modulated by minocycline alone or in add-on to haloperidol: Implications for treatment resistant schizophrenia. J. Psychopharmacol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Fell, M.J.; McKinzie, D.L.; Monn, J.A.; Svensson, K.A. Group II metabotropic glutamate receptor agonists and positive allosteric modulators as novel treatments for schizophrenia. Neuropharmacology 2012, 62, 1473–1483. [Google Scholar] [CrossRef] [PubMed]
- Balu, D.T.; Li, Y.; Takagi, S.; Presti, K.T.; Ramikie, T.S.; Rook, J.M.; Jones, C.K.; Lindsley, C.W.; Conn, P.J.; Bolshakov, V.Y.; et al. An mGlu(5)-Positive Allosteric Modulator Rescues the Neuroplasticity Deficits in a Genetic Model of NMDA Receptor Hypofunction in Schizophrenia. Neuropsychopharmacology 2016, 41, 2052–2061. [Google Scholar] [CrossRef] [PubMed]
- Iasevoli, F.; Buonaguro, E.F.; Sarappa, C.; Marmo, F.; Latte, G.; Rossi, R.; Eramo, A.; Tomasetti, C.; de Bartolomeis, A. Regulation of postsynaptic plasticity genes’ expression and topography by sustained dopamine perturbation and modulation by acute memantine: Relevance to schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2014, 54, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Goff, D.C.; Lamberti, J.S.; Leon, A.C.; Green, M.F.; Miller, A.L.; Patel, J.; Manschreck, T.; Freudenreich, O.; Johnson, S.A. A placebo-controlled add-on trial of the Ampakine, CX516, for cognitive deficits in schizophrenia. Neuropsychopharmacology 2008, 33, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Strzelecki, D.; Kaluzynska, O.; Szyburska, J.; Wysokinski, A. MMP-9 Serum Levels in Schizophrenic Patients during Treatment Augmentation with Sarcosine (Results of the PULSAR Study). Int. J. Mol. Sci. 2016, 17, 1075. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.J.; Skalak, M.; Lo Bu, R.; Bhatia, S.N. Neuron-Targeted Nanoparticle for siRNA Delivery to Traumatic Brain Injuries. ACS Nano 2016, 10, 7926–7933. [Google Scholar] [CrossRef] [PubMed]
- Aarts, M.; Liu, Y.T.; Liu, L.D.; Besshoh, S.; Arundine, M.; Gurd, J.W.; Wang, Y.T.; Salter, M.W.; Tymianski, M. Treatment of ischemic brain damage by perturbing NMDA receptor-PSD-95 protein interactions. Science 2002, 298, 846–850. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.S.; Doucette, T.A.; Liu, Y.T.; Fang, Y.; Teves, L.; Aarts, M.; Ryan, C.L.; Bernard, P.B.; Lau, A.; Forder, J.P.; et al. Effectiveness of PSD95 inhibitors in permanent and transient focal ischemia in the rat. Stroke 2008, 39, 2544–2553. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.J.; Teves, L.; Tymianski, M. Treatment of stroke with a PSD-95 inhibitor in the gyrencephalic primate brain. Nature 2012, 483, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.D.; Martin, R.H.; Mikulis, D.; Wong, J.H.; Silver, F.L.; Terbrugge, K.G.; Milot, G.; Clark, W.M.; MacDonald, R.L.; Kelly, M.E.; et al. Safety and efficacy of NA-1 in patients with iatrogenic stroke after endovascular aneurysm repair (ENACT): A phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2012, 11, 942–950. [Google Scholar] [CrossRef]
- Norton, N.; Williams, H.J.; Williams, N.M.; Spurlock, G.; Zammit, S.; Jones, G.; Jones, S.; Owen, R.; O’Donovan, M.C.; Owen, M.J. Mutation screening of the Homer gene family and association analysis in schizophrenia. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2003, 120B, 18–21. [Google Scholar] [CrossRef] [PubMed]
- Szumlinski, K.K.; Lominac, K.D.; Kleschen, M.J.; Oleson, E.B.; Dehoff, M.H.; Schwarz, M.K.; Seeburg, P.H.; Worley, P.F.; Kalivas, P.W. Behavioral and neurochemical phenotyping of Homer1 mutant mice: Possible relevance to schizophrenia. Genes Brain Behav. 2005, 4, 273–288. [Google Scholar] [CrossRef] [PubMed]
- Spellmann, I.; Rujescu, D.; Musil, R.; Mayr, A.; Giegling, I.; Genius, J.; Zill, P.; Dehning, S.; Opgen-Rhein, M.; Cerovecki, A.; et al. Homer-1 polymorphisms are associated with psychopathology and response to treatment in schizophrenic patients. J. Psychiatr. Res. 2011, 45, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Iasevoli, F.; Tomasetti, C.; Buonaguro, E.F.; de Bartolomeis, A. The Glutamatergic Aspects of Schizophrenia Molecular Pathophysiology: Role of the Postsynaptic Density, and Implications for Treatment. Curr. Neuropharmacol. 2014, 12, 219–238. [Google Scholar] [CrossRef] [PubMed]
- Rietschel, M.; Mattheisen, M.; Frank, J.; Treutlein, J.; Degenhardt, F.; Breuer, R.; Steffens, M.; Mier, D.; Esslinger, C.; Walter, H.; et al. Genome-wide association-, replication-, and neuroimaging study implicates HOMER1 in the etiology of major depression. Biol. Psychiatry 2010, 68, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Grinevich, V.; Seeburg, P.H.; Schwarz, M.K.; Jezova, D. Homer 1—A new player linking the hypothalamic-pituitary-adrenal axis activity to depression and anxiety. Endocr. Regul. 2012, 46, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.C.; Mao, L.M.; Liu, X.Y.; Parelkar, N.K.; Arora, A.; Yang, L.; Hains, M.; Fibuch, E.E.; Wang, J.Q. In vivo regulation of Homer1a expression in the striatum by cocaine. Mol. Pharmacol. 2007, 71, 1148–1158. [Google Scholar] [CrossRef] [PubMed]
- Rezvani, K.; Teng, Y.; Shim, D.; de Biasi, M. Nicotine regulates multiple synaptic proteins by inhibiting proteasomal activity. J. Neurosci. 2007, 27, 10508–10519. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, J.B.; Arpin-Bott, M.P.; Kao, D.; Dirrig-Grosch, S.; Aunis, D.; Zwiller, J. Cocaine induces the expression of homer 1b/c, homer 3a/b, and hsp 27 proteins in rat cerebellum. Synapse 2007, 61, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Nakahara, T.; Yamada, H.; Hirano, M.; Kuroki, T.; Kanba, S. A neurotoxic dose of methamphetamine induces gene expression of Homer 1a, but not Homer 1b or 1c, in the striatum and nucleus accumbens. Neurochem. Int. 2007, 51, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Kane, J.K.; Hwang, Y.; Konu, O.; Loughlin, S.E.; Leslie, F.M.; Li, M.D. Regulation of Homer and group I metabotropic glutamate receptors by nicotine. Eur. J. Neurosci. 2005, 21, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Lominac, K.D.; Oleson, E.B.; Pava, M.; Klugmann, M.; Schwarz, M.K.; Seeburg, P.H.; During, M.J.; Worley, P.F.; Kalivas, P.W.; Szumlinski, K.K. Distinct roles for different Homer1 isoforms in behaviors and associated prefrontal cortex function. J. Neurosci. 2005, 25, 11586–11594. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shahar, O.; Obara, I.; Ary, A.W.; Ma, N.; Mangiardi, M.A.; Medina, R.L.; Szumlinski, K.K. Extended daily access to cocaine results in distinct alterations in Homer 1b/c and NMDA receptor subunit expression within the medial prefrontal cortex. Synapse 2009, 63, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Knackstedt, L.A.; Moussawi, K.; Lalumiere, R.; Schwendt, M.; Klugmann, M.; Kalivas, P.W. Extinction training after cocaine self-administration induces glutamatergic plasticity to inhibit cocaine seeking. J. Neurosci. 2010, 30, 7984–7992. [Google Scholar] [CrossRef] [PubMed]
- Tappe, A.; Klugmann, M.; Luo, C.; Hirlinger, D.; Agarwal, N.; Benrath, J.; Ehrengruber, M.U.; During, M.J.; Kuner, R. Synaptic scaffolding protein Homer1a protects against chronic inflammatory pain. Nat. Med. 2006, 12, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.X.; Jiang, Z.; Zhao, Z.Q. Knockdown of synaptic scaffolding protein Homer 1b/c attenuates secondary hyperalgesia induced by complete Freund’s adjuvant in rats. Anesth. Analg. 2011, 113, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Giuffrida, R.; Musumeci, S.; D’Antoni, S.; Bonaccorso, C.M.; Giuffrida-Stella, A.M.; Oostra, B.A.; Catania, M.V. A reduced number of metabotropic glutamate subtype 5 receptors are associated with constitutive homer proteins in a mouse model of fragile X syndrome. J. Neurosci. 2005, 25, 8908–8916. [Google Scholar] [CrossRef] [PubMed]
- Roselli, F.; Hutzler, P.; Wegerich, Y.; Livrea, P.; Almeida, O.F. Disassembly of shank and homer synaptic clusters is driven by soluble β-amyloid1-40 through divergent NMDAR-dependent signalling pathways. PLoS ONE 2009, 4, e6011. [Google Scholar] [CrossRef] [PubMed]
- De Luca, V.; Annesi, G.; De Marco, E.V.; de Bartolomeis, A.; Nicoletti, G.; Pugliese, P.; Muscettola, G.; Barone, P.; Quattrone, A. Homer1 promoter analysis in Parkinson’s disease: Association study with psychotic symptoms. Neuropsychobiology 2009, 59, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Yang, Y.F.; Luo, P.; Liu, W.; Dai, S.H.; Zheng, X.R.; Fei, Z.; Jiang, X.F. Homer1 knockdown protects dopamine neurons through regulating calcium homeostasis in an in vitro model of Parkinson’s disease. Cell Signal. 2013, 25, 2863–2870. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.D.; Fei, Z.; Zhang, X. Traumatic injury induced homer-1a gene expression in cultured cortical neurons of rat. Neurosci. Lett. 2005, 389, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Rao, W.; Zhang, C.; Zhang, C.; Liu, M.D.; Han, F.; Yao, L.B.; Han, H.; Luo, P.; Su, N.; et al. Scaffolding protein Homer1a protects against NMDA-induced neuronal injury. Cell Death Dis. 2015, 6, e1843. [Google Scholar] [CrossRef] [PubMed]
- Iasevoli, F.; Ambesi-Impiombato, A.; Fiore, G.; Panariello, F.; Muscettola, G.; de Bartolomeis, A. Pattern of acute induction of Homer1a gene is preserved after chronic treatment with first- and second-generation antipsychotics: Effect of short-term drug discontinuation and comparison with Homer1a-interacting genes. J. Psychopharmacol. 2011, 25, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Iasevoli, F.; Ambesi-Impiombato, A.; Fiore, G.; Panariello, F.; Muscettola, G.; de Bartolomeis, A. Topographical and temporal distribution of Homer1a expression is correlated to antipsychotics dopaminergic profile. Eur. Neuropsychopharmacol. 2008, 18, S15–S16. [Google Scholar] [CrossRef]
- Gilks, W.P.; Allott, E.H.; Donohoe, G.; Cummings, E.; International Schizophrenia, C.; Gill, M.; Corvin, A.P.; Morris, D.W. Replicated genetic evidence supports a role for HOMER2 in schizophrenia. Neurosci. Lett. 2010, 468, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Smothers, C.T.; Szumlinski, K.K.; Worley, P.F.; Woodward, J.J. Altered NMDA receptor function in primary cultures of hippocampal neurons from mice lacking the Homer2 gene. Synapse 2016, 70, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Haider, A.; Woodward, N.C.; Lominac, K.D.; Sacramento, A.D.; Klugmann, M.; Bell, R.L.; Szumlinski, K.K. Homer2 within the nucleus accumbens core bidirectionally regulates alcohol intake by both P and Wistar rats. Alcohol 2015, 49, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Meyers, J.L.; Salling, M.C.; Almli, L.M.; Ratanatharathorn, A.; Uddin, M.; Galea, S.; Wildman, D.E.; Aiello, A.E.; Bradley, B.; Ressler, K.; et al. Frequency of alcohol consumption in humans; the role of metabotropic glutamate receptors and downstream signaling pathways. Transl. Psychiatry 2015, 5, e586. [Google Scholar] [CrossRef] [PubMed]
- Ruegsegger, C.; Stucki, D.M.; Steiner, S.; Angliker, N.; Radecke, J.; Keller, E.; Zuber, B.; Ruegg, M.A.; Saxena, S. Impaired mTORC1-Dependent Expression of Homer-3 Influences SCA1 Pathophysiology. Neuron 2016, 89, 129–146. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Wildemann, B. ‘Medusa-head ataxia’: The expanding spectrum of Purkinje cell antibodies in autoimmune cerebellar ataxia. Part 1: Anti-mGluR1, anti-Homer-3, anti-Sj/ITPR1 and anti-CARP VIII. J. Neuroinflamm. 2015, 12, 166. [Google Scholar] [CrossRef] [PubMed]
- Matosin, N.; Fernandez-Enright, F.; Lum, J.S.; Engel, M.; Andrews, J.L.; Gassen, N.C.; Wagner, K.V.; Schmidt, M.V.; Newell, K.A. Molecular evidence of synaptic pathology in the CA1 region in schizophrenia. NPJ Schizophr. 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Fujita-Jimbo, E.; Tanabe, Y.; Yu, Z.; Kojima, K.; Mori, M.; Li, H.; Iwamoto, S.; Yamagata, T.; Momoi, M.Y.; Momoi, T. The association of GPR85 with PSD-95-neuroligin complex and autism spectrum disorder: A molecular analysis. Mol. Autism 2015, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- Toro, C.; Deakin, J.F. NMDA receptor subunit NRI and postsynaptic protein PSD-95 in hippocampus and orbitofrontal cortex in schizophrenia and mood disorder. Schizophr. Res. 2005, 80, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, L.V.; Meador-Woodruff, J.H. Abnormal striatal expression of transcripts encoding NMDA interacting PSD proteins in schizophrenia, bipolar disorder and major depression. Schizophr. Res. 2005, 78, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.K.; Lee, C.H.; Cho, H.Y.; You, Y.S.; Lee, B.J.; Lee, J.G.; Park, S.W.; Kim, Y.H. Effects of antipsychotic drugs on the expression of synapse-associated proteins in the frontal cortex of rats subjected to immobilization stress. Psychiatry Res. 2015, 229, 968–974. [Google Scholar] [CrossRef] [PubMed]
- De Bartolomeis, A.; Sarappa, C.; Buonaguro, E.F.; Marmo, F.; Eramo, A.; Tomasetti, C.; Iasevoli, F. Different effects of the NMDA receptor antagonists ketamine, MK-801, and memantine on postsynaptic density transcripts and their topography: Role of Homer signaling, and implications for novel antipsychotic and pro-cognitive targets in psychosis. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 46, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Schmeisser, M.J. Translational neurobiology in Shank mutant mice—Model systems for neuropsychiatric disorders. Ann. Anat. 2015, 200, 115–117. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sun, X.Y.; Zhang, L.Y. MicroRNA-7/Shank3 axis involved in schizophrenia pathogenesis. J. Clin. Neurosci. 2015, 22, 1254–1257. [Google Scholar] [CrossRef] [PubMed]
- Sala, C.; Vicidomini, C.; Bigi, I.; Mossa, A.; Verpelli, C. Shank synaptic scaffold proteins: Keys to understanding the pathogenesis of autism and other synaptic disorders. J. Neurochem. 2015, 135, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Wang, H. SHANK1 and autism spectrum disorders. Sci. China Life Sci. 2015, 58, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Mashayekhi, F.; Mizban, N.; Bidabadi, E.; Salehi, Z. The Association of SHANK3 Gene Polymorphism and Autism. Minerva Pediatr. 2016. Available online: http://europepmc.org/abstract/med/27271042 (accessed on 20 October 2016). [Google Scholar]
- Tamura, M.; Mukai, J.; Gordon, J.A.; Gogos, J.A. Developmental Inhibition of Gsk3 Rescues Behavioral and Neurophysiological Deficits in a Mouse Model of Schizophrenia Predisposition. Neuron 2016, 89, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Dachtler, J.; Elliott, C.; Rodgers, R.J.; Baillie, G.S.; Clapcote, S.J. Missense mutation in DISC1 C-terminal coiled-coil has GSK3beta signaling and sex-dependent behavioral effects in mice. Sci. Rep. 2016, 6, 18748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del’Guidice, T.; Latapy, C.; Rampino, A.; Khlghatyan, J.; Lemasson, M.; Gelao, B.; Quarto, T.; Rizzo, G.; Barbeau, A.; Lamarre, C.; et al. FXR1P is a GSK3beta substrate regulating mood and emotion processing. Proc. Natl. Acad. Sci. USA 2015, 112, E4610–E4619. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, M.; Waheed Khan, R.A.; He, K.; Wang, Q.; Li, Z.; Shen, J.; Song, Z.; Li, W.; Wen, Z.; et al. The GSK3B gene confers risk for both major depressive disorder and schizophrenia in the Han Chinese population. J. Affect. Disord. 2015, 185, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Luca, A.; Calandra, C.; Luca, M. Gsk3 Signalling and Redox Status in Bipolar Disorder: Evidence from Lithium Efficacy. Oxidative Med. Cell. Longev. 2016, 2016, 3030547. [Google Scholar] [CrossRef] [PubMed]
- Madison, J.M.; Zhou, F.; Nigam, A.; Hussain, A.; Barker, D.D.; Nehme, R.; van der Ven, K.; Hsu, J.; Wolf, P.; Fleishman, M.; et al. Characterization of bipolar disorder patient-specific induced pluripotent stem cells from a family reveals neurodevelopmental and mRNA expression abnormalities. Mol. Psychiatry 2015, 20, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Huang, X.F.; Deng, C. Aripiprazole and Haloperidol Activate GSK3beta-Dependent Signalling Pathway Differentially in Various Brain Regions of Rats. Int. J. Mol. Sci. 2016, 17, 459. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Zhang, X.; Cui, X.; Zhu, D.; Wu, J.; Sun, D.; Yue, Q.; Li, Z.; Liu, H.; Li, G.; et al. Paliperidone protects SK-N-SH cells against glutamate toxicity via Akt1/GSK3β signaling pathway. Schizophr. Res. 2014, 157, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Sutton, L.P.; Rushlow, W.J. The effects of neuropsychiatric drugs on glycogen synthase kinase-3 signaling. Neuroscience 2011, 199, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, J.M.; Gainetdinov, R.R.; Caron, M.G. Akt/GSK3 signaling in the action of psychotropic drugs. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 327–347. [Google Scholar] [CrossRef] [PubMed]
- Xing, B.; Liang, X.P.; Liu, P.; Zhao, Y.; Chu, Z.; Dang, Y.H. Valproate Inhibits Methamphetamine Induced Hyperactivity via Glycogen Synthase Kinase 3beta Signaling in the Nucleus Accumbens Core. PLoS ONE 2015, 10, e0128068. [Google Scholar] [CrossRef] [PubMed]
- Niwa, M.; Cash-Padgett, T.; Kubo, K.I.; Saito, A.; Ishii, K.; Sumitomo, A.; Taniguchi, Y.; Ishizuka, K.; Jaaro-Peled, H.; Tomoda, T.; et al. DISC1 a key molecular lead in psychiatry and neurodevelopment: No-More Disrupted-in-Schizophrenia 1. Mol. Psychiatry 2016, 21, 1488–1489. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Golbaz, K.; Honer, W.G.; Beasley, C.L. Deficits in axon-associated proteins in prefrontal white matter in bipolar disorder but not schizophrenia. Bipolar Disord. 2016, 18, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Estrada, J.; Benitez-King, G.; Berlanga, C.; Meza, I. Altered subcellular distribution of the 75-kDa DISC1 isoform, cAMP accumulation, and decreased neuronal migration in schizophrenia and bipolar disorder: Implications for neurodevelopment. CNS Neurosci. Ther. 2015, 21, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S.; Hashimoto, R.; Hattori, S.; Yohda, M.; Lipska, B.; Weinberger, D.R.; Kunugi, H. Effect of antipsychotic drugs on DISC1 and dysbindin expression in mouse frontal cortex and hippocampus. J. Neural Transm. 2006, 113, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Mouaffak, F.; Kebir, O.; Chayet, M.; Tordjman, S.; Vacheron, M.N.; Millet, B.; Jaafari, N.; Bellon, A.; Olie, J.P.; Krebs, M.O. Association of Disrupted in Schizophrenia 1 (DISC1) missense variants with ultra-resistant schizophrenia. Pharmacogenom. J. 2011, 11, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Robison, A.J. Emerging role of CaMKII in neuropsychiatric disease. Trends Neurosci. 2014, 37, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Zhou, T.; Liao, L.; Yang, Z.; Wong, C.; Henn, F.; Malinow, R.; Yates III, J.R.; Hu, H. betaCaMKII in lateral habenula mediates core symptoms of depression. Science 2013, 341, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Purkayastha, S.; Ford, J.; Kanjilal, B.; Diallo, S.; Del Rosario Inigo, J.; Neuwirth, L.; El Idrissi, A.; Ahmed, Z.; Wieraszko, A.; Azmitia, E.C.; et al. Clozapine functions through the prefrontal cortex serotonin 1A receptor to heighten neuronal activity via calmodulin kinase II-NMDA receptor interactions. J. Neurochem. 2012, 120, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Rushlow, W.J.; Seah, C.; Sutton, L.P.; Bjelica, A.; Rajakumar, N. Antipsychotics affect multiple calcium calmodulin dependent proteins. Neuroscience 2009, 161, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Browning, J.L.; Patel, T.; Brandt, P.C.; Young, K.A.; Holcomb, L.A.; Hicks, P.B. Clozapine and the mitogen-activated protein kinase signal transduction pathway: Implications for antipsychotic actions. Biol. Psychiatry 2005, 57, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Robison, A.J.; Vialou, V.; Sun, H.S.; Labonte, B.; Golden, S.A.; Dias, C.; Turecki, G.; Tamminga, C.; Russo, S.; Mazei-Robison, M.; et al. Fluoxetine epigenetically alters the CaMKIIα promoter in nucleus accumbens to regulate DFosB binding and antidepressant effects. Neuropsychopharmacology 2014, 39, 1178–1186. [Google Scholar] [CrossRef] [PubMed]


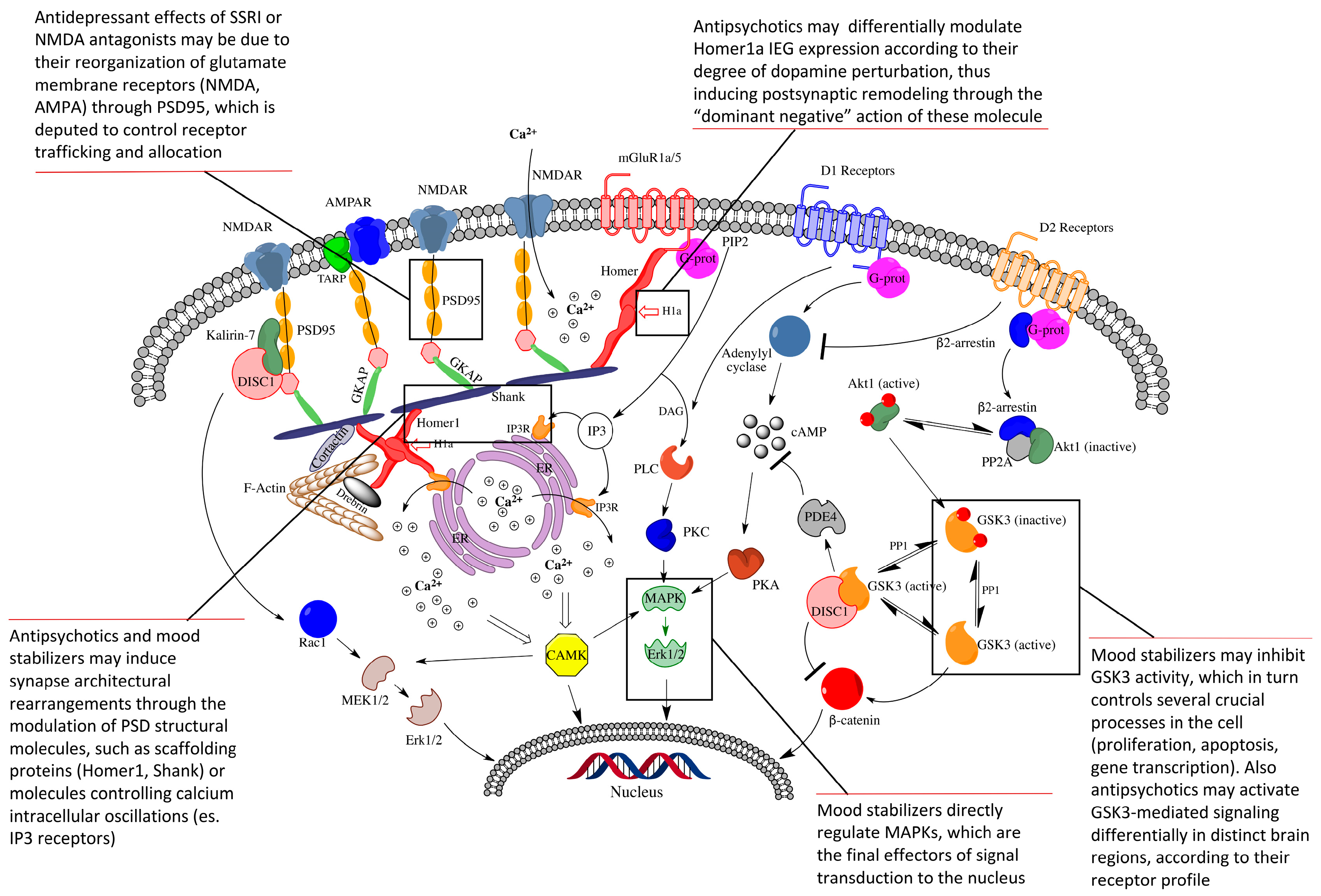{kind=link}
| PSD Molecule | Involvement in Major Neuropsychiatric Disorders | Modulation by Psychopharmacologic Drugs |
|---|---|---|
| Homer 1 | -Schizophrenia [2,65,66,115,116,117,118] -Bipolar Disorder [9] -Major Depressive Disorder [119,120] -Drug addiction [121,122,123,124,125,126,127,128] -Chronic inflammatory pain [129,130] -Fragile X Syndrome [131] -Alzheimer’s Disease [132] -Parkinson’s Disease [133,134] -Traumatic brain injury [135,136] | -Homer 1a may be differentially modulated by both first generation and second generation antipsychotics tightly depending on their own individual receptor profile [42,61,62,137,138] -The mood stabilizers lithium and valproate have scarce effects on Homer 1a expression, whereas they deeply impact synaptic structure conformation by modulating constitutive Homer 1b/c gene expression [71] -Combination of antipsychotics and mood stabilizers elicits changes in Homer 1a gene expression that are substantially different from those induced by these drugs individually administered [72] -Antidepressants and serotonin-modulating antipsychotics induce peculiar cortical expression of Homer 1a in brain regions relevant for negative and cognitive symptoms of schizophrenia [62,84] |
| Homer 2 | -Schizophrenia [139] -Alcohol abuse [140,141,142] | -Chronic haloperidol and clozapine administration may induce overexpression of Homer 2 in lateral septum in animal models [62] |
| Homer 3 | -Cerebellar ataxias [143,144] | |
| PSD-95 | -Schizophrenia [46,145] -Autism Spectrum Disorders [46,146] -Bipolar Disorder [9,147] -Major Depressive Disorder [148] | -Lurasidone and fluoxetine decrease PSD-95 expression in prefrontal cortex and hippocampus [81] -Olanzapine and aripiprazole may reverse the immobilization stress-induced decrease in PSD-95 levels in frontal cortex [149] -PSD-95 is crucial for serotonin 5HT2A and 5HT2C receptors expression and abolishing its expression in knockout animals impairs atypical antipsychotics effects [64] -Ketamine impacts PSD-95 expression in cortical and striatal regions [150], and PSD-95 seems to be crucial for ketamine antidepressant effects [95] |
| Shank | -Schizophrenia [44,151,152] -Autism Spectrum Disorders [153,154,155] | -The mood stabilizers lithium and valproate may down-regulate Shank cortical expression when chronically administered in animal models [71] |
| GSK3β | -Schizophrenia [156,157,158,159] -Major Depressive Disorder [158,159] -Bipolar Disorder [160,161] | -Aripiprazole activates GSK3β signaling in prefrontal cortex and nucleus accumbens, whereas haloperidol activates GSK3β signaling only in nucleus accumbens [162] -Paliperidone exerts protective effects on neurons via decreasing glutamate-induced overactivation of GSK3β signaling [163] -Clozapine may increase GSK3β signaling in prefrontal cortex, but not in striatum, where it is activated by haloperidol [164] -Fluoxetine and imipramine have scarce effects on GSK3β signaling [164] -The inhibition of GSK3β signaling seems to be a crucial mechanism explaining mood stabilizing effects of lithium [165] -Valproate inhibits metamphetamine-induced hyperlocomotion via decreasing GSK3β activity [166] |
| DISC1 | -Schizophrenia [157,167] -Bipolar Disorder [168,169] | -Atypical antipsychotics may increase cortical expression of DISC1, whereas typical antipsychotics have no effects [170] -Specific genomic variants in DISC1 gene in humans have been associated to ultra-resistance to antipsychotic treatment [171] |
| CAMKII | -Schizophrenia [172] -Major Depressive Disorder [173] | -Clozapine-induced increase in prefrontal cortex activity is crucially mediated by CAMKII-NMDA receptor interactions [174] -Clozapine, haloperidol and risperidone may decrease CAMKII expression in striatum in animal models [175] -CAMKII is essential for clozapine-mediated effects on conditioned avoidance responses in animal models [176] -Fluoxetine may induce changes in CAMKII promoter [177] |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomasetti, C.; Iasevoli, F.; Buonaguro, E.F.; De Berardis, D.; Fornaro, M.; Fiengo, A.L.C.; Martinotti, G.; Orsolini, L.; Valchera, A.; Di Giannantonio, M.; et al. Treating the Synapse in Major Psychiatric Disorders: The Role of Postsynaptic Density Network in Dopamine-Glutamate Interplay and Psychopharmacologic Drugs Molecular Actions. Int. J. Mol. Sci. 2017, 18, 135. https://doi.org/10.3390/ijms18010135
Tomasetti C, Iasevoli F, Buonaguro EF, De Berardis D, Fornaro M, Fiengo ALC, Martinotti G, Orsolini L, Valchera A, Di Giannantonio M, et al. Treating the Synapse in Major Psychiatric Disorders: The Role of Postsynaptic Density Network in Dopamine-Glutamate Interplay and Psychopharmacologic Drugs Molecular Actions. International Journal of Molecular Sciences. 2017; 18(1):135. https://doi.org/10.3390/ijms18010135
Chicago/Turabian StyleTomasetti, Carmine, Felice Iasevoli, Elisabetta Filomena Buonaguro, Domenico De Berardis, Michele Fornaro, Annastasia Lucia Carmela Fiengo, Giovanni Martinotti, Laura Orsolini, Alessandro Valchera, Massimo Di Giannantonio, and et al. 2017. "Treating the Synapse in Major Psychiatric Disorders: The Role of Postsynaptic Density Network in Dopamine-Glutamate Interplay and Psychopharmacologic Drugs Molecular Actions" International Journal of Molecular Sciences 18, no. 1: 135. https://doi.org/10.3390/ijms18010135
APA StyleTomasetti, C., Iasevoli, F., Buonaguro, E. F., De Berardis, D., Fornaro, M., Fiengo, A. L. C., Martinotti, G., Orsolini, L., Valchera, A., Di Giannantonio, M., & De Bartolomeis, A. (2017). Treating the Synapse in Major Psychiatric Disorders: The Role of Postsynaptic Density Network in Dopamine-Glutamate Interplay and Psychopharmacologic Drugs Molecular Actions. International Journal of Molecular Sciences, 18(1), 135. https://doi.org/10.3390/ijms18010135