
Prelysosomal Compartments in the Unconventional Secretion of Amyloidogenic Seeds


Abstract
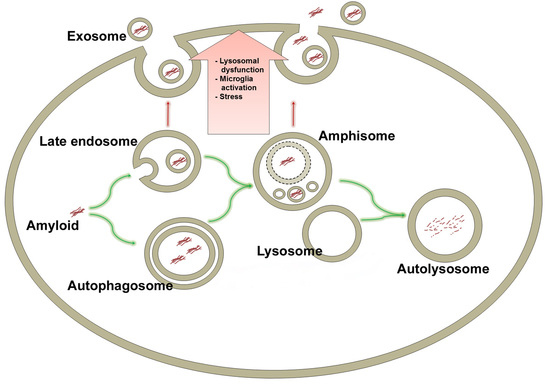
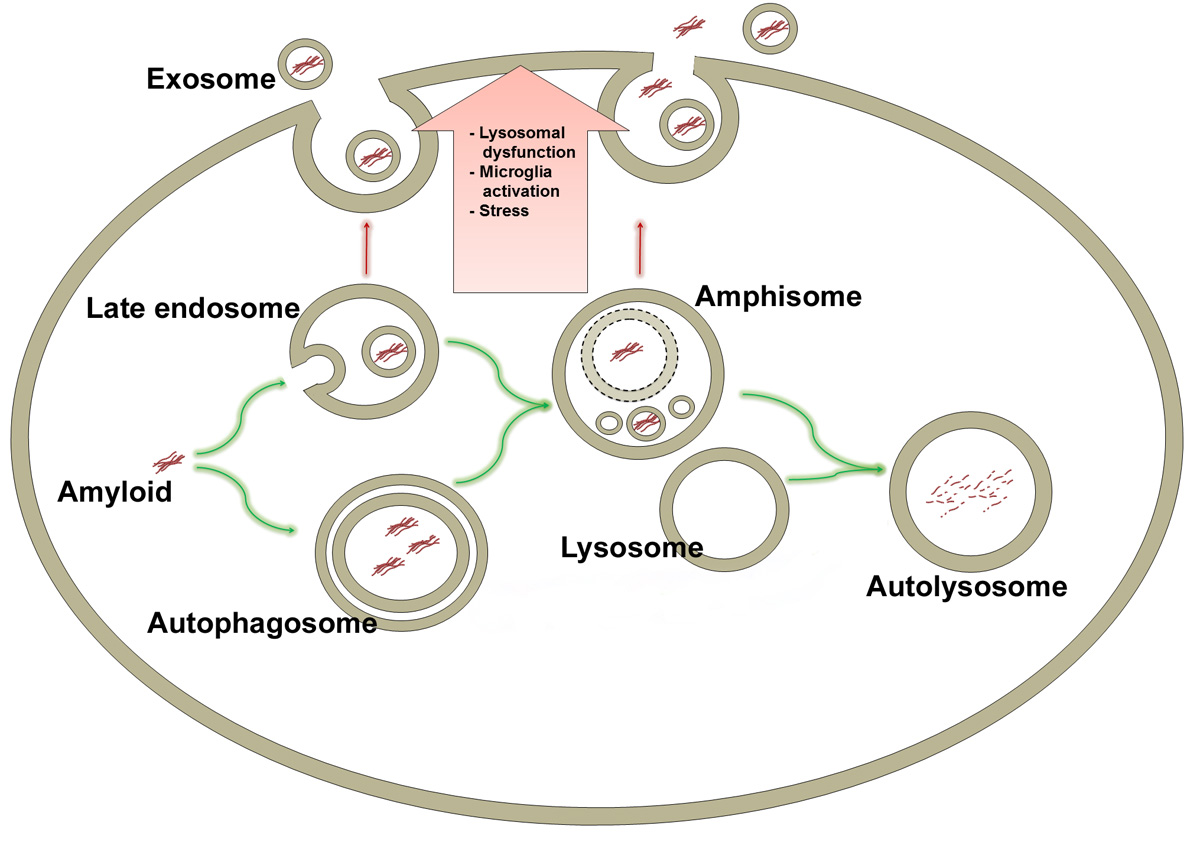
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Seeded Conversion and Transmission of Amyloid Proteopathy In Vivo and In Vitro
2.1. Seeded Conversion
2.2. The Unit Operations: Release, Transmission, and Uptake
2.3. In Vivo Transmission of Proteopathic Amyloids
3. The Endosomal Pathway
3.1. ESCRT-Dependent and -Independent Loading of Intraluminal Vesicles (~Exosomes)
3.2. Final Endosome Maturation
4. The Autophagosomal Pathway
4.1. Autophagosome Nucleation
Selective Autophagy
4.2. Autophagosome Maturation
Autophagosome Mobility
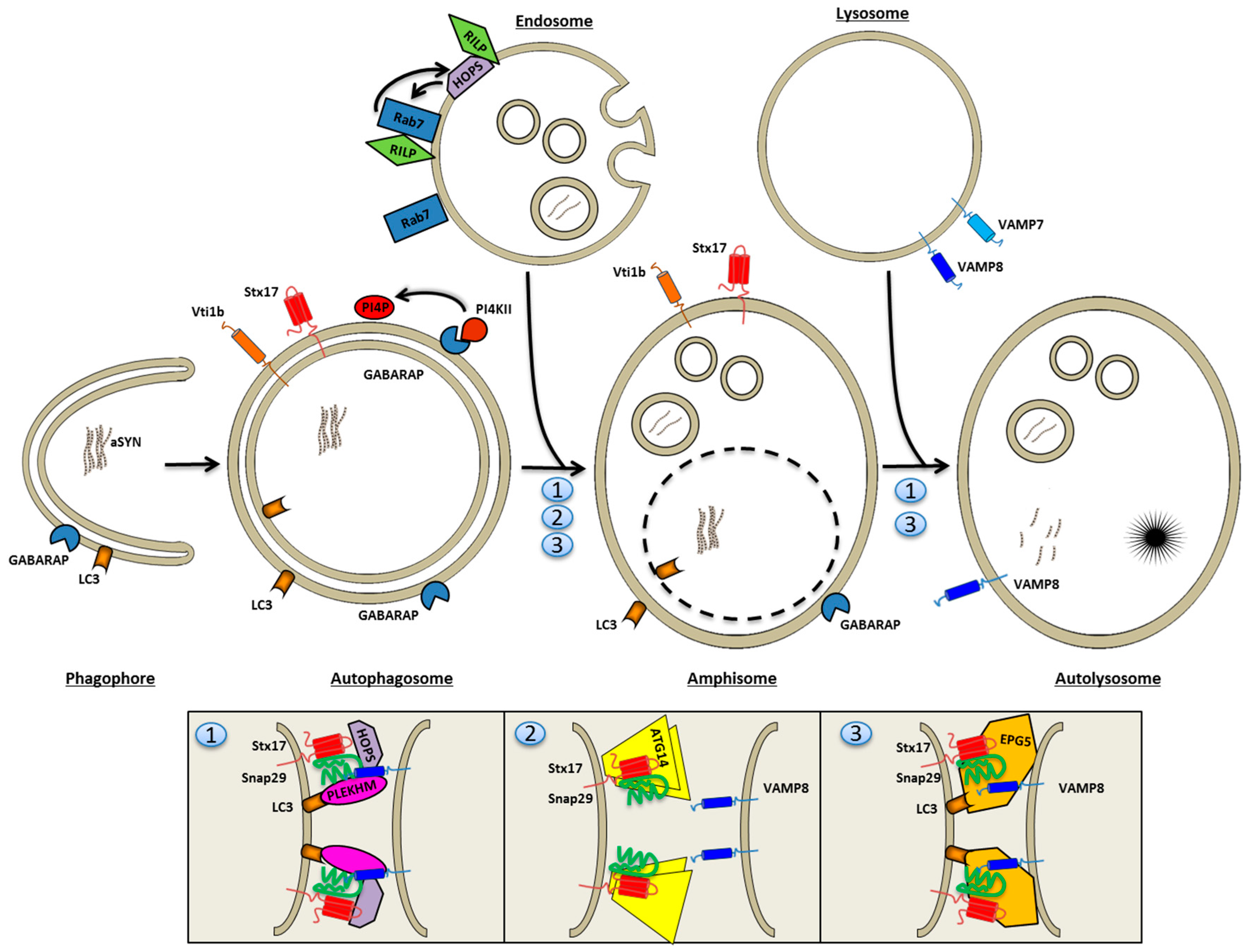
4.3. Autophagosome Fusion with Endolysosomes
5. Amyloids Are Substrates of the Autophagosomal Machinery
6. Late Endosomes and Amphisomes in Secretion of Amyloids
6.1. Unconventional Secretion
6.2. Exocytosis of Amyloid by MVBs and Late Endosomes
6.2.1. MVB and Late Endosome Secretion of Aβ
6.2.2. MVB and Late Endosome Secretion of α-Synuclein and Other Cytosolic Amyloids
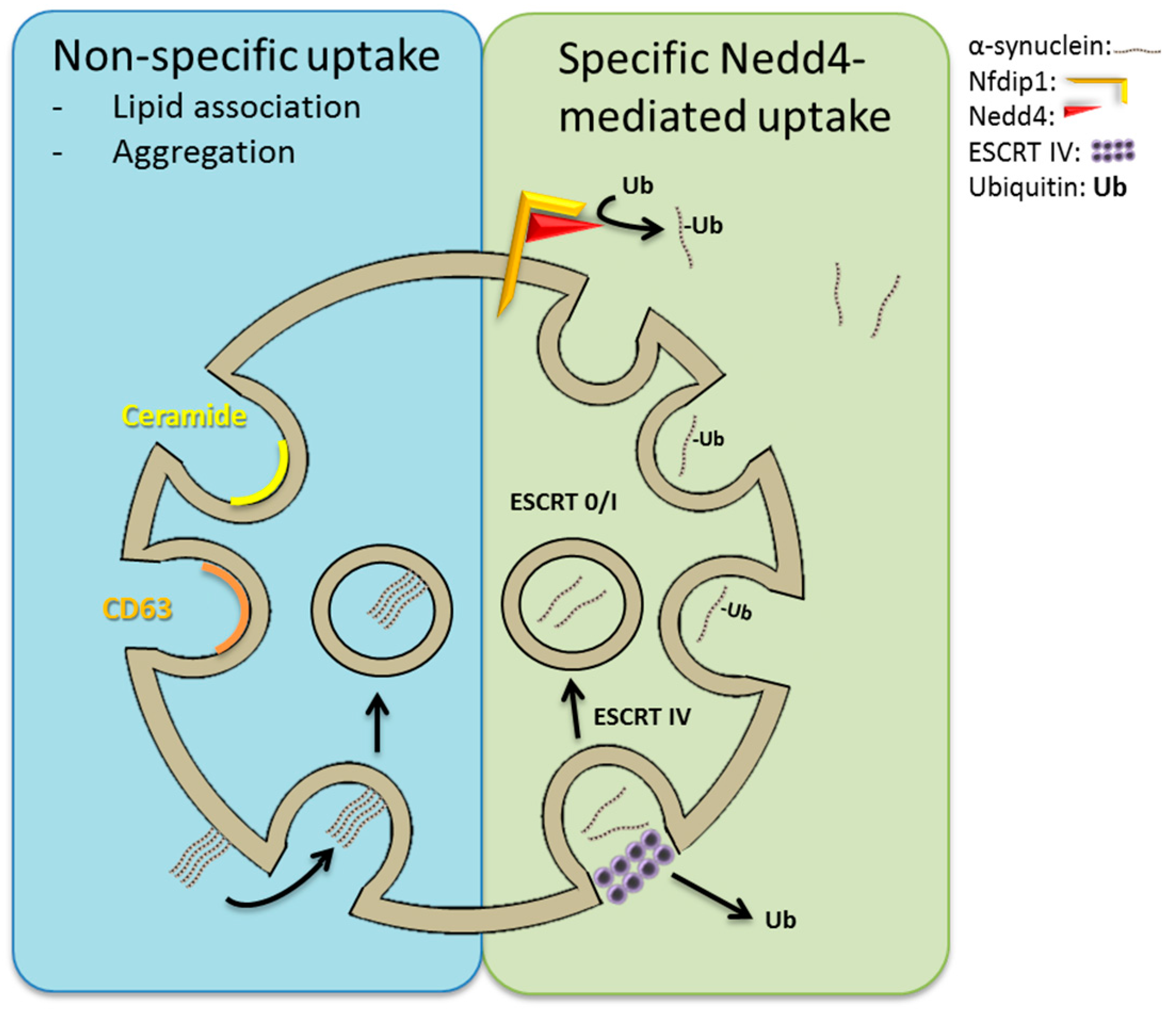
7. Neuronal Precursor Cell-Expressed Developmentally Downregulated 4 (Nedd4)-Mediated Import of α-Synuclein into ILVs?
8. Exocytosis of Amyloid by Amphisomes
Secretion of α-Synuclein by Exophagy
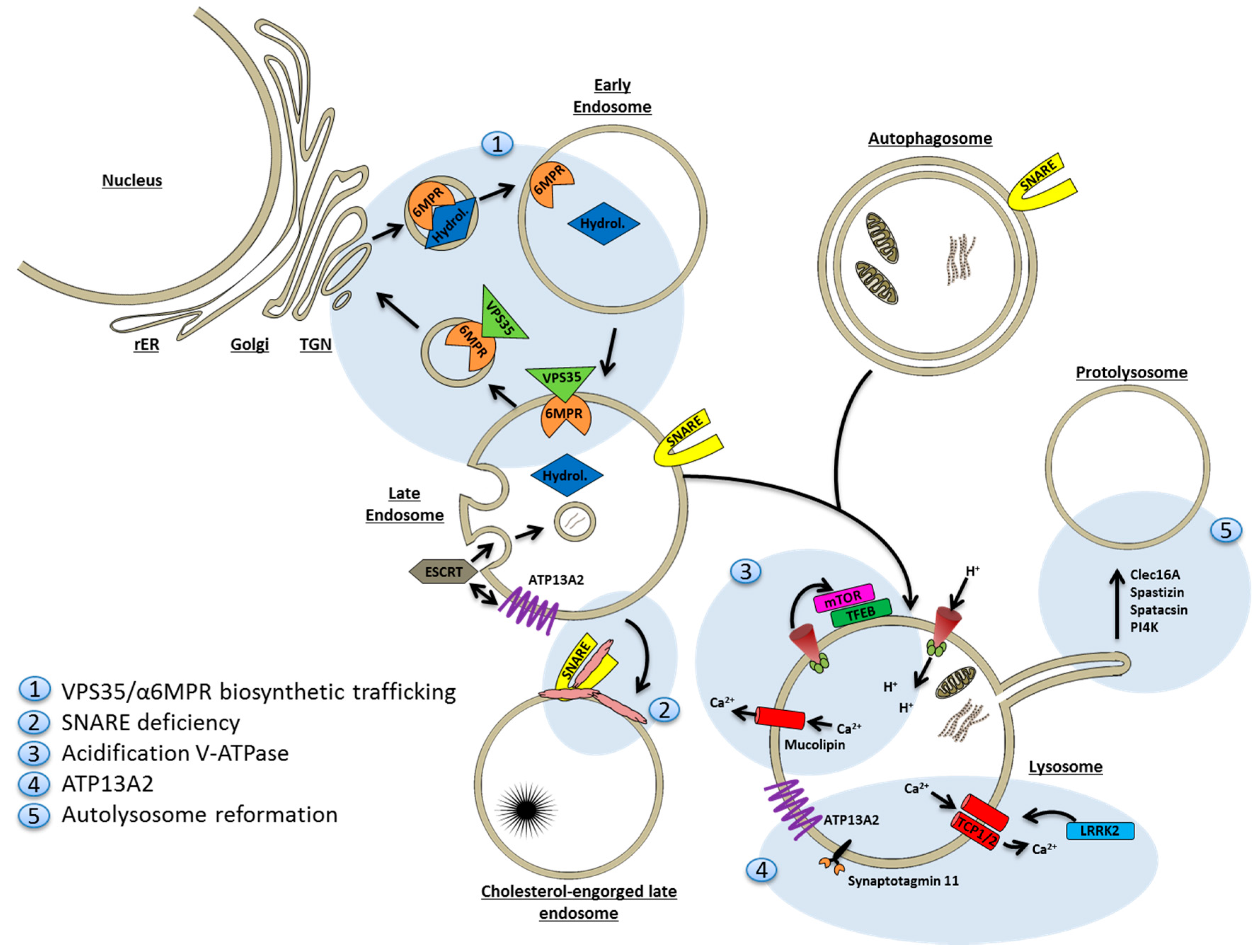
9. Lysosomal Deficiency
9.1. Lysosomes and Autophagy Are Closely Interrelated
9.2. Mechanisms of Lysosomal Deficiency in Neurodegenerative Disease
9.2.1. Deficiency of Degradative Hydrolases
9.2.2. Sequestration of Fusion Machinery Components
9.2.3. Lysosomal Proton and Calcium Imbalances
9.2.4. ATP13A2
9.2.5. Faulty Lysosome Regeneration
9.3. Exocytosis of Amyloid by Lysosomes
10. Microglia in Disease Dissemination
11. Aging, Proteostasis, and Autophagy
12. Therapeutic Implications
13. Conclusions and Perspectives
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | Amyloid β |
| Acyl-CoA | Acetyl coenzyme A |
| AD | Alzheimer’s Disease |
| AMPK | AMP-activated protein kinase |
| APP | Amyloid precursor protein |
| ARF6 | Adenosine diphosphate-ribosylation factor 6 |
| ATG | Autophagy-related |
| ATPase | Adenosine Triphosphatase |
| BAG | Bcl-2 associated athanogene |
| C. elegans | Caenorhabditis elegans |
| ER | Endoplasmic reticulum |
| EPG5 | Ectopic P-Granules Autophagy Protein 5 Homolog |
| ESCRT | Endosomal sorting complex required for transport |
| FcγR | Fc receptors for IgG |
| GABARAP | GABA(A) receptor-associated protein |
| GFP | Green fluorescent protein |
| GRASP | Golgi reassembly stacking protein |
| GTPase | Guanosine triphosphatase |
| HDAC6 | histone deacetylase 6 |
| HMBG1 | High mobility group box 1 |
| HOPS | Homotypic fusion and protein sorting |
| Hrs | Hepatocyte growth factor-regulated substrate |
| ILV | Intraluminal vesicle |
| IL-1β | Interleukin-1 β |
| LAMP | Lysosome-associated membrane protein-2 |
| LC3 | Microtubule-associated proteins 1A/1B light chain 3B |
| LRRK2 | Leucine-rich repeat kinase 2 |
| M6PR | Mannose-6-phosphate receptor |
| MTOC | Microtubule-organizing center |
| mTOR | Mechanistic target of rapamycin |
| MVB | Multivesicular body |
| NBR1 | Neighbor of BRCA1 gene 1 |
| NDP52 | Nuclear domain 10 protein 52 |
| Nedd4 | Neuronal precursor cell-expressed developmentally downregulated 4 |
| Nfdip1 | Nedd family interacting protein 1 |
| PD | Parkinson’s Disease |
| PI(3/4)P | phosphatidylinositol (3/4)-phosphate |
| PI3K | Phosphoinositide 3-kinase |
| PI4K | Phosphatidylinositol 4-kinase |
| PLEKHM | Pleckstrin homology domain-containing family M |
| PSEN | Presenilin |
| PrP(sc) | Prion protein (scrapie associated) |
| RAGE | Receptor for advanced glycation endproducts |
| Ral | Ras-related protein Ral |
| RILP | Rab interacting lysosomal protein |
| RUBICON | Run domain Beclin-1-interacting and cysteine-rich domain-containing protein |
| SNAP | Synaptosomal-associated protein |
| SNARE | Soluble N-ethylmaleimide-sensitive factor-attachment protein receptors |
| SOD | Superoxide dismutase |
| STAM | Signal-transducing adaptor molecule |
| SYT11 | Synaptotagmin 11 |
| TFEB | Transcription factor EB |
| TGN | Trans-Golgi network |
| TLRs | Toll-like receptors |
| TPC | Two-pore channel |
| TPPP | Tubulin polymerization-promoting protein |
| ULK | The ser/thr-kinase uncoordinated family member-51-like kinase |
| UVRAG | UV radiation resistance-associated gene protein |
| Vam7 | Vacuolar morphogenesis protein 7 |
| VAMP | Vesicle-associated membrane protein |
| Vti1b | Vesicle transport through interaction with t-SNAREs homolog 1B |
| (C-)VPS | (Class C-) Vacuolar protein sorting-associated protein |
References
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Sawaya, M.R.; Sambashivan, S.; Nelson, R.; Ivanova, M.I.; Sievers, S.A.; Apostol, M.I.; Thompson, M.J.; Balbirnie, M.; Wiltzius, J.J.; McFarlane, H.T.; et al. Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature 2007, 447, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Luehmann, M.; Coomaraswamy, J.; Bolmont, T.; Kaeser, S.; Schaefer, C.; Kilger, E.; Neuenschwander, A.; Abramowski, D.; Frey, P.; Jaton, A.L.; et al. Exogenous induction of cerebral β-amyloidogenesis is governed by agent and host. Science 2006, 313, 1781–1784. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.; Diamond, M.I. Prion-like mechanisms in neurodegenerative diseases. Nat. Rev. Neurosci. 2009, 11, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Diogenes, M.J.; Dias, R.B.; Rombo, D.M.; Miranda, H.V.; Maiolino, F.; Guerreiro, P.; Nasstrom, T.; Franquelim, H.G.; Oliveira, L.M.; Castanho, M.A.; et al. Extracellular α-synuclein oligomers modulate synaptic transmission and impair LTP via NMDA-receptor activation. J. Neurosci. 2012, 32, 11750–11762. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, T.; Matsuyama, S.; Iso, H.; Umeda, T.; Takuma, H.; Ohnishi, K.; Ishibashi, K.; Teraoka, R.; Sakama, N.; Yamashita, T.; et al. A mouse model of amyloid β oligomers: Their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J. Neurosci. 2010, 30, 4845–4856. [Google Scholar] [CrossRef] [PubMed]
- Peelaerts, W.; Bousset, L.; Van der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; Van den Haute, C.; Melki, R.; Baekelandt, V. Α-synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015, 522, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, J.T.; Lansbury, P.T., Jr. Seeding “one-dimensional crystallization” of amyloid: A pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 1993, 73, 1055–1058. [Google Scholar] [CrossRef]
- Knowles, T.P.; Waudby, C.A.; Devlin, G.L.; Cohen, S.I.; Aguzzi, A.; Vendruscolo, M.; Terentjev, E.M.; Welland, M.E.; Dobson, C.M. An analytical solution to the kinetics of breakable filament assembly. Science 2009, 326, 1533–1537. [Google Scholar] [CrossRef] [PubMed]
- Flagmeier, P.; Meisl, G.; Vendruscolo, M.; Knowles, T.P.; Dobson, C.M.; Buell, A.K.; Galvagnion, C. Mutations associated with familial Parkinson’s disease alter the initiation and amplification steps of α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2016, 113, 10328–10333. [Google Scholar] [CrossRef] [PubMed]
- Buell, A.K.; Galvagnion, C.; Gaspar, R.; Sparr, E.; Vendruscolo, M.; Knowles, T.P.; Linse, S.; Dobson, C.M. Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2014, 111, 7671–7676. [Google Scholar] [CrossRef] [PubMed]
- Bousset, L.; Pieri, L.; Ruiz-Arlandis, G.; Gath, J.; Jensen, P.H.; Habenstein, B.; Madiona, K.; Olieric, V.; Bockmann, A.; Meier, B.H.; et al. Structural and functional characterization of two α-synuclein strains. Nat. Commun. 2013, 4, 2575. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.X.; Qiang, W.; Yau, W.M.; Schwieters, C.D.; Meredith, S.C.; Tycko, R. Molecular structure of β-amyloid fibrils in Alzheimer’s disease brain tissue. Cell 2013, 154, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B.; Woerman, A.L.; Mordes, D.A.; Watts, J.C.; Rampersaud, R.; Berry, D.B.; Patel, S.; Oehler, A.; Lowe, J.K.; Kravitz, S.N.; et al. Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. USA 2015, 112, E5308–E5317. [Google Scholar] [CrossRef] [PubMed]
- Woerman, A.L.; Stohr, J.; Aoyagi, A.; Rampersaud, R.; Krejciova, Z.; Watts, J.C.; Ohyama, T.; Patel, S.; Widjaja, K.; Oehler, A.; et al. Propagation of prions causing synucleinopathies in cultured cells. Proc. Natl. Acad. Sci. USA 2015, 112, E4949–E4958. [Google Scholar] [CrossRef] [PubMed]
- Heilbronner, G.; Eisele, Y.S.; Langer, F.; Kaeser, S.A.; Novotny, R.; Nagarathinam, A.; Aslund, A.; Hammarstrom, P.; Nilsson, K.P.; Jucker, M. Seeded strain-like transmission of β-amyloid morphotypes in APP transgenic mice. EMBO Rep. 2013, 14, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Morales, R.; Estrada, L.D.; Diaz-Espinoza, R.; Morales-Scheihing, D.; Jara, M.C.; Castilla, J.; Soto, C. Molecular cross talk between misfolded proteins in animal models of Alzheimer’s and prion diseases. J. Neurosci. 2010, 30, 4528–4535. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Covell, D.J.; Daniels, J.P.; Iba, M.; Stieber, A.; Zhang, B.; Riddle, D.M.; Kwong, L.K.; Xu, Y.; Trojanowski, J.Q.; et al. Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell 2013, 154, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.J.; Lee, V.M.; Trojanowski, J.Q. Parkinson’s disease dementia: Convergence of α-synuclein, tau and amyloid-β pathologies. Nat. Rev. Neurosci. 2013, 14, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Langer, F.; Eisele, Y.S.; Fritschi, S.K.; Staufenbiel, M.; Walker, L.C.; Jucker, M. Soluble Aβ seeds are potent inducers of cerebral β-amyloid deposition. J. Neurosci. 2011, 31, 14488–14495. [Google Scholar] [CrossRef] [PubMed]
- Brundin, P.; Melki, R.; Kopito, R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol. 2010, 11, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Rajendran, L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 2009, 64, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Bae, E.J.; Lee, S.J. Extracellular α-synuclein-a novel and crucial factor in Lewy body diseases. Nat. Rev. Neurol. 2014, 10, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Kfoury, N.; Holmes, B.B.; Jiang, H.; Holtzman, D.M.; Diamond, M.I. Trans-cellular propagation of Tau aggregation by fibrillar species. J. Biol. Chem. 2012, 287, 19440–19451. [Google Scholar] [CrossRef] [PubMed]
- Saman, S.; Kim, W.; Raya, M.; Visnick, Y.; Miro, S.; Saman, S.; Jackson, B.; McKee, A.C.; Alvarez, V.E.; Lee, N.C.; et al. Exosome-associated Tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 2012, 287, 3842–3849. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Gardiner, C.; Sargent, I.L.; Wood, M.J.; Cooper, J.M. Lysosomal dysfunction increases exosome-mediated α-synuclein release and transmission. Neurobiol. Dis. 2011, 42, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Danzer, K.M.; Kranich, L.R.; Ruf, W.P.; Cagsal-Getkin, O.; Winslow, A.R.; Zhu, L.; Vanderburg, C.R.; McLean, P.J. Exosomal cell-to-cell transmission of α synuclein oligomers. Mol. Neurodegener. 2012, 7, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ejlerskov, P.; Rasmussen, I.; Nielsen, T.T.; Bergstrom, A.L.; Tohyama, Y.; Jensen, P.H.; Vilhardt, F. Tubulin Polymerization Promoting Protein (TPPP/p25α) promotes unconventional secretion of α-synuclein through exophagy by impairing autophagosome-lysosome fusion. J. Biol. Chem. 2013, 288, 17313. [Google Scholar] [CrossRef] [PubMed]
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-produced α-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 2010, 30, 6838–6851. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, T.; Konno, M.; Baba, T.; Sugeno, N.; Kikuchi, A.; Kobayashi, M.; Miura, E.; Tanaka, N.; Tamai, K.; Furukawa, K.; et al. The AAA-ATPase VPS4 regulates extracellular secretion and lysosomal targeting of α-synuclein. PLoS ONE 2011, 6, e29460. [Google Scholar] [CrossRef] [PubMed]
- Jang, A.; Lee, H.J.; Suk, J.E.; Jung, J.W.; Kim, K.P.; Lee, S.J. Non-classical exocytosis of α-synuclein is sensitive to folding states and promoted under stress conditions. J. Neurochem. 2010, 113, 1263–1274. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Patel, S.; Lee, S.J. Intravesicular localization and exocytosis of α-synuclein and its aggregates. J. Neurosci. 2005, 25, 6016–6024. [Google Scholar] [CrossRef] [PubMed]
- Gomes, C.; Keller, S.; Altevogt, P.; Costa, J. Evidence for secretion of Cu,Zn superoxide dismutase via exosomes from a cell model of amyotrophic lateral sclerosis. Neurosci. Lett. 2007, 428, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Grad, L.I.; Yerbury, J.J.; Turner, B.J.; Guest, W.C.; Pokrishevsky, E.; O’Neill, M.A.; Yanai, A.; Silverman, J.M.; Zeineddine, R.; Corcoran, L.; et al. Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. USA 2014, 111, 3620–3625. [Google Scholar] [CrossRef] [PubMed]
- Desplats, P.; Lee, H.J.; Bae, E.J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.; Angot, E.; Bergstrom, A.L.; Steiner, J.A.; Pieri, L.; Paul, G.; Outeiro, T.F.; Melki, R.; Kallunki, P.; Fog, K.; et al. Α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Investig. 2011, 121, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Konno, M.; Hasegawa, T.; Baba, T.; Miura, E.; Sugeno, N.; Kikuchi, A.; Fiesel, F.C.; Sasaki, T.; Aoki, M.; Itoyama, Y.; et al. Suppression of dynamin GTPase decreases α-synuclein uptake by neuronal and oligodendroglial cells: A potent therapeutic target for synucleinopathy. Mol. Neurodegener. 2012, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Suk, J.E.; Bae, E.J.; Lee, J.H.; Paik, S.R.; Lee, S.J. Assembly-dependent endocytosis and clearance of extracellular α-synuclein. Int. J. Biochem. Cell Biol. 2008, 40, 1835–1849. [Google Scholar] [CrossRef] [PubMed]
- Volpicelli-Daley, L.A.; Luk, K.C.; Patel, T.P.; Tanik, S.A.; Riddle, D.M.; Stieber, A.; Meaney, D.F.; Trojanowski, J.Q.; Lee, V.M. Exogenous α-synuclein fibrils induce lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 2009, 284, 12845–12852. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.W.; Herman, M.; Liu, L.; Simoes, S.; Acker, C.M.; Figueroa, H.; Steinberg, J.I.; Margittai, M.; Kayed, R.; Zurzolo, C.; et al. Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J. Biol. Chem. 2013, 288, 1856–1870. [Google Scholar] [CrossRef] [PubMed]
- Munch, C.; O’Brien, J.; Bertolotti, A. Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3548–3553. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Kim, K.S.; Lee, S.B.; Ryu, J.S.; Chung, K.C.; Choo, Y.K.; Jou, I.; Kim, J.; Park, S.M. On the mechanism of internalization of α-synuclein into microglia: Roles of ganglioside GM1 and lipid raft. J. Neurochem. 2009, 110, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef] [PubMed]
- Um, J.W.; Kaufman, A.C.; Kostylev, M.; Heiss, J.K.; Stagi, M.; Takahashi, H.; Kerrisk, M.E.; Vortmeyer, A.; Wisniewski, T.; Koleske, A.J.; et al. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer aβ oligomer bound to cellular prion protein. Neuron 2013, 79, 887–902. [Google Scholar] [CrossRef] [PubMed]
- Vilhardt, F. Microglia: Phagocyte and glia cell. Int. J. Biochem. Cell Biol. 2005, 37, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia 2012, 2012, 22437. [Google Scholar] [CrossRef] [PubMed]
- Stefanova, N.; Fellner, L.; Reindl, M.; Masliah, E.; Poewe, W.; Wenning, G.K. Toll-like receptor 4 promotes α-synuclein clearance and survival of nigral dopaminergic neurons. Am. J. Pathol. 2011, 179, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Standaert, D.G.; Harms, A.S. The gamma chain subunit of Fc receptors is required for α-synuclein-induced pro-inflammatory signaling in microglia. J. Neuroinflamm. 2012, 9, 259. [Google Scholar] [CrossRef] [PubMed]
- Codolo, G.; Plotegher, N.; Pozzobon, T.; Brucale, M.; Tessari, I.; Bubacco, L.; de Bernard, M. Triggering of inflammasome by aggregated α-synuclein, an inflammatory response in synucleinopathies. PLoS ONE 2013, 8, e55375. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Ho, D.H.; Suk, J.E.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.J.; et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Theodore, S.; Standaert, D.G. Fcgamma receptors are required for NF-kappab signaling, microglial activation and dopaminergic neurodegeneration in an AAV-synuclein mouse model of Parkinson’s disease. Mol. Neurodegner. 2010, 5, 42. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chu, C.H.; Stewart, T.; Ginghina, C.; Wang, Y.; Nie, H.; Guo, M.; Wilson, B.; Hong, J.S.; Zhang, J. Α-synuclein, a chemoattractant, directs microglial migration via H2O2-dependent Lyn phosphorylation. Proc. Natl. Acad. Sci. USA 2015, 112, E1926–E1935. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Cho, E.D.; Kim, H.K.; You, S.; Lee, H.J.; Hwang, D.; Lee, S.J. Β1-integrin-dependent migration of microglia in response to neuron-released α-synuclein. Exp. Mol. Med. 2014, 46, e91. [Google Scholar] [CrossRef] [PubMed]
- Coraci, I.S.; Husemann, J.; Berman, J.W.; Hulette, C.; Dufour, J.H.; Campanella, G.K.; Luster, A.D.; Silverstein, S.C.; El-Khoury, J.B. Cd36, a class B scavenger receptor, is expressed on microglia in Alzheimer’s disease brains and can mediate production of reactive oxygen species in response to β-amyloid fibrils. Am. J. Pathol. 2002, 160, 101–112. [Google Scholar] [CrossRef]
- El Khoury, J.; Hickman, S.E.; Thomas, C.A.; Cao, L.; Silverstein, S.C.; Loike, J.D. Scavenger receptor-mediated adhesion of microglia to β-amyloid fibrils. Nature 1996, 382, 716–719. [Google Scholar] [CrossRef] [PubMed]
- El Khoury, J.B.; Moore, K.J.; Means, T.K.; Leung, J.; Terada, K.; Toft, M.; Freeman, M.W.; Luster, A.D. Cd36 mediates the innate host response to β-amyloid. J. Exp. Med. 2003, 197, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Lue, L.F.; Yan, S.; Xu, H.; Luddy, J.S.; Chen, D.; Walker, D.G.; Stern, D.M.; Yan, S.; Schmidt, A.M.; et al. Rage-dependent signaling in microglia contributes to neuroinflammation, Aβ accumulation, and impaired learning/memory in a mouse model of Alzheimer’s disease. FASEB J. 2010, 24, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Liu, B.; Frost, J.L.; Hong, S.; Jin, M.; Ostaszewski, B.; Shankar, G.M.; Costantino, I.M.; Carroll, M.C.; Mayadas, T.N.; et al. Complement component C3 and complement receptor type 3 contribute to the phagocytosis and clearance of fibrillar Aβ by microglia. Glia 2012, 60, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Jana, M.; Palencia, C.A.; Pahan, K. Fibrillar amyloid-β peptides activate microglia via TLR2: Implications for Alzheimer’s disease. J. Immunol. 2008, 181, 7254–7262. [Google Scholar] [CrossRef] [PubMed]
- Koenigsknecht, J.; Landreth, G. Microglial phagocytosis of fibrillar β-amyloid through a β1 integrin-dependent mechanism. J. Neurosci. 2004, 24, 9838–9846. [Google Scholar] [CrossRef] [PubMed]
- Kounnas, M.Z.; Moir, R.D.; Rebeck, G.W.; Bush, A.I.; Argraves, W.S.; Tanzi, R.E.; Hyman, B.T.; Strickland, D.K. Ldl receptor-related protein, a multifunctional ApoE receptor, binds secreted β-amyloid precursor protein and mediates its degradation. Cell 1995, 82, 331–340. [Google Scholar] [CrossRef]
- Origlia, N.; Bonadonna, C.; Rosellini, A.; Leznik, E.; Arancio, O.; Yan, S.S.; Domenici, L. Microglial receptor for advanced glycation end product-dependent signal pathway drives β-amyloid-induced synaptic depression and long-term depression impairment in entorhinal cortex. J. Neurosci. 2010, 30, 11414–11425. [Google Scholar] [CrossRef] [PubMed]
- Paresce, D.M.; Richik, N.G.; Maxfield, F.R. Microglial cells internalize aggregates of the Alzheimer disease amyloid β-protein via a scavenger receptor. Neuron 1996, 17, 553–565. [Google Scholar] [CrossRef]
- Reed-Geaghan, E.G.; Savage, J.C.; Hise, A.G.; Landreth, G.E. Cd14 and toll-like receptors 2 and 4 are required for fibrillar Aβ-stimulated microglial activation. J. Neurosci. 2009, 29, 11982–11992. [Google Scholar] [CrossRef] [PubMed]
- Richard, K.L.; Filali, M.; Prefontaine, P.; Rivest, S. Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid β 1–42 and delay the cognitive decline in a mouse model of Alzheimer’s disease. J. Neurosci. 2008, 28, 5784–5793. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar, S.; Jiang, Q.; Lee, C.Y.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Landreth, G.E. Microglia mediate the clearance of soluble Aβ through fluid phase macropinocytosis. J. Neurosci. 2009, 29, 4252–4262. [Google Scholar] [CrossRef] [PubMed]
- Yuyama, K.; Sun, H.; Mitsutake, S.; Igarashi, Y. Sphingolipid-modulated exosome secretion promotes clearance of amyloid-β by microglia. J. Biol. Chem. 2012, 287, 10977–10989. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Bjorklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Kehm, V.M.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J. Exp. Med. 2012, 209, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef]
- De Calignon, A.; Polydoro, M.; Suarez-Calvet, M.; William, C.; Adamowicz, D.H.; Kopeikina, K.J.; Pitstick, R.; Sahara, N.; Ashe, K.H.; Carlson, G.A.; et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 2012, 73, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Iba, M.; Guo, J.L.; McBride, J.D.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J. Neurosci. 2013, 33, 1024–1037. [Google Scholar] [CrossRef]
- Liu, L.; Drouet, V.; Wu, J.W.; Witter, M.P.; Small, S.A.; Clelland, C.; Duff, K. Trans-synaptic spread of tau pathology in vivo. PLoS ONE 2012, 7, e31302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, H.F.; Ridley, R.M.; Duchen, L.W.; Crow, T.J.; Bruton, C.J. Induction of β (A4)-amyloid in primates by injection of Alzheimer’s disease brain homogenate. Comparison with transmission of spongiform encephalopathy. Mol. Neurobiol. 1994, 8, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Eisele, Y.S.; Obermuller, U.; Heilbronner, G.; Baumann, F.; Kaeser, S.A.; Wolburg, H.; Walker, L.C.; Staufenbiel, M.; Heikenwalder, M.; Jucker, M. Peripherally applied Aβ-containing inoculates induce cerebral β-amyloidosis. Science 2010, 330, 980–982. [Google Scholar] [CrossRef]
- Nath, S.; Agholme, L.; Kurudenkandy, F.R.; Granseth, B.; Marcusson, J.; Hallbeck, M. Spreading of neurodegenerative pathology via neuron-to-neuron transmission of β-amyloid. J. Neurosci. 2012, 32, 8767–8777. [Google Scholar] [CrossRef] [PubMed]
- Stöhr, J.; Watts, J.; Mensinger, Z.; Oehler, A.; Grillo, S.; DeArmond, S.; Prusiner, S.B.K.G. Purified and synthetic Alzheimer’s amyloid β (Aβ) prions. Proc. Natl. Acad. Sci. USA 2012, 109, 11025–11030. [Google Scholar] [CrossRef] [PubMed]
- Grad, L.I.; Guest, W.C.; Yanai, A.; Pokrishevsky, E.; O’Neill, M.A.; Gibbs, E.; Semenchenko, V.; Yousefi, M.; Wishart, D.S.; Plotkin, S.S.; et al. Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16398–16403. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Freeman, T.B.; Olanow, C.W. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 2008, 14, 504–506. [Google Scholar] [CrossRef] [PubMed]
- Ahn, T.B.; Langston, J.W.; Aachi, V.R.; Dickson, D.W. Relationship of neighboring tissue and gliosis to α-synuclein pathology in a fetal transplant for Parkinson’s disease. Am. J. Neurodegener. Dis. 2012, 1, 49–59. [Google Scholar] [PubMed]
- Kane, M.D.; Lipinski, W.J.; Callahan, M.J.; Bian, F.; Durham, R.A.; Schwarz, R.D.; Roher, A.E.; Walker, L.C. Evidence for seeding of β-amyloid by intracerebral infusion of Alzheimer brain extracts in β-amyloid precursor protein-transgenic mice. J. Neurosci. 2000, 20, 3606–3611. [Google Scholar] [PubMed]
- Clavaguera, F.; Akatsu, H.; Fraser, G.; Crowther, R.A.; Frank, S.; Hench, J.; Probst, A.; Winkler, D.T.; Reichwald, J.; Staufenbiel, M.; et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc. Natl. Acad. Sci. USA 2013, 110, 9535–9540. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K.; Rub, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Rey, N.L.; Steiner, J.A.; Maroof, N.; Luk, K.C.; Madaj, Z.; Trojanowski, J.Q.; Lee, V.M.; Brundin, P. Widespread transneuronal propagation of α-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J. Exp. Med. 2016, 213, 1759–1778. [Google Scholar] [CrossRef] [PubMed]
- Pan-Montojo, F.; Schwarz, M.; Winkler, C.; Arnhold, M.; O’Sullivan, G.A.; Pal, A.; Said, J.; Marsico, G.; Verbavatz, J.M.; Rodrigo-Angulo, M.; et al. Environmental toxins trigger PD-like progression via increased α-synuclein release from enteric neurons in mice. Sci. Rep. 2012, 2, 898. [Google Scholar] [CrossRef] [PubMed]
- Jaunmuktane, Z.; Mead, S.; Ellis, M.; Wadsworth, J.D.; Nicoll, A.J.; Kenny, J.; Launchbury, F.; Linehan, J.; Richard-Loendt, A.; Walker, A.S.; et al. Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature 2015, 525, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.D.; Donaldson, J.G. Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 2009, 10, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Escola, J.M.; Kleijmeer, M.J.; Stoorvogel, W.; Griffith, J.M.; Yoshie, O.; Geuze, H.J. Selective enrichment of tetraspan proteins on the internal vesicles of multivesicular endosomes and on exosomes secreted by human B-lymphocytes. J. Biol. Chem. 1998, 273, 20121–20127. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.L.; Urbe, S. The emerging shape of the ESCRT machinery. Nat. Rev. Mol. Cell Biol. 2007, 8, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; Porto-Carreiro, I.; Simoes, S.; Raposo, G. Exosomes: A common pathway for a specialized function. J. Biochem. 2006, 140, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Simons, M. Exosomes: Vesicular carriers for intercellular communication in neurodegenerative disorders. Cell Tissue Res. 2012, 352, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Gross, J.C.; Chaudhary, V.; Bartscherer, K.; Boutros, M. Active Wnt proteins are secreted on exosomes. Nat. Cell Biol. 2012, 14, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Shields, S.B.; Piper, R.C. How ubiquitin functions with ESCRTs. Traffic 2011, 12, 1306–1317. [Google Scholar] [CrossRef] [PubMed]
- Shields, S.B.; Oestreich, A.J.; Winistorfer, S.; Nguyen, D.; Payne, J.A.; Katzmann, D.J.; Piper, R. Escrt ubiquitin-binding domains function cooperatively during MVB cargo sorting. J. Cell Biol. 2009, 185, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Sachse, M.; Urbe, S.; Oorschot, V.; Strous, G.J.; Klumperman, J. Bilayered clathrin coats on endosomal vacuoles are involved in protein sorting toward lysosomes. Mol. Biol. Cell 2002, 13, 1313–1328. [Google Scholar] [CrossRef] [PubMed]
- Baietti, M.F.; Zhang, Z.; Mortier, E.; Melchior, A.; Degeest, G.; Geeraerts, A.; Ivarsson, Y.; Depoortere, F.; Coomans, C.; Vermeiren, E.; et al. Syndecan-syntenin-Alix regulates the biogenesis of exosomes. Nat. Cell Biol. 2012, 14, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Ghossoub, R.; Lembo, F.; Rubio, A.; Gaillard, C.B.; Bouchet, J.; Vitale, N.; Slavik, J.; Machala, M.; Zimmermann, P. Syntenin-Alix exosome biogenesis and budding into multivesicular bodies are controlled by ARF6 and PLD2. Nat. Commun. 2014, 5, 3477. [Google Scholar] [CrossRef] [PubMed]
- Sharples, R.A.; Vella, L.J.; Nisbet, R.M.; Naylor, R.; Perez, K.; Barnham, K.J.; Masters, C.L.; Hill, A.F. Inhibition of gamma-secretase causes increased secretion of amyloid precursor protein C-terminal fragments in association with exosomes. FASEB J. 2008, 22, 1469–1478. [Google Scholar] [CrossRef] [PubMed]
- Vingtdeux, V.; Hamdane, M.; Loyens, A.; Gele, P.; Drobeck, H.; Begard, S.; Galas, M.C.; Delacourte, A.; Beauvillain, J.C.; Buee, L.; et al. Alkalizing drugs induce accumulation of amyloid precursor protein by-products in luminal vesicles of multivesicular bodies. J. Biol. Chem. 2007, 282, 18197–18205. [Google Scholar] [CrossRef] [PubMed]
- Pashkova, N.; Gakhar, L.; Winistorfer, S.C.; Sunshine, A.B.; Rich, M.; Dunham, M.J.; Yu, L.; Piper, R.C. The yeast Alix homolog Bro1 functions as a ubiquitin receptor for protein sorting into multivesicular endosomes. Dev. Cell 2013, 25, 520–533. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; Charrin, S.; Simoes, S.; Romao, M.; Rochin, L.; Saftig, P.; Marks, M.S.; Rubinstein, E.; Raposo, G. The tetraspanin CD63 regulates ESCRT-independent and -dependent endosomal sorting during melanogenesis. Dev. Cell 2011, 21, 708–721. [Google Scholar] [CrossRef] [PubMed]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brugger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Wu, N.; Gan, X.; Yan, W.; Morrell, J.C.; Gould, S.J. Higher-order oligomerization targets plasma membrane proteins and HIV gag to exosomes. PLoS Biol. 2007, 5, e158. [Google Scholar] [CrossRef] [PubMed]
- Edgar, J.R.; Eden, E.R.; Futter, C.E. Hrs- and CD63-dependent competing mechanisms make different sized endosomal intraluminal vesicles. Traffic 2014, 15, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Pryor, P.R.; Mullock, B.M.; Bright, N.A.; Lindsay, M.R.; Gray, S.R.; Richardson, S.C.; Stewart, A.; James, D.E.; Piper, R.C.; Luzio, J.P. Combinatorial SNARE complexes with VAMP7 or VAMP8 define different late endocytic fusion events. EMBO Rep. 2004, 5, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef] [PubMed]
- Kraft, C.; Peter, M.; Hofmann, K. Selective autophagy: Ubiquitin-mediated recognition and beyond. Nat. Cell Biol. 2010, 12, 836–841. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Klionsky, D.J. Autophagosome formation: Core machinery and adaptations. Nat. Cell Biol. 2007, 9, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Sahu, R.; Kaushik, S.; Clement, C.C.; Cannizzo, E.S.; Scharf, B.; Follenzi, A.; Potolicchio, I.; Nieves, E.; Cuervo, A.M.; Santambrogio, L. Microautophagy of cytosolic proteins by late endosomes. Dev. Cell 2011, 20, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef] [PubMed]
- Mehrpour, M.; Esclatine, A.; Beau, I.; Codogno, P. Overview of macroautophagy regulation in mammalian cells. Cell 2010, 20, 748–762. [Google Scholar] [CrossRef] [PubMed]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009, 11, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Tooze, S.A.; Yoshimori, T. The origin of the autophagosomal membrane. Nat. Cell Biol. 2010, 12, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Longatti, A.; Lamb, C.A.; Razi, M.; Yoshimura, S.; Barr, F.A.; Tooze, S.A. Tbc1d14 regulates autophagosome formation via Rab11- and Ulk1-positive recycling endosomes. J. Cell Biol. 2012, 197, 659–675. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 2010, 12, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Puri, C.; Renna, M.; Bento, C.F.; Moreau, K.; Rubinsztein, D.C. ATG16L1 meets ATG9 in recycling endosomes: Additional roles for the plasma membrane and endocytosis in autophagosome biogenesis. Autophagy 2014, 10, 182–184. [Google Scholar] [CrossRef] [PubMed]
- Puri, C.; Renna, M.; Bento, C.F.; Moreau, K.; Rubinsztein, D.C. Diverse autophagosome membrane sources coalesce in recycling endosomes. Cell 2013, 154, 1285–1299. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; Komatsu, M.; Otsu, K.; Tsujimoto, Y.; Shimizu, S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009, 461, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Jiang, S.; Dupont, N. Autophagy intersections with conventional and unconventional secretion in tissue development, remodeling and inflammation. Trends Cell Biol. 2012, 22, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Biazik, J.; Yla-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.L. Ultrastructural relationship of the phagophore with surrounding organelles. Autophagy 2015, 11, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Mi, N.; Chen, Y.; Wang, S.; Chen, M.; Zhao, M.; Yang, G.; Ma, M.; Su, Q.; Luo, S.; Shi, J.; et al. CapZ regulates autophagosomal membrane shaping by promoting actin assembly inside the isolation membrane. Nat. Cell Biol. 2015, 17, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Coutts, A.S.; La Thangue, N.B. Actin nucleation by WH2 domains at the autophagosome. Nat. Commun. 2015, 6, 7888. [Google Scholar] [CrossRef] [PubMed]
- Kast, D.J.; Zajac, A.L.; Holzbaur, E.L.; Ostap, E.M.; Dominguez, R. Whamm directs the Arp2/3 complex to the ER for autophagosome biogenesis through an actin comet tail mechanism. Curr. Biol. 2015, 25, 1791–1797. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Nair, U.; Klionsky, D.J. Atg8 controls phagophore expansion during autophagosome formation. Mol. Biol. Cell. 2008, 19, 3290–3298. [Google Scholar] [CrossRef] [PubMed]
- Weidberg, H.; Shvets, E.; Shpilka, T.; Shimron, F.; Shinder, V.; Elazar, Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010, 29, 1792–1802. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Kakuta, S.; Watanabe, T.M.; Kitamura, A.; Sekito, T.; Kondo-Kakuta, C.; Ichikawa, R.; Kinjo, M.; Ohsumi, Y. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J. Cell Biol. 2012, 198, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Moreau, K.; Ravikumar, B.; Renna, M.; Puri, C.; Rubinsztein, D.C. Autophagosome precursor maturation requires homotypic fusion. Cell 2011, 146, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Szalai, P.; Hagen, L.K.; Saetre, F.; Luhr, M.; Sponheim, M.; Overbye, A.; Mills, I.G.; Seglen, P.O.; Engedal, N. Autophagic bulk sequestration of cytosolic cargo is independent of LC3, but requires GABARAPs. Exp. Cell Res. 2015, 333, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, H.Q.; Zhu, X.; Zhang, L.; Albanesi, J.; Levine, B.; Yin, H. Gabaraps regulate PI4P-dependent autophagosome:Lysosome fusion. Proc. Natl. Acad. Sci. USA 2015, 112, 7015–7020. [Google Scholar] [CrossRef] [PubMed]
- Behrends, C.; Sowa, M.E.; Gygi, S.P.; Harper, J.W. Network organization of the human autophagy system. Nature 2010, 466, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Bauvy, C.; Souquere, S.; Tonelli, G.; Liu, L.; Zhu, Y.; Qiao, Z.; Bakula, D.; Proikas-Cezanne, T.; Pierron, G.; et al. The Bcl-2 homology domain 3 mimetic gossypol induces both Beclin 1-dependent and Beclin 1-independent cytoprotective autophagy in cancer cells. J. Biol. Chem. 2010, 285, 25570–25581. [Google Scholar] [CrossRef] [PubMed]
- Codogno, P.; Mehrpour, M.; Proikas-Cezanne, T. Canonical and non-canonical autophagy: Variations on a common theme of self-eating? Nat. Rev. Mol. Cell Biol. 2012, 13, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and α-synuclein. J. Biol. Chem. 2007, 282, 5641–5652. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, A.S.; Miller, J.; Arrasate, M.; Wong, J.S.; Pleiss, M.A.; Finkbeiner, S. A small-molecule scaffold induces autophagy in primary neurons and protects against toxicity in a Huntington disease model. Proc. Natl. Acad. Sci. USA 2010, 107, 16982–16987. [Google Scholar] [CrossRef] [PubMed]
- Ktistakis, N.T.; Tooze, S.A. Digesting the expanding mechanisms of autophagy. Trends Cell Biol. 2016, 26, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. P62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Birgisdottir, A.B.; Lamark, T.; Johansen, T. The LIR motif—Crucial for selective autophagy. J. Cell Sci. 2013, 126, 3237–3247. [Google Scholar] [PubMed]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014, 16, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Mandell, M.A.; Jain, A.; Arko-Mensah, J.; Chauhan, S.; Kimura, T.; Dinkins, C.; Silvestri, G.; Munch, J.; Kirchhoff, F.; Simonsen, A.; et al. Trim proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev. Cell 2014, 30, 394–409. [Google Scholar] [CrossRef] [PubMed]
- Pandey, U.B.; Nie, Z.; Batlevi, Y.; McCray, B.A.; Ritson, G.P.; Nedelsky, N.B.; Schwartz, S.L.; DiProspero, N.A.; Knight, M.A.; Schuldiner, O.; et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007, 447, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Gamerdinger, M.; Hajieva, P.; Kaya, A.M.; Wolfrum, U.; Hartl, F.U.; Behl, C. Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO J. 2009, 28, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Liou, W.; Geuze, H.J.; Geelen, M.J.; Slot, J.W. The autophagic and endocytic pathways converge at the nascent autophagic vacuoles. J. Cell Biol. 1997, 136, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Berg, T.O.; Fengsrud, M.; Stromhaug, P.E.; Berg, T.; Seglen, P.O. Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes. J. Biol. Chem. 1998, 273, 21883–21892. [Google Scholar] [CrossRef] [PubMed]
- Fader, C.M.; Sanchez, D.; Furlan, M.; Colombo, M.I. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic 2008, 9, 230–250. [Google Scholar] [CrossRef] [PubMed]
- Kochl, R.; Hu, X.W.; Chan, E.Y.; Tooze, S.A. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic 2006, 7, 129–145. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Acevedo-Arozena, A.; Imarisio, S.; Berger, Z.; Vacher, C.; O’Kane, C.J.; Brown, S.D.; Rubinsztein, D.C. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat. Genet. 2005, 37, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Maday, S.; Wallace, K.E.; Holzbaur, E.L. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J. Cell Biol. 2012, 196, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Katsumata, K.; Nishiyama, J.; Inoue, T.; Mizushima, N.; Takeda, J.; Yuzaki, M. Dynein- and activity-dependent retrograde transport of autophagosomes in neuronal axons. Autophagy 2010, 6, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.T.; Zhou, B.; Lin, M.Y.; Cai, Q.; Sheng, Z.H. Axonal autophagosomes recruit dynein for retrograde transport through fusion with late endosomes. J. Cell Biol. 2015, 209, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Lee, J.S.; Inn, K.S.; Gack, M.U.; Li, Q.; Roberts, E.A.; Vergne, I.; Deretic, V.; Feng, P.; Akazawa, C.; et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 2008, 10, 776–787. [Google Scholar] [CrossRef] [PubMed]
- Tabata, K.; Matsunaga, K.; Sakane, A.; Sasaki, T.; Noda, T.; Yoshimori, T. Rubicon and PLEKHM1 negatively regulate the endocytic/autophagic pathway via a novel Rab7-binding domain. Mol. Biol. Cell 2010, 21, 4162–4172. [Google Scholar] [CrossRef] [PubMed]
- Progida, C.; Malerod, L.; Stuffers, S.; Brech, A.; Bucci, C.; Stenmark, H. Rilp is required for the proper morphology and function of late endosomes. J. Cell Sci. 2007, 120, 3729–3737. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.; Rocha, N.; Zwart, W.; Jordens, I.; Janssen, L.; Kuijl, C.; Olkkonen, V.M.; Neefjes, J. Activation of endosomal dynein motors by stepwise assembly of Rab7-RILP-p150Glued, ORP1L, and the receptor βlll spectrin. J. Cell Biol. 2007, 176, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Alemu, E.A.; Brech, A.; Bruun, J.A.; Lamark, T.; Overvatn, A.; Bjorkoy, G.; Johansen, T. Fyco1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J. Cell Biol. 2010, 188, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Lebrand, C.; Corti, M.; Goodson, H.; Cosson, P.; Cavalli, V.; Mayran, N.; Faure, J.; Gruenberg, J. Late endosome motility depends on lipids via the small GTPase Rab7. EMBO J. 2002, 21, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Li, L.; Chudaev, M.V.; Chen, J.; Perez, F.A.; Palmiter, R.D.; Chin, L.S. Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J. Cell Biol. 2007, 178, 1025–1038. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.G.; Munafo, D.B.; Beron, W.; Colombo, M.I. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci. 2004, 117, 2687–2697. [Google Scholar] [CrossRef] [PubMed]
- Ganley, I.G.; Wong, P.M.; Gammoh, N.; Jiang, X. Distinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol. Cell 2011, 42, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Jager, S.; Bucci, C.; Tanida, I.; Ueno, T.; Kominami, E.; Saftig, P.; Eskelinen, E.L. Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 2004, 117, 4837–4848. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Westphal, W.; Wong, K.N.; Tan, I.; Zhong, Q. Rubicon controls endosome maturation as a Rab7 effector. Proc. Natl. Acad. Sci. USA 2010, 107, 19338–19343. [Google Scholar] [CrossRef] [PubMed]
- Bucci, C.; Thomsen, P.; Nicoziani, P.; McCarthy, J.; van Deurs, B. Rab7: A key to lysosome biogenesis. Mol. Biol. Cell 2000, 11, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- Furuta, N.; Fujita, N.; Noda, T.; Yoshimori, T.; Amano, A. Combinational soluble N-ethylmaleimide-sensitive factor attachment protein receptor proteins VAMP8 and Vti1b mediate fusion of antimicrobial and canonical autophagosomes with lysosomes. Mol. Biol. Cell 2010, 21, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.M.; Mizushima, N. At the end of the autophagic road: An emerging understanding of lysosomal functions in autophagy. Trends Biochem. Sci. 2014, 39, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Moreau, K.; Renna, M.; Rubinsztein, D.C. Connections between SNAREs and autophagy. Trends Biochem. Sci. 2013, 38, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Takats, S.; Nagy, P.; Varga, A.; Pircs, K.; Karpati, M.; Varga, K.; Kovacs, A.L.; Hegedus, K.; Juhasz, G. Autophagosomal Syntaxin17-dependent lysosomal degradation maintains neuronal function in Drosophila. J. Cell Biol. 2013, 201, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Takats, S.; Pircs, K.; Nagy, P.; Varga, A.; Karpati, M.; Hegedus, K.; Kramer, H.; Kovacs, A.L.; Sass, M.; Juhasz, G. Interaction of the HOPS complex with Syntaxin 17 mediates autophagosome clearance in Drosophila. Mol. Biol. Cell 2014, 25, 1338–1354. [Google Scholar] [CrossRef] [PubMed]
- McLelland, G.L.; Lee, S.A.; McBride, H.M.; Fon, E.A. Syntaxin-17 delivers PINK1/parkin-dependent mitochondrial vesicles to the endolysosomal system. J. Cell Biol. 2016, 214, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Atlashkin, V.; Kreykenbohm, V.; Eskelinen, E.L.; Wenzel, D.; Fayyazi, A.; Fischer von Mollard, G. Deletion of the SNARE vti1b in mice results in the loss of a single SNARE partner, syntaxin 8. Mol. Cell. Biol. 2003, 23, 5198–5207. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Liang, Q.; Li, L.; Hu, Z.; Wu, F.; Zhang, P.; Ma, Y.; Zhao, B.; Kovacs, A.L.; Zhang, Z.; et al. O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation. Nat. Cell Biol. 2014, 16, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009, 11, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Wang, Q.J.; Li, X.; Yan, Y.; Backer, J.M.; Chait, B.T.; Heintz, N.; Yue, Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 2009, 11, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Diao, J.; Liu, R.; Rong, Y.; Zhao, M.; Zhang, J.; Lai, Y.; Zhou, Q.; Wilz, L.M.; Li, J.; Vivona, S.; et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015, 520, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Miao, G.; Xue, X.; Guo, X.; Yuan, C.; Wang, Z.; Zhang, G.; Chen, Y.; Feng, D.; Hu, J.; et al. The Vici Syndrome Protein EPG5 Is a Rab7 Effector that Determines the Fusion Specificity of Autophagosomes with Late Endosomes/Lysosomes. Mol. Cell 2016, 63, 781–795. [Google Scholar] [CrossRef] [PubMed]
- McEwan, D.G.; Popovic, D.; Gubas, A.; Terawaki, S.; Suzuki, H.; Stadel, D.; Coxon, F.P.; Miranda de Stegmann, D.; Bhogaraju, S.; Maddi, K.; et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol. Cell 2015, 57, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Mao, K.; Yu, A.Y.; Omairi-Nasser, A.; Austin, J., 2nd; Glick, B.S.; Yip, C.K.; Klionsky, D.J. The Atg17-Atg31-Atg29 Complex Coordinates with Atg11 to Recruit the Vam7 SNARE and Mediate Autophagosome-Vacuole Fusion. Curr. Biol. 2016, 26, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Wartosch, L.; Gunesdogan, U.; Graham, S.C.; Luzio, J.P. Recruitment of VPS33A to HOPS by VPS16 Is Required for Lysosome Fusion with Endosomes and Autophagosomes. Traffic 2015, 16, 727–742. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Yang, T.; Wang, S.; Wang, Z.; Yun, Y.; Sun, L.; Zhou, Y.; Xu, X.; Akazawa, C.; Hong, W.; et al. RILP interacts with HOPS complex via VPS41 subunit to regulate endocytic trafficking. Sci. Rep. 2014, 4, 7282. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Y.; Li, W. Impairment of autophagosome-lysosome fusion in the buff mutant mice with the VPS33A(D251E) mutation. Autophagy 2015, 11, 1608–1622. [Google Scholar] [CrossRef] [PubMed]
- Rusten, T.E.; Filimonenko, M.; Rodahl, L.M.; Stenmark, H.; Simonsen, A. ESCRTing autophagic clearance of aggregating proteins. Autophagy 2007, 4, 2. [Google Scholar] [CrossRef]
- Lee, J.A.; Beigneux, A.; Ahmad, S.T.; Young, S.G.; Gao, F.B. ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr. Biol. 2007, 17, 1561–1567. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A.; Gao, F.B. Inhibition of autophagy induction delays neuronal cell loss caused by dysfunctional ESCRT-III in frontotemporal dementia. J. Neurosci. 2009, 29, 8506–8511. [Google Scholar] [CrossRef] [PubMed]
- Filimonenko, M.; Stuffers, S.; Raiborg, C.; Yamamoto, A.; Malerod, L.; Fisher, E.M.; Isaacs, A.; Brech, A.; Stenmark, H.; Simonsen, A. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J. Cell Biol. 2007, 179, 485–500. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Koga, H.; Kawaguchi, Y.; Tang, W.; Wong, E.; Gao, Y.S.; Pandey, U.B.; Kaushik, S.; Tresse, E.; Lu, J.; et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010, 29, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Tokesi, N.; Lehotzky, A.; Horvath, I.; Szabo, B.; Olah, J.; Lau, P.; Ovadi, J. TPPP/p25 promotes tubulin acetylation by inhibiting histone deacetylase 6. J. Biol. Chem. 2010, 285, 17896–17906. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. Α-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [PubMed]
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild type α-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008, 283, 23542–23556. [Google Scholar] [CrossRef] [PubMed]
- Mak, S.K.; McCormack, A.L.; Manning-Bog, A.B.; Cuervo, A.M.; di Monte, D.A. Lysosomal degradation of α-synuclein in vivo. J. Biol. Chem. 2010, 285, 13621–13629. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M.; Talloczy, Z.; Kaushik, S.; Massey, A.C.; Mazzulli, J.; Mosharov, E.V.; Hodara, R.; Fredenburg, R.; Wu, D.C.; Follenzi, A.; et al. Dopamine-modified α-synuclein blocks chaperone-mediated autophagy. J. Clin. Investig. 2008, 118, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Bauer, P.O.; Goswami, A.; Wong, H.K.; Okuno, M.; Kurosawa, M.; Yamada, M.; Miyazaki, H.; Matsumoto, G.; Kino, Y.; Nagai, Y.; et al. Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat. Biotechnol. 2010, 28, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Li, H.; Li, S. Clearance of mutant huntingtin. Autophagy 2011, 6, 5. [Google Scholar] [CrossRef]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; de Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, A.; Magri, A.; Medina, D.X.; Wisely, E.V.; Lopez-Aranda, M.F.; Silva, A.J.; Oddo, S. mTOR regulates tau phosphorylation and degradation: Implications for Alzheimer’s disease and other tauopathies. Aging Cell 2013, 12, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, A.; Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-β, and Tau: Effects on cognitive impairments. J. Biol. Chem. 2010, 285, 13107–13120. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.M.; Wong, E.S.; Kirkpatrick, D.S.; Pletnikova, O.; Ko, H.S.; Tay, S.P.; Ho, M.W.; Troncoso, J.; Gygi, S.P.; Lee, M.K.; et al. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum. Mol. Genet. 2008, 17, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Moreau, K.; Fleming, A.; Imarisio, S.; Lopez Ramirez, A.; Mercer, J.L.; Jimenez-Sanchez, M.; Bento, C.F.; Puri, C.; Zavodszky, E.; Siddiqi, F.; et al. PICALM modulates autophagy activity and tau accumulation. Nat. Commun. 2014, 5, 4998. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, P.A.; Pickford, F.; Sun, C.H.; Lucin, K.M.; Masliah, E.; Wyss-Coray, T. Regulation of amyloid precursor protein processing by the Beclin 1 complex. PLoS ONE 2010, 5, e11102. [Google Scholar] [CrossRef] [PubMed]
- Boland, B.; Smith, D.A.; Mooney, D.; Jung, S.S.; Walsh, D.M.; Platt, F.M. Macroautophagy is not directly involved in the metabolism of amyloid precursor protein. J. Biol. Chem. 2010, 285, 37415–37426. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, P.; Loganathan, K.; Sekiguchi, M.; Matsuba, Y.; Hui, K.; Tsubuki, S.; Tanaka, M.; Iwata, N.; Saito, T.; Saido, T.C. Aβ Secretion and Plaque Formation Depend on Autophagy. Cell Rep. 2013, 5, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Maejima, I.; Takahashi, A.; Omori, H.; Kimura, T.; Takabatake, Y.; Saitoh, T.; Yamamoto, A.; Hamasaki, M.; Noda, T.; Isaka, Y.; et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 2013, 32, 2336–2347. [Google Scholar] [CrossRef] [PubMed]
- Hung, Y.H.; Chen, L.M.; Yang, J.Y.; Yang, W.Y. Spatiotemporally controlled induction of autophagy-mediated lysosome turnover. Nat. Commun. 2013, 4, 2111. [Google Scholar] [CrossRef] [PubMed]
- Winslow, A.R.; Chen, C.W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. α-Synuclein impairs macroautophagy: Implications for Parkinson’s disease. J. Cell Biol. 2010, 190, 1023–1037. [Google Scholar] [CrossRef] [PubMed]
- Tanik, S.A.; Schultheiss, C.E.; Volpicelli-Daley, L.A.; Brunden, K.R.; Lee, V.M. Lewy body-like α-synuclein aggregates resist degradation and impair macroautophagy. J. Biol. Chem. 2013, 288, 15194–15210. [Google Scholar] [CrossRef] [PubMed]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Grey, M.; Dunning, C.J.; Gaspar, R.; Grey, C.; Brundin, P.; Sparr, E.; Linse, S. Acceleration of α-synuclein aggregation by exosomes. J. Biol. Chem. 2015, 290, 2969–2982. [Google Scholar] [CrossRef] [PubMed]
- Rabouille, C.; Malhotra, V.; Nickel, W. Diversity in unconventional protein secretion. J. Cell Sci. 2012, 125, 5251–5255. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, S.N.; Zheng, D.; Sabbagh, J.J.; Martin, M.D.; Chaput, D.; Darling, A.; Trotter, J.H.; Stothert, A.R.; Nordhues, B.A.; Lussier, A.; et al. DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. EMBO J. 2016, 35, 1537–1549. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.G.; Takahama, S.; Zhang, G.; Tomarev, S.I.; Ye, Y. Unconventional secretion of misfolded proteins promotes adaptation to proteasome dysfunction in mammalian cells. Nat. Cell Biol. 2016, 18, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Dupont, N.; Jiang, S.; Pilli, M.; Ornatowski, W.; Bhattacharya, D.; Deretic, V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 2011, 30, 4701–4711. [Google Scholar] [CrossRef] [PubMed]
- Savina, A.; Fader, C.M.; Damiani, M.T.; Colombo, M.I. Rab11 promotes docking and fusion of multivesicular bodies in a calcium-dependent manner. Traffic 2005, 6, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010, 12, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.; Morohashi, Y.; Yoshimura, S.; Manrique-Hoyos, N.; Jung, S.; Lauterbach, M.A.; Bakhti, M.; Gronborg, M.; Mobius, W.; Rhee, J.; et al. Regulation of exosome secretion by Rab35 and its GTPase-activating proteins TBC1D10A-C. J. Cell Biol. 2010, 189, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Hyenne, V.; Apaydin, A.; Rodriguez, D.; Spiegelhalter, C.; Hoff-Yoessle, S.; Diem, M.; Tak, S.; Lefebvre, O.; Schwab, Y.; Goetz, J.G.; et al. RAL-1 controls multivesicular body biogenesis and exosome secretion. J. Cell Biol. 2015, 211, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Choy, R.W.; Cheng, Z.; Schekman, R. Amyloid precursor protein (APP) traffics from the cell surface via endosomes for amyloid β (Aβ) production in the trans-Golgi network. Proc. Natl. Acad. Sci. USA 2012, 109, E2077–E2082. [Google Scholar] [CrossRef] [PubMed]
- Fjorback, A.W.; Seaman, M.; Gustafsen, C.; Mehmedbasic, A.; Gokool, S.; Wu, C.; Militz, D.; Schmidt, V.; Madsen, P.; Nyengaard, J.R.; et al. Retromer binds the FANSHY sorting motif in SorLA to regulate amyloid precursor protein sorting and processing. J. Neurosci. 2012, 32, 1467–1480. [Google Scholar] [CrossRef] [PubMed]
- Burgos, P.V.; Mardones, G.A.; Rojas, A.L.; daSilva, L.L.; Prabhu, Y.; Hurley, J.H.; Bonifacino, J.S. Sorting of the Alzheimer’s disease amyloid precursor protein mediated by the AP-4 complex. Dev. Cell 2010, 18, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Das, U.; Scott, D.A.; Ganguly, A.; Koo, E.H.; Tang, Y.; Roy, S. Activity-Induced Convergence of APP and BACE-1 in Acidic Microdomains via an Endocytosis-Dependent Pathway. Neuron 2013, 79, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, L.; Honsho, M.; Zahn, T.R.; Keller, P.; Geiger, K.D.; Verkade, P.; Simons, K. Alzheimer’s disease β-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 11172–11177. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.H.; Milner, T.A.; Li, F.; Nam, E.E.; Edgar, M.A.; Yamaguchi, H.; Beal, M.F.; Xu, H.; Greengard, P.; Gouras, G.K. Intraneuronal Alzheimer Aβ42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am. J. Pathol. 2002, 161, 1869–1879. [Google Scholar] [CrossRef]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Bordi, M.; Berg, M.J.; Mohan, P.S.; Peterhoff, C.M.; Alldred, M.J.; Che, S.; Ginsberg, S.D.; Nixon, R.A. Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 2016, 12, 2467–2483. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.M.; Chan, B.K.; Park, J.S.; Hill, K.J.; Aitken, J.B.; Cottle, L.; Farghaian, H.; Cole, A.R.; Lay, P.A.; Sue, C.M.; et al. Parkinson’s disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes α-Synuclein externalization via exosomes. Hum. Mol. Genet. 2014, 23, 2816–2833. [Google Scholar] [CrossRef] [PubMed]
- Tsunemi, T.; Hamada, K.; Krainc, D. ATP13A2/PARK9 regulates secretion of exosomes and α-synuclein. J. Neurosci. 2014, 34, 15281–15287. [Google Scholar] [CrossRef] [PubMed]
- Fevrier, B.; Vilette, D.; Archer, F.; Loew, D.; Faigle, W.; Vidal, M.; Laude, H.; Raposo, G. Cells release prions in association with exosomes. Proc. Natl. Acad. Sci. USA 2004, 101, 9683–9688. [Google Scholar] [CrossRef] [PubMed]
- Vella, L.J.; Sharples, R.A.; Lawson, V.A.; Masters, C.L.; Cappai, R.; Hill, A.F. Packaging of prions into exosomes is associated with a novel pathway of PrP processing. J. Pathol. 2007, 211, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, H.J.; Hartfield, E.M.; Christian, H.C.; Emmanoulidou, E.; Zheng, Y.; Booth, H.; Bogetofte, H.; Lang, C.; Ryan, B.J.; Sardi, S.P.; et al. ER Stress and Autophagic Perturbations Lead to Elevated Extracellular α-Synuclein in GBA-N370S Parkinson’s iPSC-Derived Dopamine Neurons. Stem Cell Rep. 2016, 6, 342–356. [Google Scholar] [CrossRef] [PubMed]
- Rind, H.B.; Butowt, R.; von Bartheld, C.S. Synaptic targeting of retrogradely transported trophic factors in motoneurons: Comparison of glial cell line-derived neurotrophic factor, brain-derived neurotrophic factor, and cardiotrophin-1 with tetanus toxin. J. Neurosci. 2005, 25, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Cooney, J.R.; Hurlburt, J.L.; Selig, D.K.; Harris, K.M.; Fiala, J.C. Endosomal compartments serve multiple hippocampal dendritic spines from a widespread rather than a local store of recycling membrane. J. Neurosci. 2002, 22, 2215–2224. [Google Scholar] [PubMed]
- Popov, V.I.; Medvedev, N.I.; Kraev, I.V.; Gabbott, P.L.; Davies, H.A.; Lynch, M.; Cowley, T.R.; Berezin, V.; Bock, E.; Stewart, M.G. A cell adhesion molecule mimetic, FGL peptide, induces alterations in synapse and dendritic spine structure in the dentate gyrus of aged rats: A three-dimensional ultrastructural study. Eur. J. Neurosci. 2008, 27, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Koles, K.; Nunnari, J.; Korkut, C.; Barria, R.; Brewer, C.; Li, Y.; Leszyk, J.; Zhang, B.; Budnik, V. Mechanism of evenness interrupted (Evi)-exosome release at synaptic boutons. J. Biol. Chem. 2012, 287, 16820–16834. [Google Scholar] [CrossRef] [PubMed]
- Korkut, C.; Ataman, B.; Ramachandran, P.; Ashley, J.; Barria, R.; Gherbesi, N.; Budnik, V. Trans-synaptic transmission of vesicular Wnt signals through Evi/Wntless. Cell 2009, 139, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Maskos, U.; Kissa, K.; St Cloment, C.; Brulet, P. Retrograde trans-synaptic transfer of green fluorescent protein allows the genetic mapping of neuronal circuits in transgenic mice. Proc. Natl. Acad. Sci. USA 2002, 99, 10120–10125. [Google Scholar] [CrossRef] [PubMed]
- Lachenal, G.; Pernet-Gallay, K.; Chivet, M.; Hemming, F.J.; Belly, A.; Bodon, G.; Blot, B.; Haase, G.; Goldberg, Y.; Sadoul, R. Release of exosomes from differentiated neurons and its regulation by synaptic glutamatergic activity. Mol. Cell. Neurosci. 2011, 46, 409–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tofaris, G.K.; Kim, H.T.; Hourez, R.; Jung, J.W.; Kim, K.P.; Goldberg, A.L. Ubiquitin ligase Nedd4 promotes α-synuclein degradation by the endosomal-lysosomal pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 17004–17009. [Google Scholar] [CrossRef] [PubMed]
- Sugeno, N.; Hasegawa, T.; Tanaka, N.; Fukuda, M.; Wakabayashi, K.; Oshima, R.; Konno, M.; Miura, E.; Kikuchi, A.; Baba, T.; et al. Lys-63-linked ubiquitination by E3 ubiquitin ligase Nedd4-1 facilitates endosomal sequestration of internalized α-synuclein. J. Biol. Chem. 2014, 289, 18137–18151. [Google Scholar] [CrossRef] [PubMed]
- Tardiff, D.F.; Jui, N.T.; Khurana, V.; Tambe, M.A.; Thompson, M.L.; Chung, C.Y.; Kamadurai, H.B.; Kim, H.T.; Lancaster, A.K.; Caldwell, K.A.; et al. Yeast Reveal a “Druggable” Rsp5/Nedd4 Network that Ameliorates α-Synuclein Toxicity in Neurons. Science 2013, 24, 24. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.Y.; Khurana, V.; Auluck, P.K.; Tardiff, D.F.; Mazzulli, J.R.; Soldner, F.; Baru, V.; Lou, Y.; Freyzon, Y.; Cho, S.; et al. Identification and rescue of α-synuclein toxicity in Parkinson patient-derived neurons. Science 2013, 342, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.E.; Hallett, P.J.; Moens, T.; Smith, G.; Mangano, E.; Kim, H.T.; Goldberg, A.L.; Liu, J.L.; Isacson, O.; Tofaris, G.K. Enhanced ubiquitin-dependent degradation by Nedd4 protects against α-synuclein accumulation and toxicity in animal models of Parkinson’s disease. Neurobiol. Dis. 2014, 64, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Sette, P.; Jadwin, J.A.; Dussupt, V.; Bello, N.F.; Bouamr, F. The ESCRT-associated protein Alix recruits the ubiquitin ligase Nedd4-1 to facilitate HIV-1 release through the LYPXnL L domain motif. J. Virol. 2010, 84, 8181–8192. [Google Scholar] [CrossRef] [PubMed]
- Blot, V.; Perugi, F.; Gay, B.; Prevost, M.C.; Briant, L.; Tangy, F.; Abriel, H.; Staub, O.; Dokhelar, M.C.; Pique, C. Nedd4.1-mediated ubiquitination and subsequent recruitment of Tsg101 ensure HTLV-1 Gag trafficking towards the multivesicular body pathway prior to virus budding. J. Cell Sci. 2004, 117, 2357–2367. [Google Scholar] [PubMed]
- Medina, G.; Zhang, Y.; Tang, Y.; Gottwein, E.; Vana, M.L.; Bouamr, F.; Leis, J.; Carter, C.A. The functionally exchangeable L domains in RSV and HIV-1 Gag direct particle release through pathways linked by Tsg101. Traffic 2005, 6, 880–894. [Google Scholar] [CrossRef] [PubMed]
- Putz, U.; Howitt, J.; Doan, A.; Goh, C.P.; Low, L.H.; Silke, J.; Tan, S.S. The tumor suppressor PTEN is exported in exosomes and has phosphatase activity in recipient cells. Sci. Signal. 2012, 5, ra70. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Wu, N.; Yang, J.M.; Gould, S.J. Protein targeting to exosomes/microvesicles by plasma membrane anchors. J. Biol. Chem. 2011, 286, 14383–14395. [Google Scholar] [CrossRef] [PubMed]
- Duran, J.M.; Anjard, C.; Stefan, C.; Loomis, W.F.; Malhotra, V. Unconventional secretion of Acb1 is mediated by autophagosomes. J. Cell Biol. 2010, 188, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Manjithaya, R.; Subramani, S. Autophagy: A broad role in unconventional protein secretion? Trends Cell Biol. 2011, 21, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Bruns, C.; McCaffery, J.M.; Curwin, A.J.; Duran, J.M.; Malhotra, V. Biogenesis of a novel compartment for autophagosome-mediated unconventional protein secretion. J. Cell Biol. 2011, 195, 979–992. [Google Scholar] [CrossRef] [PubMed]
- Gee, H.Y.; Noh, S.H.; Tang, B.L.; Kim, K.H.; Lee, M.G. Rescue of DeltaF508-CFTR trafficking via a GRASP-dependent unconventional secretion pathway. Cell 2011, 146, 746–760. [Google Scholar] [CrossRef] [PubMed]
- Bodemann, B.O.; Orvedahl, A.; Cheng, T.; Ram, R.R.; Ou, Y.H.; Formstecher, E.; Maiti, M.; Hazelett, C.C.; Wauson, E.M.; Balakireva, M.; et al. RalB and the exocyst mediate the cellular starvation response by direct activation of autophagosome assembly. Cell 2011, 144, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Lindersson, E.; Lundvig, D.; Petersen, C.; Madsen, P.; Nyengaard, J.R.; Hojrup, P.; Moos, T.; Otzen, D.; Gai, W.P.; Blumbergs, P.C.; et al. p25α Stimulates α-synuclein aggregation and is co-localized with aggregated α-synuclein in α-synucleinopathies. J. Biol. Chem. 2005, 280, 5703–5715. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Cordero, J.J.; Marrero-Diaz, R.; Megias, D.; Genis, L.; Garcia-Grande, A.; Garcia, M.A.; Arroyo, A.G.; Montoya, M.C. MT1-MMP proinvasive activity is regulated by a novel Rab8-dependent exocytic pathway. EMBO J. 2007, 26, 1499–1510. [Google Scholar] [CrossRef] [PubMed]
- Grigoriev, I.; Yu, K.L.; Martinez-Sanchez, E.; Serra-Marques, A.; Smal, I.; Meijering, E.; Demmers, J.; Peranen, J.; Pasterkamp, R.J.; van der Sluijs, P.; et al. Rab6, Rab8, and MICAL3 cooperate in controlling docking and fusion of exocytotic carriers. Curr. Biol. 2011, 21, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Hattula, K.; Furuhjelm, J.; Tikkanen, J.; Tanhuanpaa, K.; Laakkonen, P.; Peranen, J. Characterization of the Rab8-specific membrane traffic route linked to protrusion formation. J. Cell Sci. 2006, 119, 4866–4877. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Mushiake, S.; Kato, Y.; Sato, K.; Sato, M.; Takeda, N.; Ozono, K.; Miki, K.; Kubo, Y.; Tsuji, A.; et al. The Rab8 GTPase regulates apical protein localization in intestinal cells. Nature 2007, 448, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Galvez-Santisteban, M.; Rodriguez-Fraticelli, A.E.; Bryant, D.M.; Vergarajauregui, S.; Yasuda, T.; Banon-Rodriguez, I.; Bernascone, I.; Datta, A.; Spivak, N.; Young, K.; et al. Synaptotagmin-like proteins control the formation of a single apical membrane domain in epithelial cells. Nat. Cell Biol. 2012, 14, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Bevis, B.J.; Shorter, J.; Strathearn, K.E.; Hamamichi, S.; Su, L.J.; Caldwell, K.A.; Caldwell, G.A.; Rochet, J.C.; McCaffery, J.M.; et al. The Parkinson’s disease protein α-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Manjithaya, R.; Anjard, C.; Loomis, W.F.; Subramani, S. Unconventional secretion of Pichia pastoris Acb1 is dependent on GRASP protein, peroxisomal functions, and autophagosome formation. J. Cell Biol. 2010, 188, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Nair, U.; Jotwani, A.; Geng, J.; Gammoh, N.; Richerson, D.; Yen, W.L.; Griffith, J.; Nag, S.; Wang, K.; Moss, T.; et al. SNARE proteins are required for macroautophagy. Cell 2011, 146, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012, 5, ra42. [Google Scholar] [CrossRef] [PubMed]
- Jewell, J.L.; Russell, R.C.; Guan, K.L. Amino acid signalling upstream of mTOR. Nat. Rev. Mol. Cell Biol. 2013, 14, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Korolchuk, V.I.; Saiki, S.; Lichtenberg, M.; Siddiqi, F.H.; Roberts, E.A.; Imarisio, S.; Jahreiss, L.; Sarkar, S.; Futter, M.; Menzies, F.M.; et al. Lysosomal positioning coordinates cellular nutrient responses. Nat. Cell Biol. 2011, 13, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Ostrowski, P.; Jaumouille, V.; Grinstein, S. The position of lysosomes within the cell determines their luminal pH. J. Cell Biol. 2016, 212, 677–692. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Manzoni, C.; Lewis, P.A. Dysfunction of the autophagy/lysosomal degradation pathway is a shared feature of the genetic synucleinopathies. FASEB J. 2013, 27, 3424–3429. [Google Scholar] [CrossRef] [PubMed]
- Avrahami, L.; Farfara, D.; Shaham-Kol, M.; Vassar, R.; Frenkel, D.; Eldar-Finkelman, H. Inhibition of glycogen synthase kinase-3 ameliorates β-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: In vivo and in vitro studies. J. Biol. Chem. 2013, 288, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Bove, J.; Rodriguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 2010, 30, 12535–12544. [Google Scholar] [CrossRef] [PubMed]
- Decressac, M.; Mattsson, B.; Weikop, P.; Lundblad, M.; Jakobsson, J.; Bjorklund, A. TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc. Natl. Acad. Sci. USA 2013, 22, 22. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Sato, Y.; Nixon, R.A. Lysosomal Proteolysis Inhibition Selectively Disrupts Axonal Transport of Degradative Organelles and Causes an Alzheimer’s-Like Axonal Dystrophy. J. Neurosci. 2011, 31, 7817–7830. [Google Scholar] [CrossRef] [PubMed]
- Schondorf, D.C.; Aureli, M.; McAllister, F.E.; Hindley, C.J.; Mayer, F.; Schmid, B.; Sardi, S.P.; Valsecchi, M.; Hoffmann, S.; Schwarz, L.K.; et al. iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 2014, 5, 4028. [Google Scholar] [CrossRef] [PubMed]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Small, S.A.; Petsko, G.A. Retromer in Alzheimer disease, Parkinson disease and other neurological disorders. Nat. Rev. Neurosci. 2015, 16, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.A.M. VPS35 in Dopamine Neurons Is Required for Endosome-to-Golgi Retrieval of Lamp2a, a Receptor of Chaperone-Mediated Autophagy That Is Critical for α-Synuclein Degradation and Prevention of Pathogenesis of Parkinson’s Disease. J. Neurosci. 2015, 35, 10613–10628. [Google Scholar] [CrossRef] [PubMed]
- Miura, E.; Hasegawa, T.; Konno, M.; Suzuki, M.; Sugeno, N.; Fujikake, N.; Geisler, S.; Tabuchi, M.; Oshima, R.; Kikuchi, A.; et al. VPS35 dysfunction impairs lysosomal degradation of α-synuclein and exacerbates neurotoxicity in a Drosophila model of Parkinson’s disease. Neurobiol. Dis. 2014, 71, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Matrone, C.; Dzamko, N.; Madsen, P.; Nyegaard, M.; Pohlmann, R.; Sondergaard, R.V.; Lassen, L.B.; Andresen, T.L.; Halliday, G.M.; Jensen, P.H.; et al. Mannose 6-Phosphate Receptor Is Reduced in α-Synuclein Overexpressing Models of Parkinsons Disease. PLoS ONE 2016, 11, e0160501. [Google Scholar] [CrossRef] [PubMed]
- Mazzulli, J.R.; Zunke, F.; Isacson, O.; Studer, L.; Krainc, D. α-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc. Natl. Acad. Sci. USA 2016, 113, 1931–1936. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. Α-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Zavodszky, E.; Seaman, M.N.; Moreau, K.; Jimenez-Sanchez, M.; Breusegem, S.Y.; Harbour, M.E.; Rubinsztein, D.C. Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat. Commun. 2014, 5, 3828. [Google Scholar] [CrossRef] [PubMed]
- Follett, J.; Bugarcic, A.; Yang, Z.; Ariotti, N.; Norwood, S.J.; Collins, B.M.; Parton, R.G.; Teasdale, R.D. Parkinson Disease-linked Vps35 R524W Mutation Impairs the Endosomal Association of Retromer and Induces α-Synuclein Aggregation. J. Biol. Chem. 2016, 291, 18283–18298. [Google Scholar] [CrossRef] [PubMed]
- Follett, J.; Norwood, S.J.; Hamilton, N.A.; Mohan, M.; Kovtun, O.; Tay, S.; Zhe, Y.; Wood, S.A.; Mellick, G.D.; Silburn, P.A.; et al. The Vps35 D620N mutation linked to Parkinson’s disease disrupts the cargo sorting function of retromer. Traffic 2014, 15, 230–244. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Carroll, B.; Buganim, Y.; Maetzel, D.; Ng, A.H.; Cassady, J.P.; Cohen, M.A.; Chakraborty, S.; Wang, H.; Spooner, E.; et al. Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease. Cell Rep. 2013, 5, 1302–1315. [Google Scholar] [CrossRef] [PubMed]
- Fraldi, A.; Annunziata, F.; Lombardi, A.; Kaiser, H.J.; Medina, D.L.; Spampanato, C.; Fedele, A.O.; Polishchuk, R.; Sorrentino, N.C.; Simons, K.; et al. Lysosomal fusion and SNARE function are impaired by cholesterol accumulation in lysosomal storage disorders. EMBO J. 2010, 29, 3607–3620. [Google Scholar] [CrossRef] [PubMed]
- Coen, K.; Flannagan, R.S.; Baron, S.; Carraro-Lacroix, L.R.; Wang, D.; Vermeire, W.; Michiels, C.; Munck, S.; Baert, V.; Sugita, S.; et al. Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. J. Cell Biol. 2012, 198, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; McBrayer, M.K.; Wolfe, D.M.; Haslett, L.J.; Kumar, A.; Sato, Y.; Lie, P.P.; Mohan, P.; Coffey, E.E.; Kompella, U.; et al. Presenilin 1 Maintains Lysosomal Ca2+ Homeostasis via TRPML1 by Regulating vATPase-Mediated Lysosome Acidification. Cell Rep. 2015, 12, 1430–1444. [Google Scholar] [CrossRef] [PubMed]
- Manzoni, C.; Mamais, A.; Dihanich, S.; Abeti, R.; Soutar, M.P.; Plun-Favreau, H.; Giunti, P.; Tooze, S.A.; Bandopadhyay, R.; Lewis, P.A. Inhibition of LRRK2 kinase activity stimulates macroautophagy. Biochim. Biophys. Acta 2013, 1833, 2900–2910. [Google Scholar] [CrossRef] [PubMed]
- Bravo-San Pedro, J.M.; Niso-Santano, M.; Gomez-Sanchez, R.; Pizarro-Estrella, E.; Aiastui-Pujana, A.; Gorostidi, A.; Climent, V.; Lopez de Maturana, R.; Sanchez-Pernaute, R.; Lopez de Munain, A.; et al. The LRRK2 G2019S mutant exacerbates basal autophagy through activation of the MEK/ERK pathway. Cell. Mol. Life Sci. 2013, 70, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Rivero-Rios, P.; Gomez-Suaga, P.; Fernandez, B.; Madero-Perez, J.; Schwab, A.J.; Ebert, A.D.; Hilfiker, S. Alterations in late endocytic trafficking related to the pathobiology of LRRK2-linked Parkinson’s disease. Biochem. Soc. Trans. 2015, 43, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Hockey, L.N.; Kilpatrick, B.S.; Eden, E.R.; Lin-Moshier, Y.; Brailoiu, G.C.; Brailoiu, E.; Futter, C.E.; Schapira, A.H.; Marchant, J.S.; Patel, S. Dysregulation of lysosomal morphology by pathogenic LRRK2 is corrected by TPC2 inhibition. J. Cell Sci. 2015, 128, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Henry, A.G.; Aghamohammadzadeh, S.; Samaroo, H.; Chen, Y.; Mou, K.; Needle, E.; Hirst, W.D. Pathogenic LRRK2 mutations, through increased kinase activity, produce enlarged lysosomes with reduced degradative capacity and increase ATP13A2 expression. Hum. Mol. Genet. 2015, 24, 6013–6028. [Google Scholar] [CrossRef] [PubMed]
- Linhart, R.; Wong, S.A.; Cao, J.; Tran, M.; Huynh, A.; Ardrey, C.; Park, J.M.; Hsu, C.; Taha, S.; Peterson, R.; et al. Vacuolar protein sorting 35 (Vps35) rescues locomotor deficits and shortened lifespan in Drosophila expressing a Parkinson’s disease mutant of Leucine-Rich Repeat Kinase 2 (LRRK2). Mol. Neurodegener. 2014, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Ramirez, A.; Martinez-Vicente, M.; Perier, C.; Canron, M.H.; Doudnikoff, E.; Vital, A.; Vila, M.; Klein, C.; Bezard, E. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 9611–9616. [Google Scholar] [CrossRef] [PubMed]
- Usenovic, M.; Tresse, E.; Mazzulli, J.R.; Taylor, J.P.; Krainc, D. Deficiency of ATP13A2 leads to lysosomal dysfunction, α-synuclein accumulation, and neurotoxicity. J. Neurosci. 2012, 32, 4240–4246. [Google Scholar] [CrossRef] [PubMed]
- Holemans, T.; Sorensen, D.M.; van Veen, S.; Martin, S.; Hermans, D.; Kemmer, G.C.; Van den Haute, C.; Baekelandt, V.; Gunther Pomorski, T.; Agostinis, P.; et al. A lipid switch unlocks Parkinson’s disease-associated ATP13A2. Proc. Natl. Acad. Sci. USA 2015, 112, 9040–9045. [Google Scholar] [CrossRef] [PubMed]
- Bento, C.F.; Ashkenazi, A.; Jimenez-Sanchez, M.; Rubinsztein, D.C. The Parkinson’s disease-associated genes ATP13A2 and SYT11 regulate autophagy via a common pathway. Nat. Commun. 2016, 7, 11803. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; McPhee, C.K.; Deng, S.; Huang, L.; Chen, L.; Liu, M.; Tracy, K.; Baehrecke, E.H.; Yu, L.; Lenardo, M.J. Spinster is required for autophagic lysosome reformation and mTOR reactivation following starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 7826–7831. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Su, Q.P.; Chen, Y.; Zhu, Y.; Jiang, D.; Rong, Y.; Zhang, S.; Zhang, Y.; Ren, H.; Zhang, C.; et al. Kinesin 1 Drives Autolysosome Tubulation. Dev. Cell 2016, 37, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Liu, M.; Ma, L.; Du, W.; Zhang, H.; Tian, Y.; Cao, Z.; Li, Y.; Ren, H.; Zhang, C.; et al. C Clathrin and phosphatidylinositol-4,5-bisphosphate regulate autophagic lysosome reformation. Nat. Cell Biol. 2012, 14, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, S.; Patel, B.; Aphkhazava, D.; Macian, F.; Santambrogio, L.; Shields, D.; Cuervo, A.M. The lipid kinase PI4KIIIβ preserves lysosomal identity. EMBO J. 2013, 32, 324–339. [Google Scholar] [CrossRef] [PubMed]
- Redmann, V.; Lamb, C.A.; Hwang, S.; Orchard, R.C.; Kim, S.; Razi, M.; Milam, A.; Park, S.; Yokoyama, C.C.; Kambal, A.; et al. Clec16a is Critical for Autolysosome Function and Purkinje Cell Survival. Sci. Rep. 2016, 6, 23326. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Lee, S.; Blackstone, C. Spastic paraplegia proteins spastizin and spatacsin mediate autophagic lysosome reformation. J. Clin. Investig. 2014, 124, 5249–5262. [Google Scholar] [CrossRef] [PubMed]
- Varga, R.E.; Khundadze, M.; Damme, M.; Nietzsche, S.; Hoffmann, B.; Stauber, T.; Koch, N.; Hennings, J.C.; Franzka, P.; Huebner, A.K.; et al. In Vivo Evidence for Lysosome Depletion and Impaired Autophagic Clearance in Hereditary Spastic Paraplegia Type SPG11. PLoS Genet. 2015, 11, e1005454. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.; Caler, E.V.; Andrews, N.W. Plasma membrane repair is mediated by Ca2+-regulated exocytosis of lysosomes. Cell 2001, 106, 157–169. [Google Scholar] [CrossRef]
- Rodriguez, A.; Webster, P.; Ortego, J.; Andrews, N.W. Lysosomes behave as Ca2+-regulated exocytotic vesicles in fibroblasts and epithelial cells. J. Cell Biol. 1997, 137, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Polishchuk, E.V.; Concilli, M.; Iacobacci, S.; Chesi, G.; Pastore, N.; Piccolo, P.; Paladino, S.; Baldantoni, D.; van, I.S.C.; Chan, J.; et al. Wilson disease protein ATP7B utilizes lysosomal exocytosis to maintain copper homeostasis. Dev. Cell 2014, 29, 686–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, I.; Chakrabarti, S.; Hellevik, T.; Morehead, J.; Fowler, K.; Andrews, N.W. Synaptotagmin VII regulates Ca2+-dependent exocytosis of lysosomes in fibroblasts. J. Cell Biol. 2000, 148, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.K.; Huynh, C.; Proux-Gillardeaux, V.; Galli, T.; Andrews, N.W. Identification of SNAREs involved in synaptotagmin VII-regulated lysosomal exocytosis. J. Biol. Chem. 2004, 279, 20471–20479. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.L.; Fraldi, A.; Bouche, V.; Annunziata, F.; Mansueto, G.; Spampanato, C.; Puri, C.; Pignata, A.; Martina, J.A.; Sardiello, M.; et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev. Cell 2011, 21, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Gowrishankar, S.; Yuan, P.; Wu, Y.; Schrag, M.; Paradise, S.; Grutzendler, J.; de Camilli, P.; Ferguson, S.M. Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer’s disease amyloid plaques. Proc. Natl. Acad. Sci. USA 2015, 112, E3699–E3708. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and amyloid-b peptide neurotoxicity in Alzheimer´s disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhou, Y.; Hong, J.S.; Zhang, J. Aggregated α-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Dallas, S.; Zhang, D.; Guo, J.P.; Pang, H.; Wilson, B.; Miller, D.S.; Chen, B.; Zhang, W.; McGeer, P.L.; et al. Microglial PHOX and Mac-1 are essential to the enhanced dopaminergic neurodegeneration elicited by A30P and A53T mutant α-synuclein. Glia 2007, 55, 1178–1188. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Priller, J.; Sisodia, S.S.; Ransohoff, R.M. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat. Neurosci. 2011, 14, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.P.; Ejlerskov, P.; Rasmussen, I.; Vilhardt, F. Reciprocal signals between microglia and neurons regulate α-synuclein secretion by exophagy through a neuronal cJUN-N-terminal kinase-signaling axis. J. Neuroinflamm. 2016, 13, 59. [Google Scholar] [CrossRef] [PubMed]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kugler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- Reis-Rodrigues, P.; Czerwieniec, G.; Peters, T.W.; Evani, U.S.; Alavez, S.; Gaman, E.A.; Vantipalli, M.; Mooney, S.D.; Gibson, B.W.; Lithgow, G.J.; et al. Proteomic analysis of age-dependent changes in protein solubility identifies genes that modulate lifespan. Aging Cell 2012, 11, 120–127. [Google Scholar] [CrossRef] [PubMed]
- David, D.C.; Ollikainen, N.; Trinidad, J.C.; Cary, M.P.; Burlingame, A.L.; Kenyon, C. Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biol. 2010, 8, e1000450. [Google Scholar] [CrossRef] [PubMed]
- Alavez, S.; Vantipalli, M.C.; Zucker, D.J.; Klang, I.M.; Lithgow, G.J. Amyloid-binding compounds maintain protein homeostasis during ageing and extend lifespan. Nature 2011, 472, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Toth, M.L.; Sigmond, T.; Borsos, E.; Barna, J.; Erdelyi, P.; Takacs-Vellai, K.; Orosz, L.; Kovacs, A.L.; Csikos, G.; Sass, M.; et al. Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans. Autophagy 2008, 4, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G. Autophagy: A druggable process that is deregulated in aging and human disease. J. Clin. Investig. 2015, 125, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Cuanalo-Contreras, K.; Mukherjee, A.; Soto, C. Role of protein misfolding and proteostasis deficiency in protein misfolding diseases and aging. Int. J. Cell Biol. 2013, 2013, 638083. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Marino, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, N.; Zhu, J.; Zhu, Y.; Fenik, P.; Lian, J.; Galante, R.; Veasey, S. Endoplasmic reticulum stress in wake-active neurons progresses with aging. Aging Cell 2011, 10, 640–649. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, M.M.; Zheng, B.; Lu, T.; Yan, Z.; Py, B.F.; Ng, A.; Xavier, R.J.; Li, C.; Yankner, B.A.; Scherzer, C.R.; et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169. [Google Scholar] [CrossRef] [PubMed]
- Spencer, B.; Potkar, R.; Trejo, M.; Rockenstein, E.; Patrick, C.; Gindi, R.; Adame, A.; Wyss-Coray, T.; Masliah, E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in α-synuclein models of Parkinson’s and Lewy body diseases. J. Neurosci. 2009, 29, 13578–13588. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H.V. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, L.R.; De Magalhaes Filho, C.D.; McQuary, P.R.; Chu, C.C.; Visvikis, O.; Chang, J.T.; Gelino, S.; Ong, B.; Davis, A.E.; Irazoqui, J.E.; et al. The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat. Commun. 2013, 4, 2267. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.K.; Lim, H.S.; Kawasaki, I.; Shim, Y.H.; Vaikath, N.N.; El-Agnaf, O.M.; Lee, H.J.; Lee, S.J. Anti-aging treatments slow propagation of synucleinopathy by restoring lysosomal function. Autophagy 2016, 12, 1849–1863. [Google Scholar] [CrossRef] [PubMed]
- Polito, V.A.; Li, H.; Martini-Stoica, H.; Wang, B.; Yang, L.; Xu, Y.; Swartzlander, D.B.; Palmieri, M.; di Ronza, A.; Lee, V.M.; et al. Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Mol. Med. 2014, 6, 1142–1160. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borland, H.; Vilhardt, F. Prelysosomal Compartments in the Unconventional Secretion of Amyloidogenic Seeds. Int. J. Mol. Sci. 2017, 18, 227. https://doi.org/10.3390/ijms18010227
Borland H, Vilhardt F. Prelysosomal Compartments in the Unconventional Secretion of Amyloidogenic Seeds. International Journal of Molecular Sciences. 2017; 18(1):227. https://doi.org/10.3390/ijms18010227
Chicago/Turabian StyleBorland, Helena, and Frederik Vilhardt. 2017. "Prelysosomal Compartments in the Unconventional Secretion of Amyloidogenic Seeds" International Journal of Molecular Sciences 18, no. 1: 227. https://doi.org/10.3390/ijms18010227
APA StyleBorland, H., & Vilhardt, F. (2017). Prelysosomal Compartments in the Unconventional Secretion of Amyloidogenic Seeds. International Journal of Molecular Sciences, 18(1), 227. https://doi.org/10.3390/ijms18010227