Fascaplysin Exerts Anti-Cancer Effects through the Downregulation of Survivin and HIF-1α and Inhibition of VEGFR2 and TRKA


Abstract
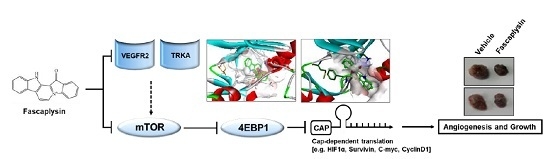
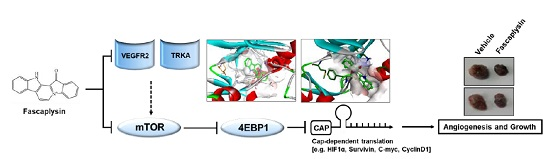
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
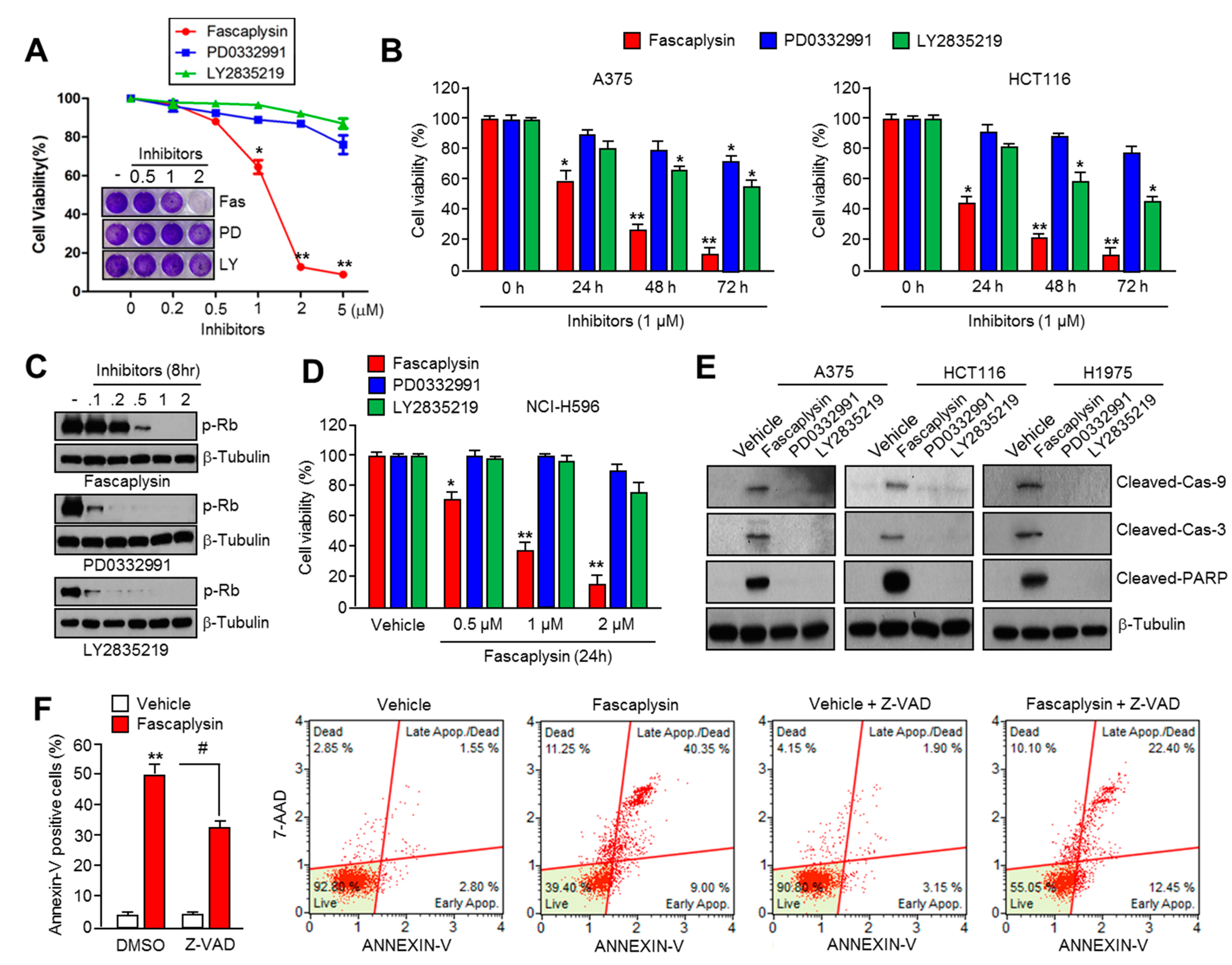
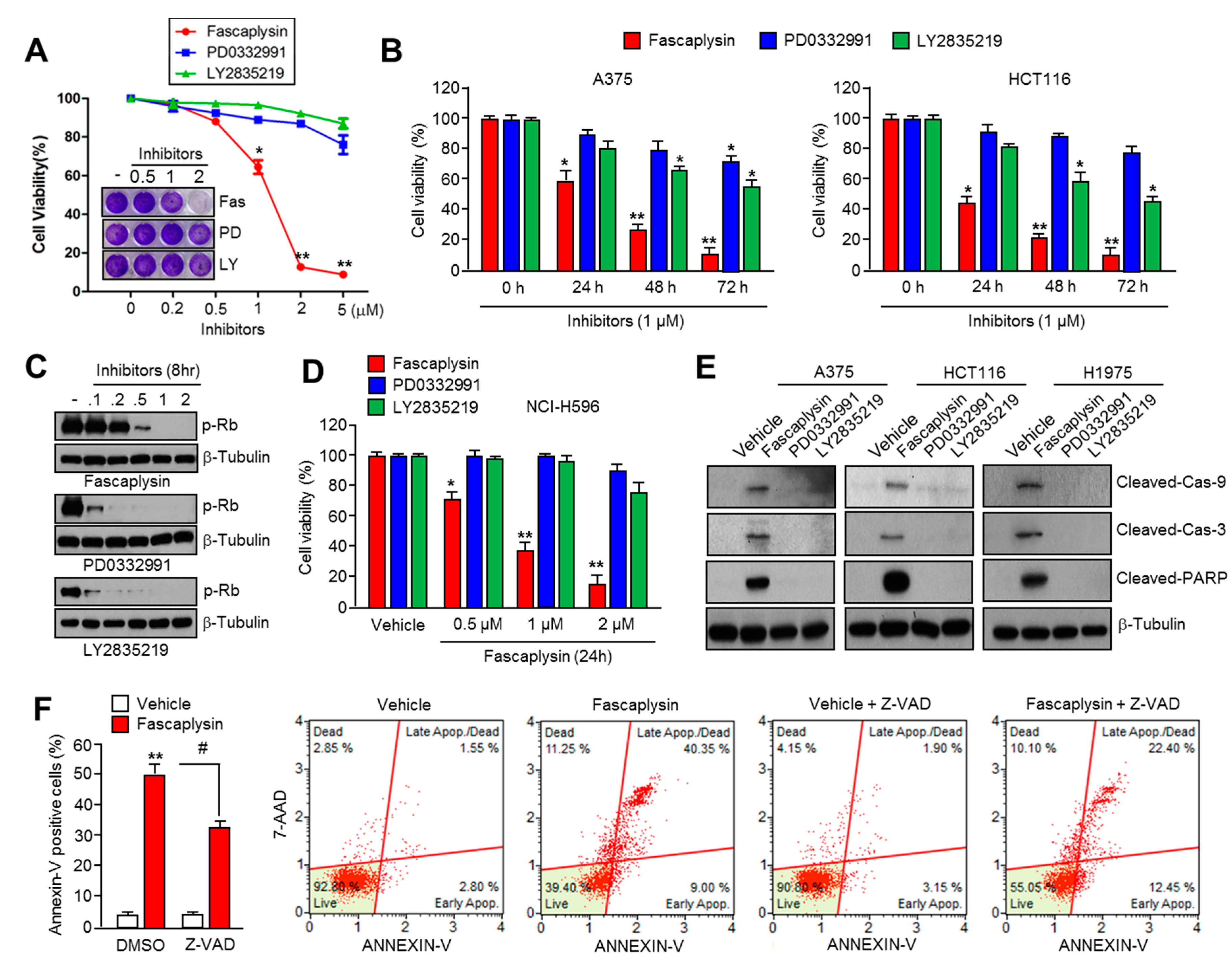
2.1. Fascaplysin Rapidly Decreases Cell Viability and Induces Apoptosis Independently of CDK4-RB Inhibition
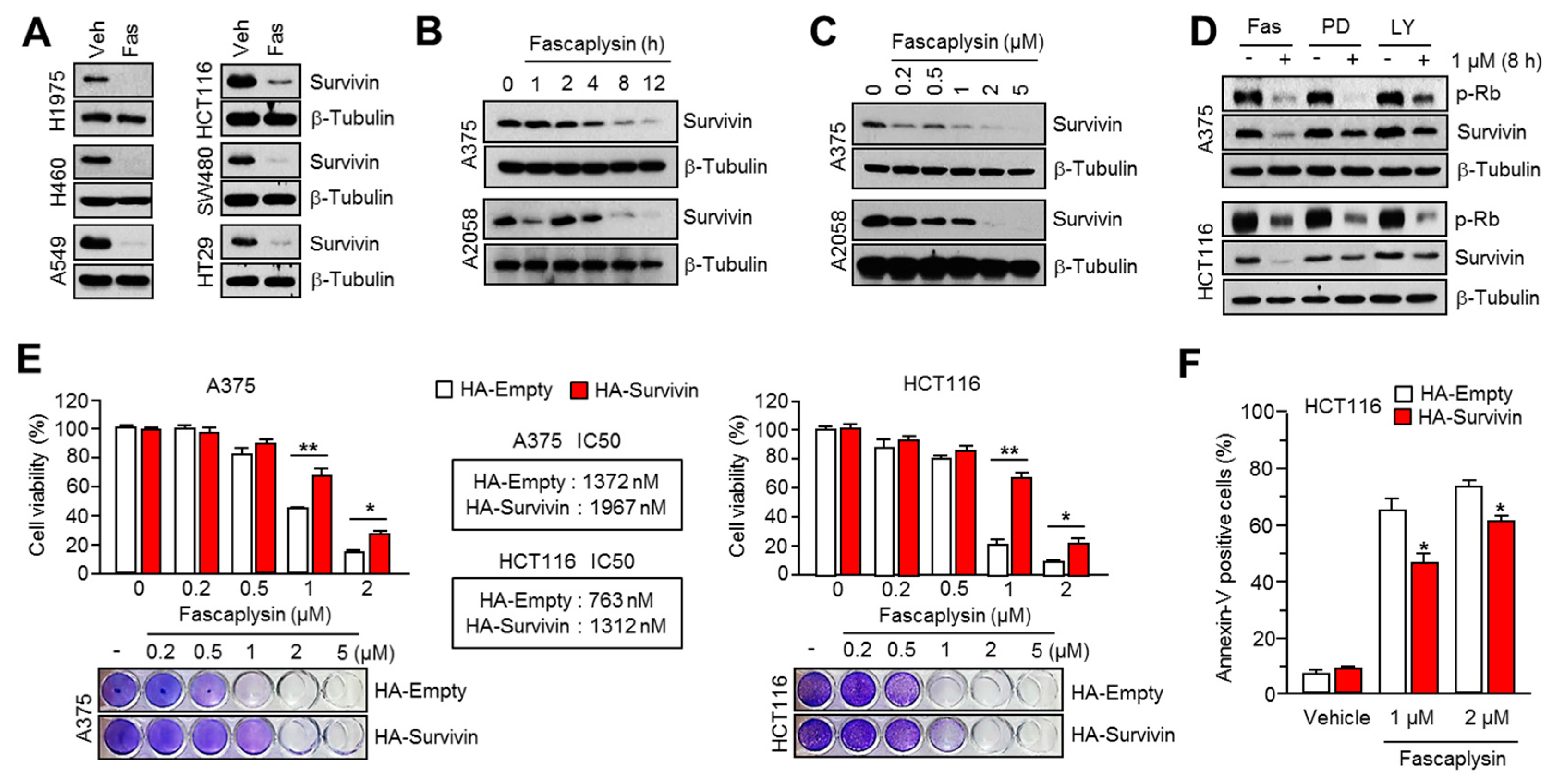
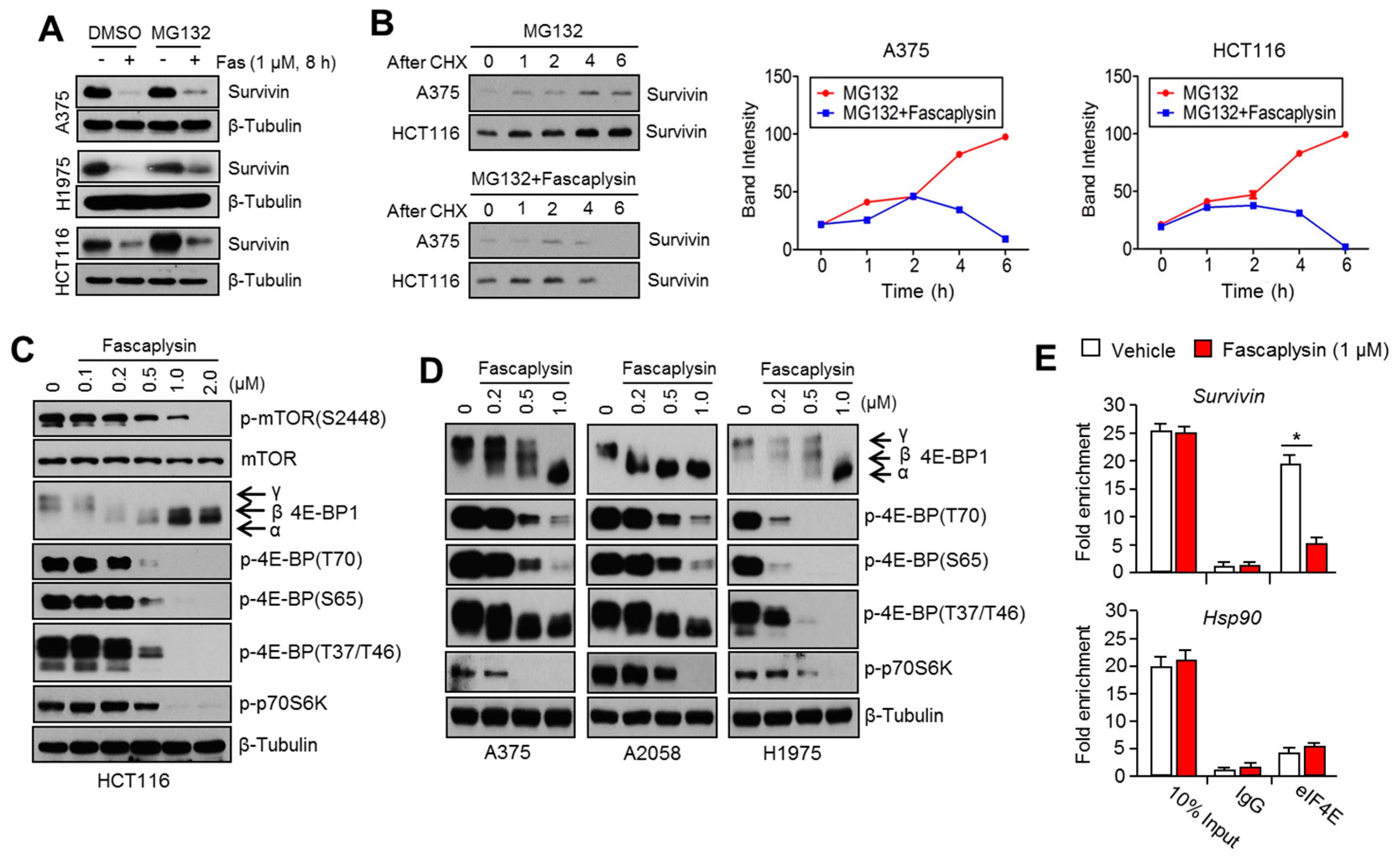
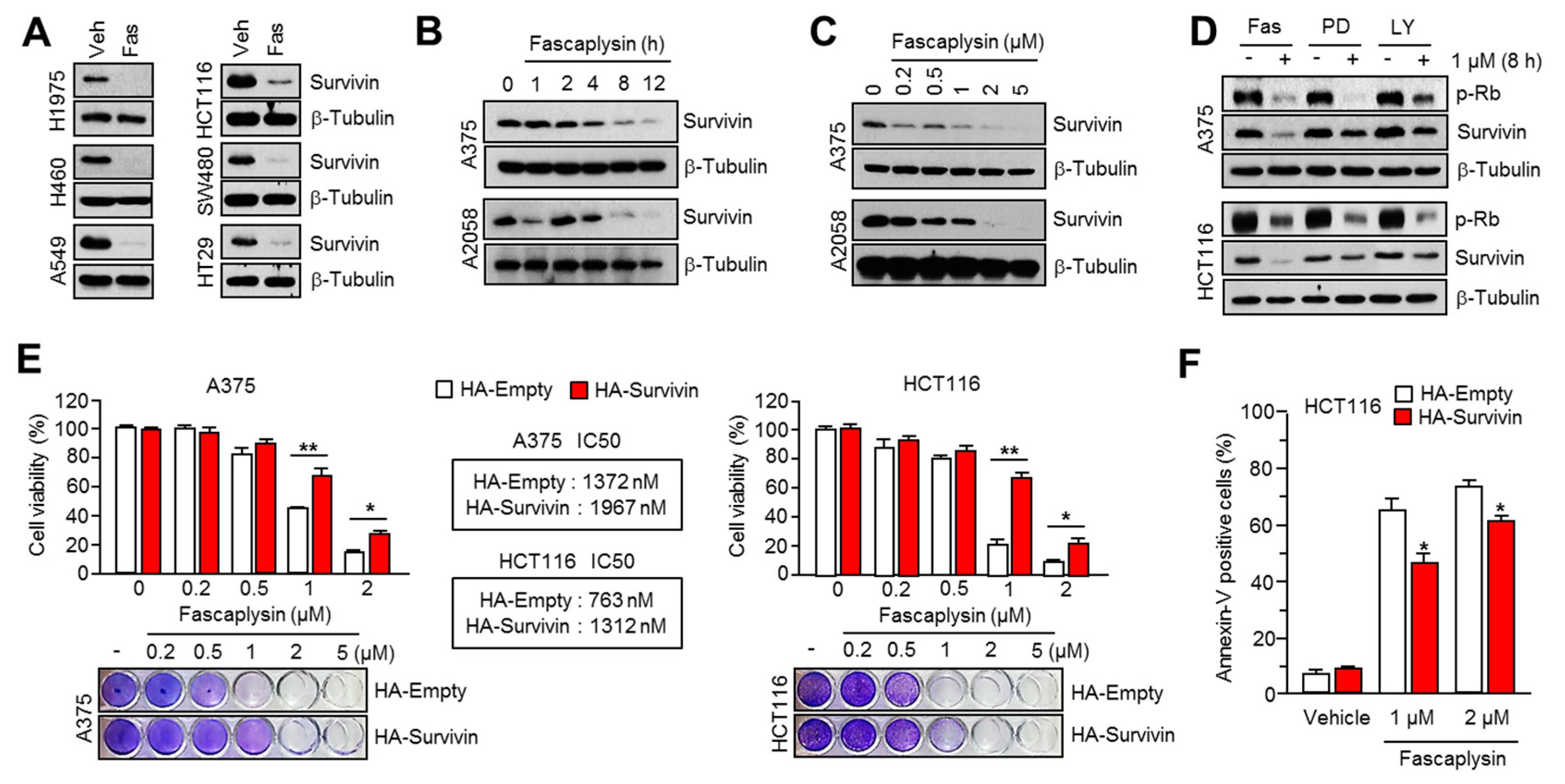
2.2. Survivin Is Involved in Fascaplysin-Induced Apoptosis
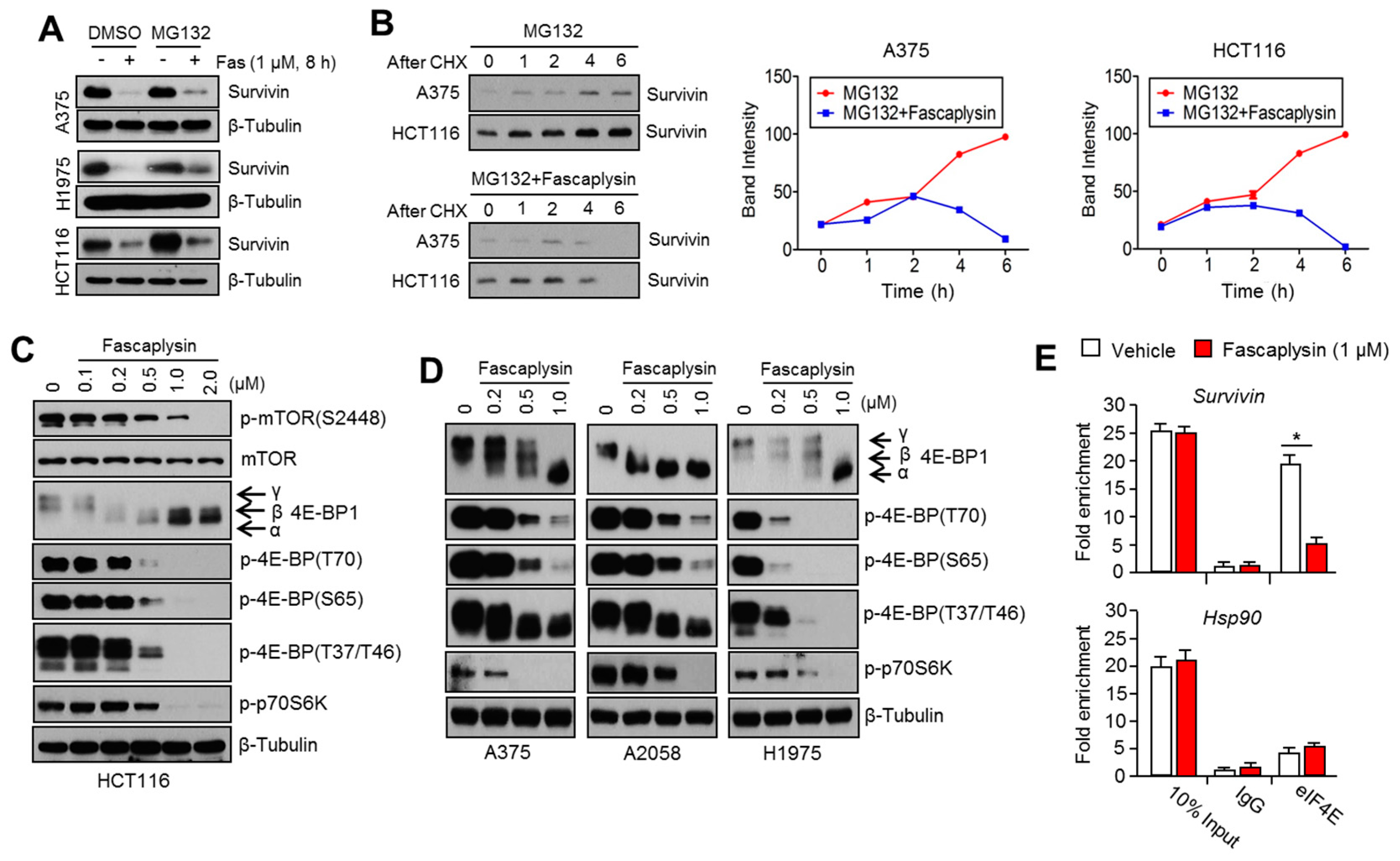
2.3. Fascaplysin Downregulates De Novo Synthesis of Survivin Protein by Inhibiting Cap-Dependent Translation Controlled by 4EBP1
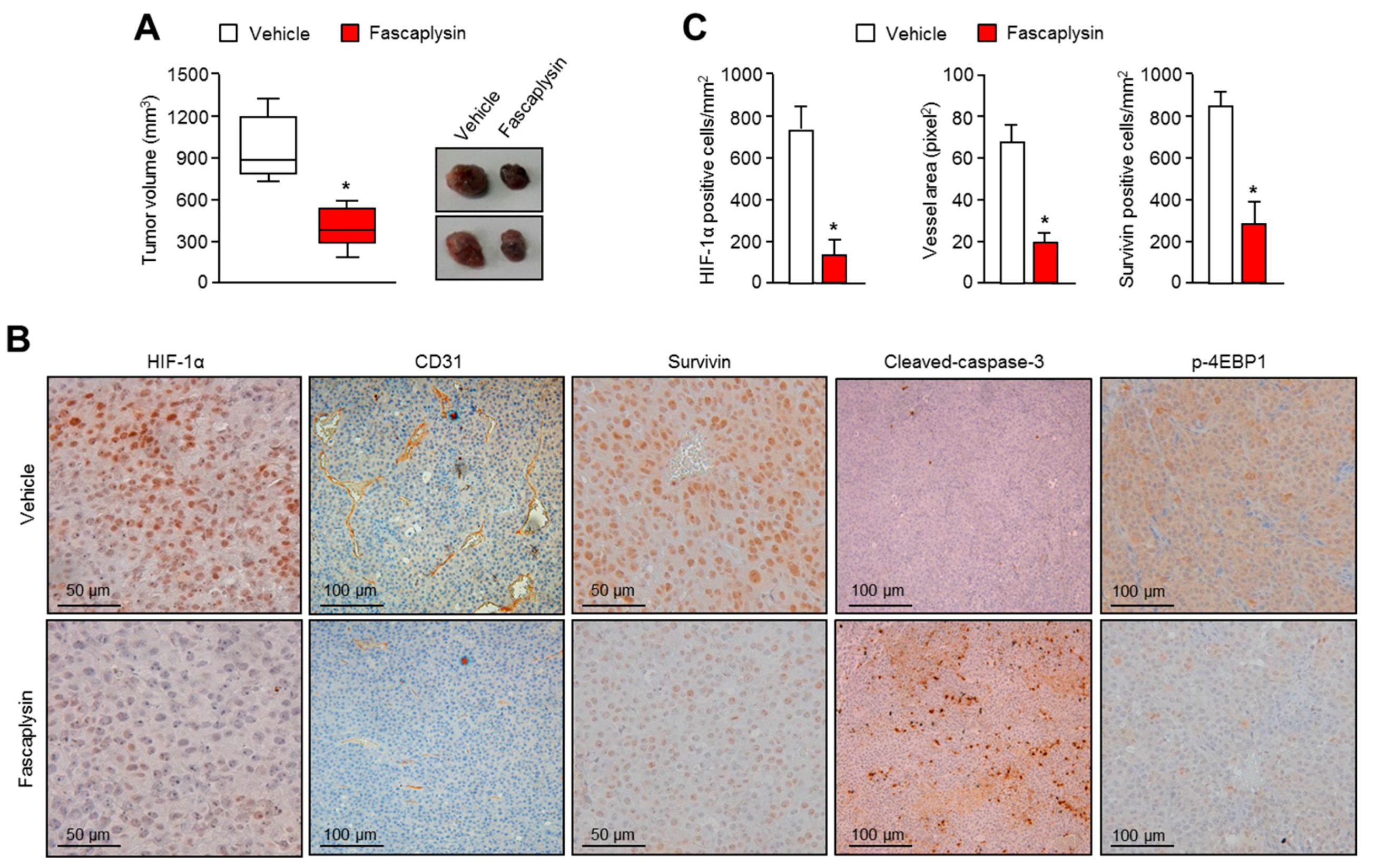
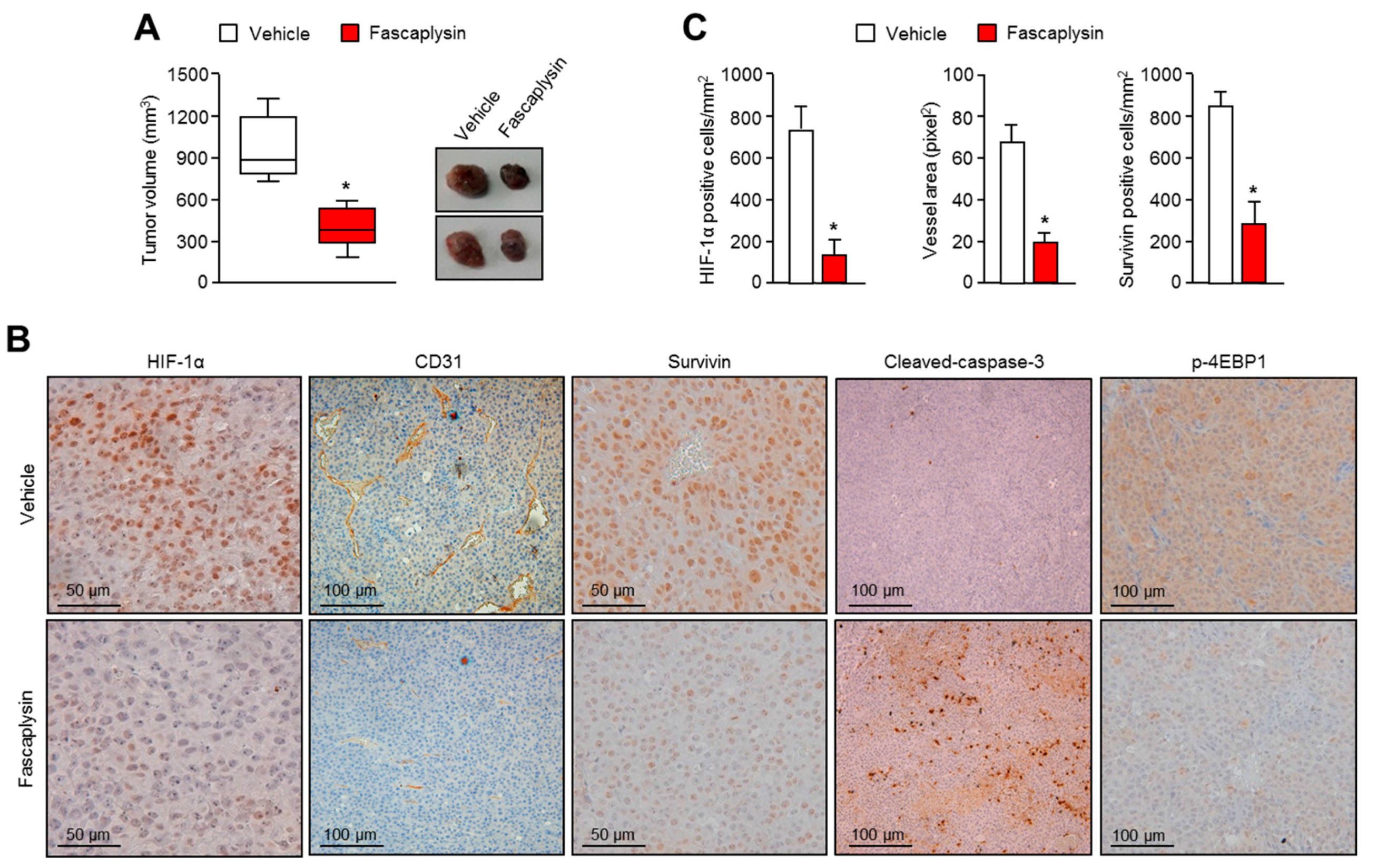
2.4. Anti-Angiogenic and Pro-Apoptotic Effects of Fascaplysin In Vivo
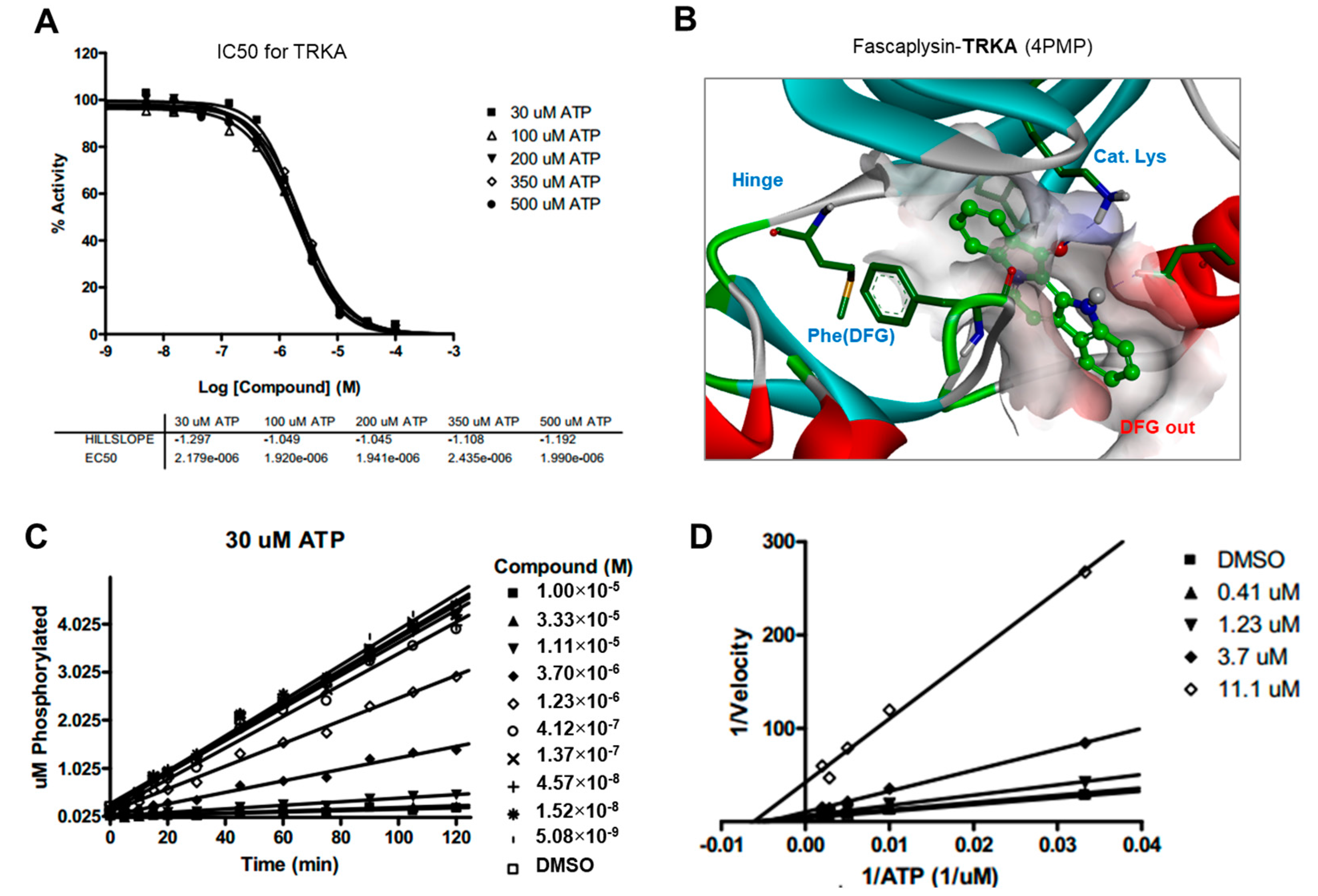
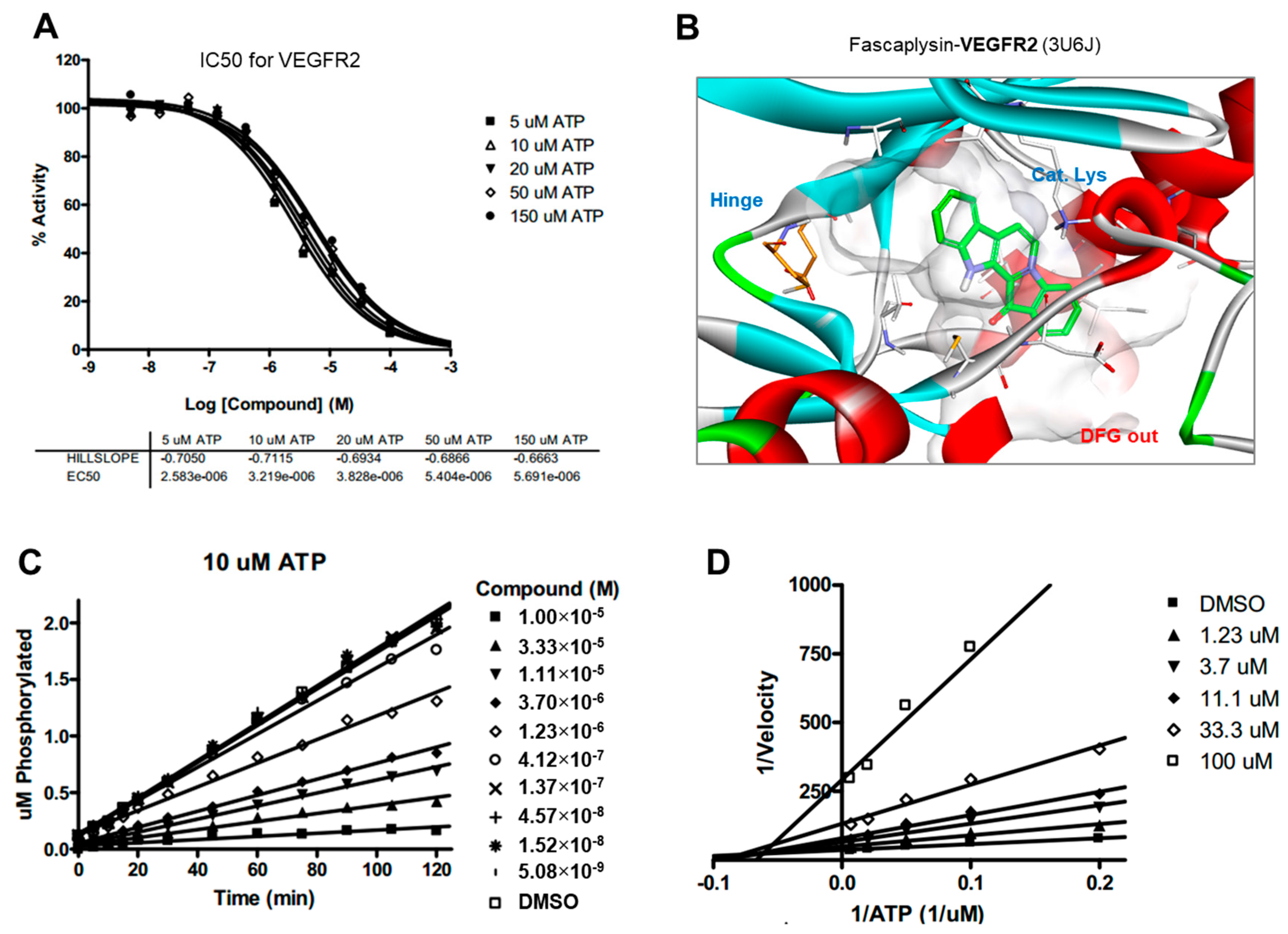
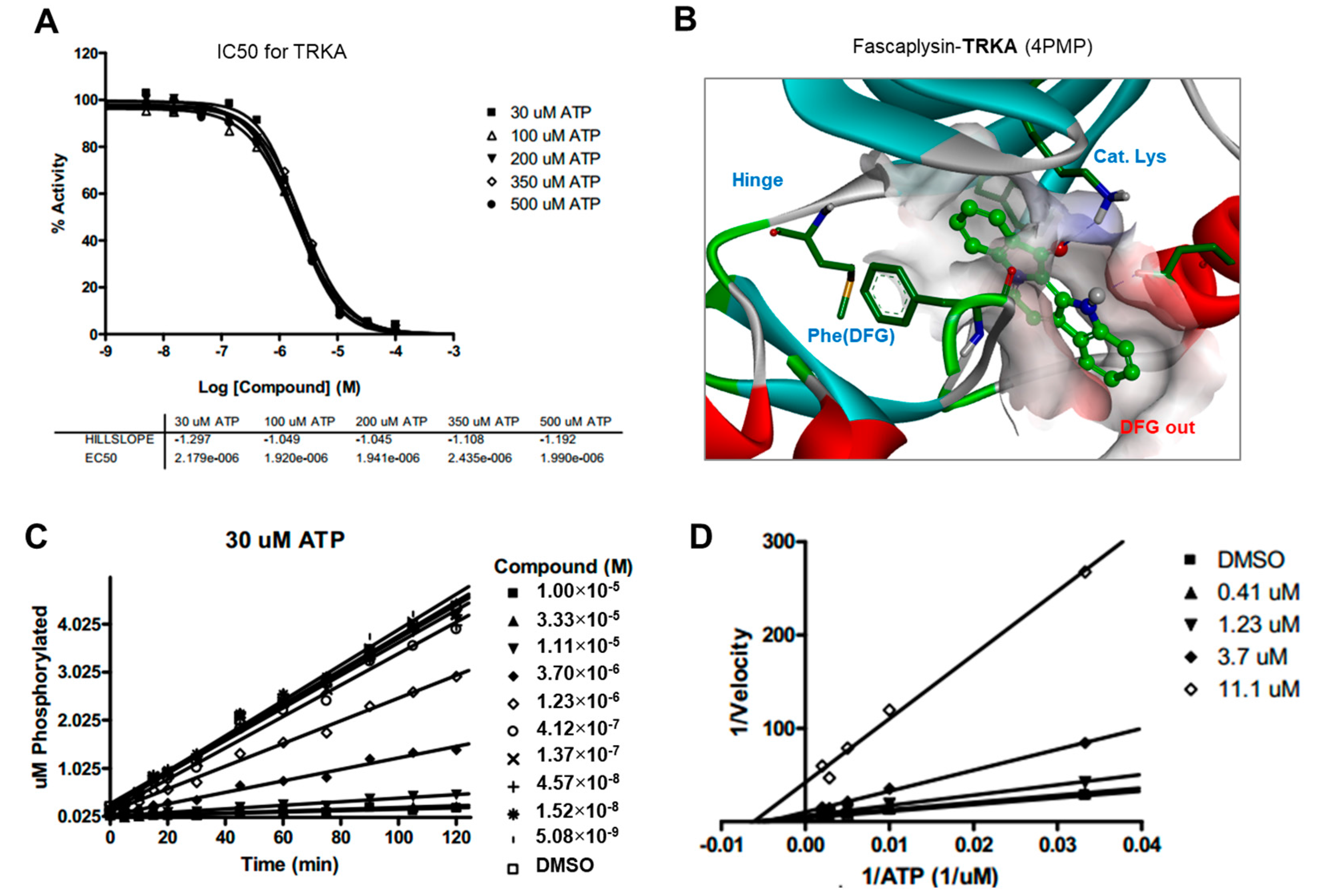
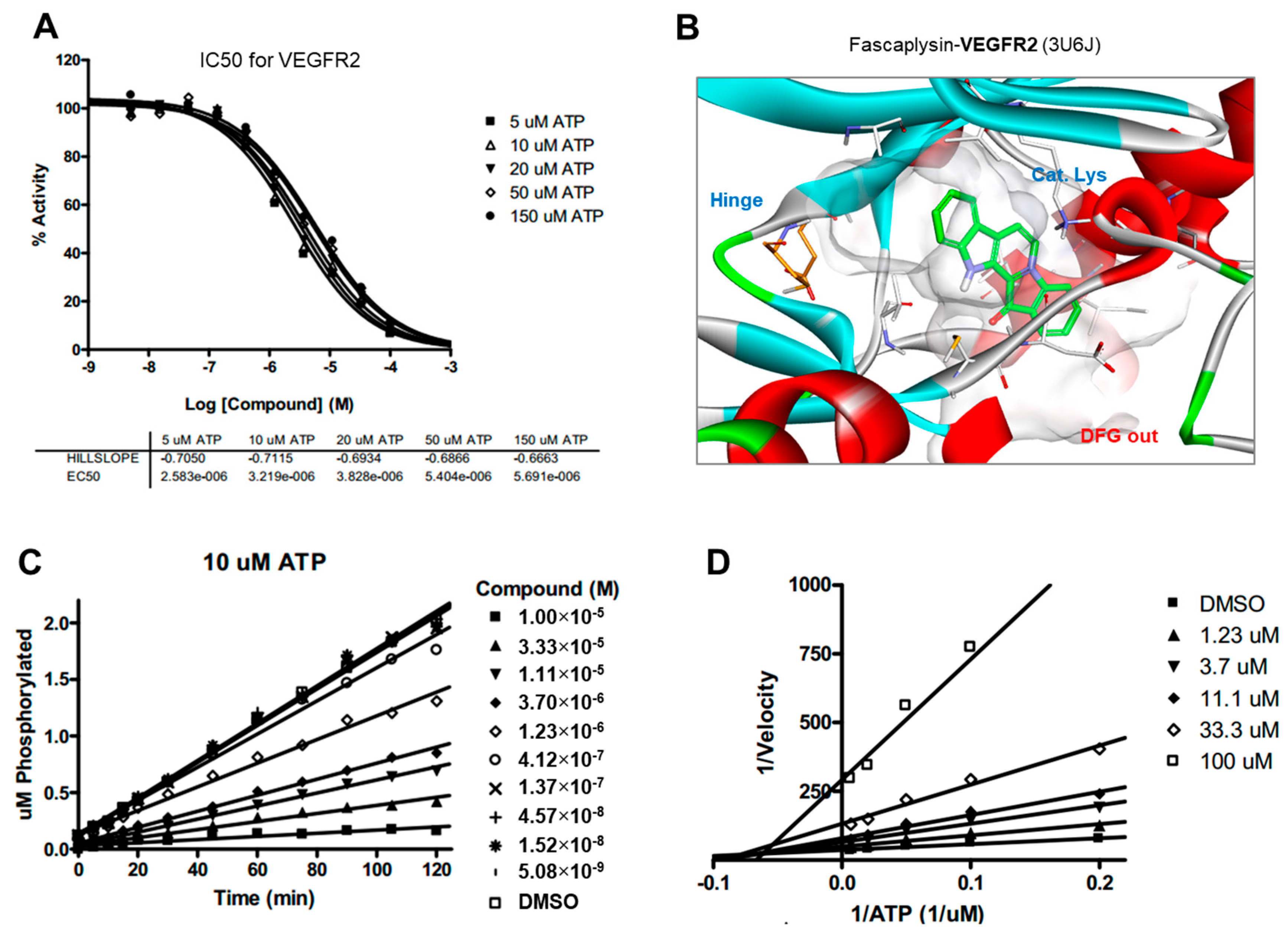
2.5. Kinase Activity of TRKA and VEGFR2 Was Inhibited by Fascaplysin through Asp-Phe-Gly (DFG)-Out Non-Competitive Inhibition
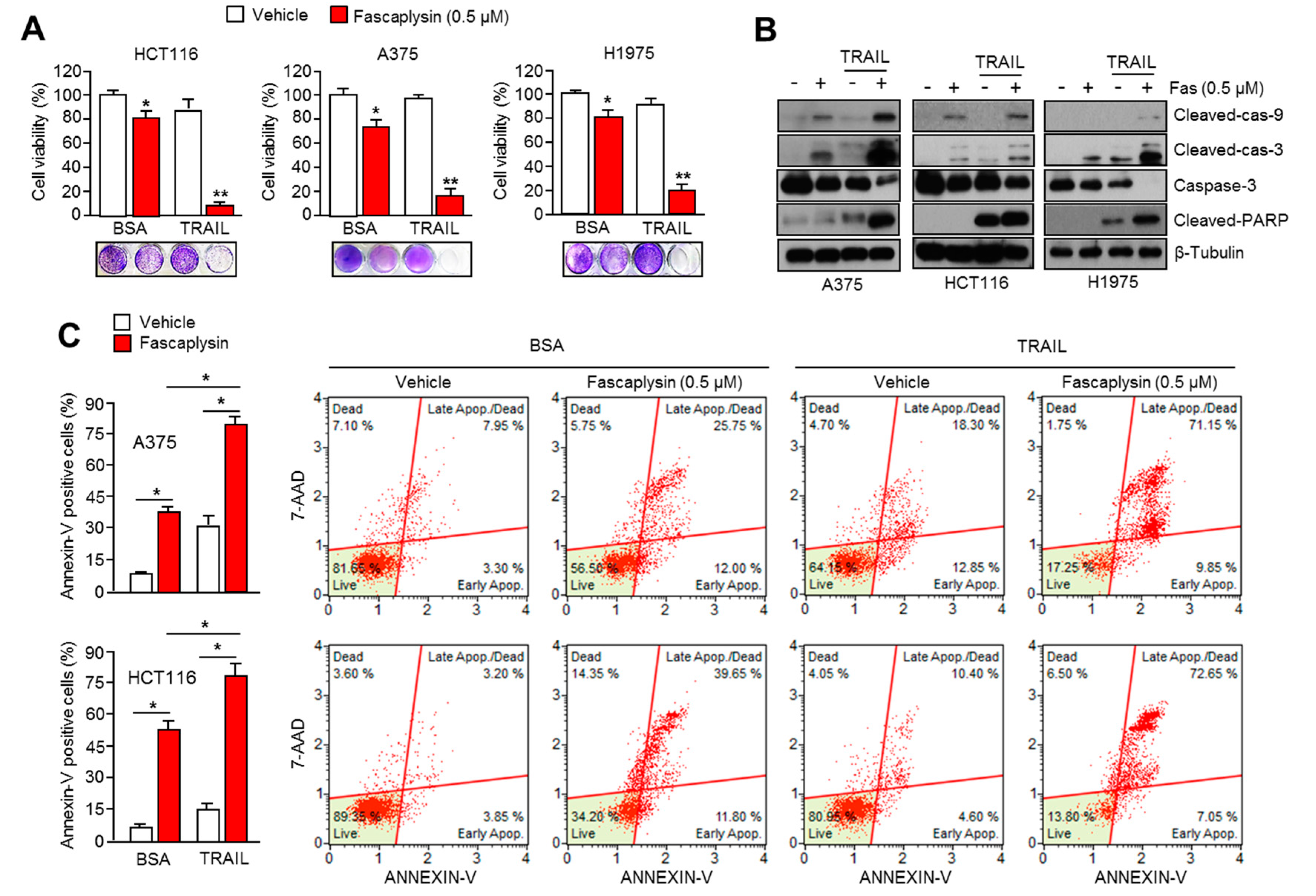
2.6. Fascaplysin Sensitizes the Anti-Cancer Effect of TRAIL
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture and Generation of Stable Cell Lines
4.3. Western Blotting
4.4. RNA-Binding Protein Immunoprecipitation (RIP) Assay
4.5. Quantitative Real Time-PCR
4.6. Tumor Xenograft Assay and Immunohistochemistry
4.7. Docking Simulation Studies
4.8. Kinase Inhibition Assay
4.9. Measurement of Cell Viability
4.10. Apoptosis Assays
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CDK4 | Cyclin-dependent kinase 4 |
| RB | Retinoblastoma |
| TRKA | Tropomyosin-related kinase A |
| VEGFR2 | Vascular endothelial growth factor receptor 2 |
| HIF-1α | Hypoxia-inducible factor-1α |
| mTOR | Mammalian target of rapamycin |
| 4EBP1 | Eukaryotic translation initiation factor 4E-binding protein 1 |
| RTK | Receptor tyrosine kinase |
| EGFR | Epidermal growth factor receptor |
| PDGFR | Platelet-derived growth factor receptor |
| TRAIL | TNF-related apoptosis-inducing ligand |
References
- Gschwind, A.; Fischer, O.M.; Ullrich, A. The discovery of receptor tyrosine kinases: Targets for cancer therapy. Nat. Rev. Cancer 2004, 4, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Khotskaya, Y.B.; Holla, V.R.; Farago, A.F.; Mills Shaw, K.R.; Meric-Bernstam, F.; Hong, D.S. Targeting TRK family proteins in cancer. Pharmacol. Ther. 2017, 173, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.H.; Jung, K.H.; Son, M.K.; Yan, H.H.; Ryu, Y.L.; Kim, J.; Lee, J.K.; Hong, S.; Hong, S.S. Anti-cancer effect of HS-345, a new tropomyosin-related kinase A inhibitor, on human pancreatic cancer. Cancer Lett. 2013, 338, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M. VEGFR and type-V RTK activation and signaling. Cold Spring Harb. Perspect. Biol. 2013, 5, a009092. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Korbel, C.; Scheuer, C.; Nenicu, A.; Menger, M.D.; Laschke, M.W. Tubeimoside-1 suppresses tumor angiogenesis by stimulation of proteasomal VEGFR2 and Tie2 degradation in a non-small cell lung cancer xenograft model. Oncotarget 2016, 7, 5258–5272. [Google Scholar] [CrossRef] [PubMed]
- Shafiq, M.I.; Steinbrecher, T.; Schmid, R. Fascaplysin as a specific inhibitor for CDK4: Insights from molecular modelling. PLoS ONE 2012, 7, e42612. [Google Scholar] [CrossRef] [PubMed]
- Aubry, C.; Wilson, A.J.; Emmerson, D.; Murphy, E.; Chan, Y.Y.; Dickens, M.P.; Garcia, M.D.; Jenkins, P.R.; Mahale, S.; Chaudhuri, B. Fascaplysin-inspired diindolyls as selective inhibitors of CDK4/cyclin D1. Bioorg. Med. Chem. 2009, 17, 6073–6084. [Google Scholar] [CrossRef] [PubMed]
- Mahale, S.; Aubry, C.; Jenkins, P.R.; Marechal, J.D.; Sutcliffe, M.J.; Chaudhuri, B. Inhibition of cancer cell growth by cyclin dependent kinase 4 inhibitors synthesized based on the structure of fascaplysin. Bioorg. Chem. 2006, 34, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Guru, S.K.; Pathania, A.S.; Manda, S.; Kumar, A.; Bharate, S.B.; Vishwakarma, R.A.; Malik, F.; Bhushan, S. Fascaplysin induces caspase mediated crosstalk between apoptosis and autophagy through the inhibition of PI3K/AKT/mTOR signaling cascade in human leukemia HL-60 cells. J. Cell. Biochem. 2015, 116, 985–997. [Google Scholar] [CrossRef] [PubMed]
- Altieri, D.C. Validating survivin as a cancer therapeutic target. Nat. Rev. Cancer 2003, 3, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Altieri, D.C. Survivin and IAP proteins in cell-death mechanisms. Biochem. J. 2010, 430, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Peery, R.C.; Liu, J.Y.; Zhang, J.T. Targeting survivin for therapeutic discovery: Past, present, and future promises. Drug Discov. Today 2017. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Kwan, T.; Yu, C.; Chen, F.; Freedman, B.; Schafer, J.M.; Lee, E.J.; Jameson, J.L.; Jordan, V.C.; Cryns, V.L. Peroxisome proliferator-activated receptor gamma agonists promote TRAIL-induced apoptosis by reducing survivin levels via cyclin D3 repression and cell cycle arrest. J. Biol. Chem. 2005, 280, 6742–6751. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Debatin, K.M. Sensitization for tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by the chemopreventive agent resveratrol. Cancer Res. 2004, 64, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Kim, S.U.; Shin, D.Y.; Choi, K.S. Roscovitine sensitizes glioma cells to TRAIL-mediated apoptosis by downregulation of survivin and XIAP. Oncogene 2004, 23, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Strohecker, A.; Chen, F.; Kwan, T.; Bosman, J.; Jordan, V.C.; Cryns, V.L. Aspirin sensitizes cancer cells to TRAIL-induced apoptosis by reducing survivin levels. Clin. Cancer Res. 2008, 14, 3168–3176. [Google Scholar] [CrossRef] [PubMed]
- Von Karstedt, S.; Montinaro, A.; Walczak, H. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat. Rev. Cancer 2017, 17, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, A.C.; Ruggero, D. Targeting eukaryotic translation initiation factor 4E (eIF4E) in cancer. Clin. Cancer Res. 2010, 16, 4914–4920. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.; Robichaud, N.; Hulea, L.; Sonenberg, N.; Pelletier, J.; Topisirovic, I. Targeting the translation machinery in cancer. Nat. Rev. Drug Discov. 2015, 14, 261–278. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Chen, H.; Lu, X.; Wang, F.; Xu, W.; Jin, H.; Zhu, P. Fascaplysin exert anti-tumor effects through apoptotic and anti-angiogenesis pathways in sarcoma mice model. Eur. J. Pharm. Sci. 2011, 43, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Soni, R.; Muller, L.; Furet, P.; Schoepfer, J.; Stephan, C.; Zumstein-Mecker, S.; Fretz, H.; Chaudhuri, B. Inhibition of cyclin-dependent kinase 4 (Cdk4) by fascaplysin, a marine natural product. Biochem. Biophys. Res. Commun. 2000, 275, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.L.; Lu, X.L.; Lin, J.; Chen, H.M.; Yan, X.J.; Wang, F.; Xu, W.F. Direct effects of fascaplysin on human umbilical vein endothelial cells attributing the anti-angiogenesis activity. Biomed. Pharmacother. 2010, 64, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.R.; Konicek, B.W.; Vincent, T.M.; Lynch, R.L.; Monteith, D.; Weir, S.N.; Schwier, P.; Capen, A.; Goode, R.L.; Dowless, M.S.; et al. Therapeutic suppression of translation initiation factor eIF4E expression reduces tumor growth without toxicity. J. Clin. Investig. 2007, 117, 2638–2648. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, H.; Lawrence, D.; Varfolomeev, E.; Totpal, K.; Morlan, J.; Schow, P.; Fong, S.; Schwall, R.; Sinicropi, D.; Ashkenazi, A. Tumor-cell resistance to death receptor—Induced apoptosis through mutational inactivation of the proapoptotic Bcl-2 homolog Bax. Nat. Med. 2002, 8, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Lee, Y.M.; Lee, G.; Choi, Y.J.; Lim, B.O.; Kim, Y.J.; Choi, D.K.; Park, J.W. PRMT5 is essential for the eIF4E-mediated 5’-cap dependent translation. Biochem. Biophys. Res. Commun. 2014, 452, 1016–1021. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Luo, C.; Vazquez, F.; Puigserver, P. Targeting mitochondrial oxidative metabolism in melanoma causes metabolic compensation through glucose and glutamine utilization. Cancer Res. 2014, 74, 3535–3545. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.S.; Cho, S.J.; Park, J.; Kim, Y.; Choi, Y.J.; Pyo, J.S.; Jang, B.G.; Park, J.W.; Kim, W.H.; Lee, B.L. Loss of FOXO1 promotes gastric tumour growth and metastasis through upregulation of human epidermal growth factor receptor 2/neu expression. Br. J. Cancer 2015, 113, 1186–1196. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, T.-I.; Lee, Y.-M.; Nam, T.-J.; Ko, Y.-S.; Mah, S.; Kim, J.; Kim, Y.; Reddy, R.H.; Kim, Y.J.; Hong, S.; et al. Fascaplysin Exerts Anti-Cancer Effects through the Downregulation of Survivin and HIF-1α and Inhibition of VEGFR2 and TRKA. Int. J. Mol. Sci. 2017, 18, 2074. https://doi.org/10.3390/ijms18102074
Oh T-I, Lee Y-M, Nam T-J, Ko Y-S, Mah S, Kim J, Kim Y, Reddy RH, Kim YJ, Hong S, et al. Fascaplysin Exerts Anti-Cancer Effects through the Downregulation of Survivin and HIF-1α and Inhibition of VEGFR2 and TRKA. International Journal of Molecular Sciences. 2017; 18(10):2074. https://doi.org/10.3390/ijms18102074
Chicago/Turabian StyleOh, Taek-In, Yoon-Mi Lee, Taek-Jin Nam, Young-San Ko, Shinmee Mah, Jinhee Kim, Younghoon Kim, Rallabandi Harikrishna Reddy, Young Jun Kim, Sungwoo Hong, and et al. 2017. "Fascaplysin Exerts Anti-Cancer Effects through the Downregulation of Survivin and HIF-1α and Inhibition of VEGFR2 and TRKA" International Journal of Molecular Sciences 18, no. 10: 2074. https://doi.org/10.3390/ijms18102074
APA StyleOh, T.-I., Lee, Y.-M., Nam, T.-J., Ko, Y.-S., Mah, S., Kim, J., Kim, Y., Reddy, R. H., Kim, Y. J., Hong, S., & Lim, J.-H. (2017). Fascaplysin Exerts Anti-Cancer Effects through the Downregulation of Survivin and HIF-1α and Inhibition of VEGFR2 and TRKA. International Journal of Molecular Sciences, 18(10), 2074. https://doi.org/10.3390/ijms18102074