Neuromuscular Junction Dismantling in Amyotrophic Lateral Sclerosis

{kind=link}
Abstract
:1. The Neuromuscular Junction as a Tripartite Synapse
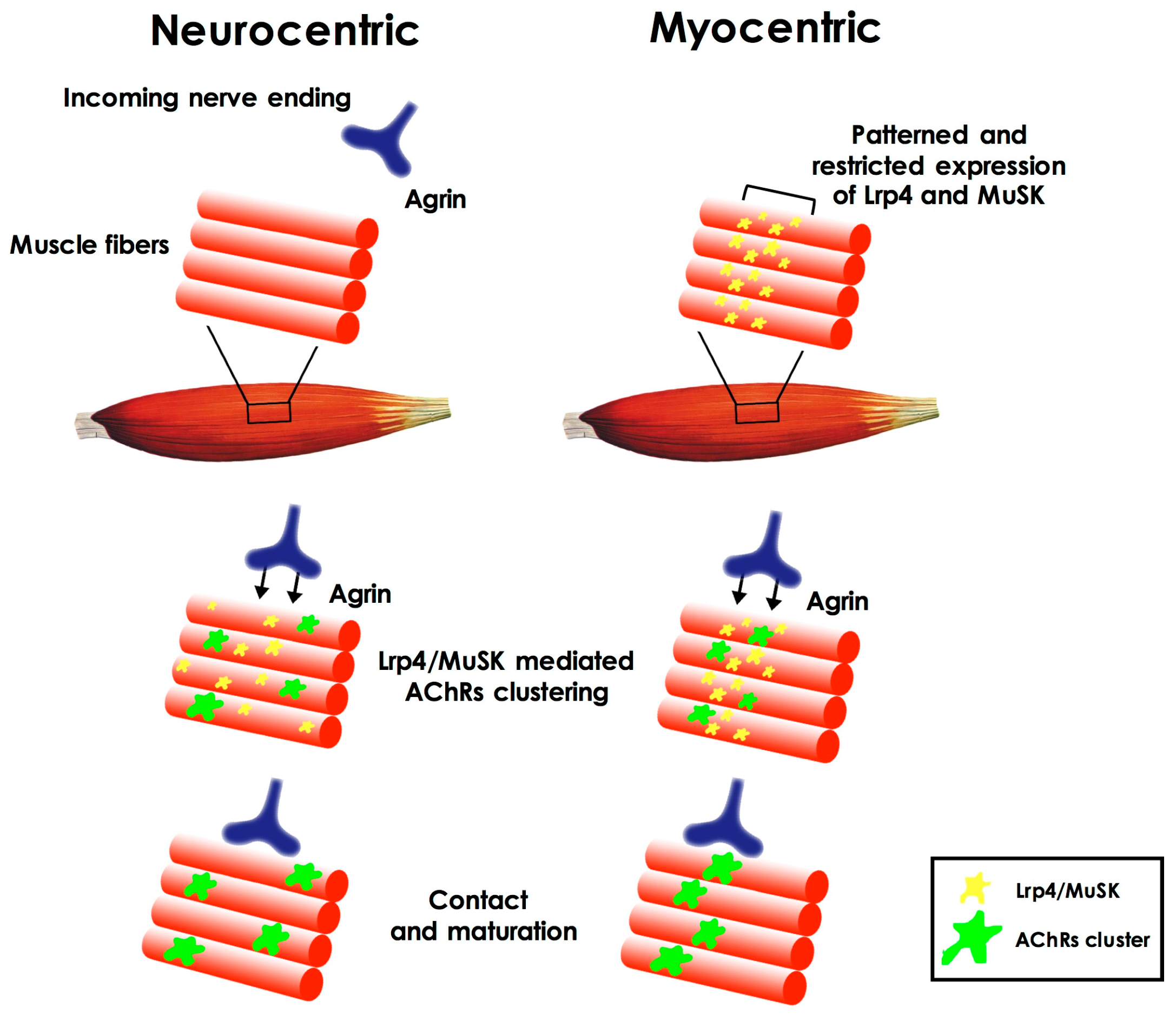
2. Neuromuscular Junction Assembly and Plasticity
Cross-Talk during Neuromuscular Junction Assembly
3. The Neuromuscular Junction in Aging
4. Amyotrophic Lateral Sclerosis
5. Neuromuscular Junction Degeneration in Amyotrophic Lateral Sclerosis
6. The Role of Skeletal Muscle in NMJ Dismantling in Amyotrophic Lateral Sclerosis
7. Glial Cells, Schwann Cells, and Terminal Schwann Cells in Amyotrophic Lateral Sclerosis Pathogenesis
8. Conclusions
Conflicts of Interest
References
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Tripartite synapses: Glia, the unacknowledged partner. Trends Neurosci. 1999, 22, 208–215. [Google Scholar] [CrossRef]
- Araque, A.; Carmignoto, G.; Haydon, P.G.; Oliet, S.H.; Robitaille, R.; Volterra, A. Gliotransmitters travel in time and space. Neuron 2014, 81, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Robitaille, R. Modulation of synaptic efficacy and synaptic depression by glial cells at the frog neuromuscular junction. Neuron 1998, 21, 847–855. [Google Scholar] [CrossRef]
- Wu, H.; Xiong, W.C.; Mei, L. To build a synapse: Signaling pathways in neuromuscular junction assembly. Development 2010, 137, 1017–1033. [Google Scholar] [CrossRef] [PubMed]
- Hanson, M.G.; Niswander, L.A. An explant muscle model to examine the refinement of the synaptic landscape. J. Neurosci. Methods 2014, 238, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Burgess, R.W.; Dominguez, B.; Pfaff, S.L.; Sanes, J.R.; Lee, K.F. Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature 2001, 410, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Lomo, T. What controls the position, number, size, and distribution of neuromuscular junctions on rat muscle fibers? J. Neurocytol. 2003, 32, 835–848. [Google Scholar] [CrossRef] [PubMed]
- Witzemann, V. Development of the neuromuscular junction. Cell Tissue Res. 2006, 326, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Arber, S.; William, C.; Li, L.; Tanabe, Y.; Jessell, T.M.; Birchmeier, C.; Burden, S.J. Patterning of muscle acetylcholine receptor gene expression in the absence of motor innervation. Neuron 2001, 30, 399–410. [Google Scholar] [CrossRef]
- Burden, S.J.; Yumoto, N.; Zhang, W. The role of MuSK in synapse formation and neuromuscular disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a009167. [Google Scholar] [CrossRef] [PubMed]
- Singhal, N.; Martin, P.T. Role of extracellular matrix proteins and their receptors in the development of the vertebrate neuromuscular junction. Dev. Neurobiol. 2011, 71, 982–1005. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.P.; Robitaille, R. Perisynaptic Schwann cells at the neuromuscular synapse: Adaptable, multitasking glial cells. Cold Spring Harb. Perspect. Biol. 2015, 7, a020503. [Google Scholar] [CrossRef] [PubMed]
- Darabid, H.; Perez-Gonzalez, A.P.; Robitaille, R. Neuromuscular synaptogenesis: Coordinating partners with multiple functions. Nat. Rev. Neurosci. 2014, 15, 703–718. [Google Scholar] [CrossRef] [PubMed]
- Barik, A.; Li, L.; Sathyamurthy, A.; Xiong, W.C.; Mei, L. Schwann cells in neuromuscular junction formation and maintenance. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 9770–9781. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.G.; Shen, X.M.; Selcen, D.; Sine, S.M. Congenital myasthenic syndromes: Pathogenesis, diagnosis, and treatment. Lancet Neurol. 2015, 14, 420–434, Erratum in 2015, 14, 461. [Google Scholar] [CrossRef]
- Tintignac, L.A.; Brenner, H.R.; Ruegg, M.A. Mechanisms regulating neuromuscular junction development and function and causes of muscle wasting. Physiol. Rev. 2015, 95, 809–852. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Freire, M.; de Cabo, R.; Studenski, S.A.; Ferrucci, L. The neuromuscular junction: Aging at the crossroad between nerves and muscle. Front. Aging Neurosci. 2014, 6, 208. [Google Scholar] [CrossRef] [PubMed]
- Rowan, S.L.; Rygiel, K.; Purves-Smith, F.M.; Solbak, N.M.; Turnbull, D.M.; Hepple, R.T. Denervation causes fiber atrophy and myosin heavy chain co-expression in senescent skeletal muscle. PLoS ONE 2012, 7, e29082. [Google Scholar] [CrossRef] [PubMed]
- Valdez, G.; Tapia, J.C.; Kang, H.; Clemenson, G.D., Jr.; Gage, F.H.; Lichtman, J.W.; Sanes, J.R. Attenuation of age-related changes in mouse neuromuscular synapses by caloric restriction and exercise. Proc. Natl. Acad. Sci. USA 2010, 107, 14863–14868. [Google Scholar] [CrossRef] [PubMed]
- Nishimune, H. Active zones of mammalian neuromuscular junctions: Formation, density, and aging. Ann. N. Y. Acad. Sci. 2012, 1274, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Ibebunjo, C.; Chick, J.M.; Kendall, T.; Eash, J.K.; Li, C.; Zhang, Y.; Vickers, C.; Wu, Z.; Clarke, B.A.; Shi, J.; et al. Genomic and proteomic profiling reveals reduced mitochondrial function and disruption of the neuromuscular junction driving rat sarcopenia. Mol. Cell. Biol. 2013, 33, 194–212. [Google Scholar] [CrossRef] [PubMed]
- Aare, S.; Spendiff, S.; Vuda, M.; Elkrief, D.; Perez, A.; Wu, Q.; Mayaki, D.; Hussain, S.N.; Hettwer, S.; Hepple, R.T. Failed reinnervation in aging skeletal muscle. Skelet. Muscle 2016, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Butikofer, L.; Zurlinden, A.; Bolliger, M.F.; Kunz, B.; Sonderegger, P. Destabilization of the neuromuscular junction by proteolytic cleavage of agrin results in precocious sarcopenia. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 4378–4393. [Google Scholar] [CrossRef] [PubMed]
- Sugita, S.; Fleming, L.L.; Wood, C.; Vaughan, S.K.; Gomes, M.P.; Camargo, W.; Naves, L.A.; Prado, V.F.; Prado, M.A.; Guatimosim, C.; et al. VAChT overexpression increases acetylcholine at the synaptic cleft and accelerates aging of neuromuscular junctions. Skelet. Muscle 2016, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Taetzsch, T.; Tenga, M.J.; Valdez, G. Muscle fibers secrete FGFBP1 to slow degeneration of neuromuscular synapses during aging and progression of ALS. J. Neurosci. Off. J. Soc. Neurosci. 2017, 37, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Rygiel, K.A.; Picard, M.; Turnbull, D.M. The ageing neuromuscular system and sarcopenia: A mitochondrial perspective. J. Physiol. 2016, 594, 4499–4512. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.L.; Fernandez, A.; Solas, M.T. Mitochondria, motor neurons and aging. J. Neurol. Sci. 2013, 330, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Deschenes, M.R.; Roby, M.A.; Eason, M.K.; Harris, M.B. Remodeling of the neuromuscular junction precedes sarcopenia related alterations in myofibers. Exp. Gerontol. 2010, 45, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, L.; Baroni, M.; Ranchelli, A.; Lauretani, F.; Maggio, M.; Mecocci, P.; Ruggiero, C. Interaction between bone and muscle in older persons with mobility limitations. Curr. Pharm. Des. 2014, 20, 3178–3197. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, L.; Schrack, J.A.; Knuth, N.D.; Simonsick, E.M. Aging and the energetic cost of life. J. Am. Geriatr. Soc. 2012, 60, 1768–1769. [Google Scholar] [CrossRef] [PubMed]
- Manini, T.M.; Hong, S.L.; Clark, B.C. Aging and muscle: A neuron’s perspective. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Klose, A.; Forman, S.; Paris, N.D.; Wei-LaPierre, L.; Cortes-Lopez, M.; Tan, A.; Flaherty, M.; Miura, P.; Dirksen, R.T.; et al. Loss of adult skeletal muscle stem cells drives age-related neuromuscular junction degeneration. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wei-LaPierre, L.; Klose, A.; Dirksen, R.T.; Chakkalakal, J.V. Inducible depletion of adult skeletal muscle stem cells impairs the regeneration of neuromuscular junctions. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Chio, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, M.; Brown, R.H., Jr. Genetics of Amyotrophic Lateral Sclerosis. Cold Spring Harb. Perspect. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bastos, A.F.; Orsini, M.; Machado, D.; Mello, M.P.; Nader, S.; Silva, J.G.; da Silva Catharino, A.M.; de Freitas, M.R.; Pereira, A.; Pessoa, L.L.; et al. Amyotrophic lateral sclerosis: One or multiple causes? Neurol. Int. 2011, 3, e4. [Google Scholar] [CrossRef] [PubMed]
- Blasco, H.; Mavel, S.; Corcia, P.; Gordon, P.H. The glutamate hypothesis in ALS: Pathophysiology and drug development. Curr. Med. Chem. 2014, 21, 3551–3575. [Google Scholar] [CrossRef] [PubMed]
- Anand, A.; Thakur, K.; Gupta, P.K. ALS and oxidative stress: The neurovascular scenario. Oxid. Med. Cell. Longev. 2013, 2013, 635831. [Google Scholar] [CrossRef] [PubMed]
- Cappello, V.; Vezzoli, E.; Righi, M.; Fossati, M.; Mariotti, R.; Crespi, A.; Patruno, M.; Bentivoglio, M.; Pietrini, G.; Francolini, M. Analysis of neuromuscular junctions and effects of anabolic steroid administration in the SOD1G93A mouse model of ALS. Mol. Cell. Neurosci. 2012, 51, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Berger, J.R. ALS syndrome in patients with HIV-1 infection. J. Neurol. Sci. 2006, 240, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Ravits, J. Sporadic amyotrophic lateral sclerosis: A hypothesis of persistent (non-lytic) enteroviral infection. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2005, 6, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Kollewe, K.; Wurster, U.; Sinzenich, T.; Korner, S.; Dengler, R.; Mohammadi, B.; Petri, S. Anti-ganglioside antibodies in amyotrophic lateral sclerosis revisited. PLoS ONE 2015, 10, e0125339. [Google Scholar] [CrossRef] [PubMed]
- Lall, D.; Baloh, R.H. Microglia and C9orf72 in neuroinflammation and ALS and frontotemporal dementia. J. Clin. Investig. 2017, 127, 3250–3258. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; Hardiman, O. The epidemiology of ALS: A conspiracy of genes, environment and time. Nat. Rev. Neurol. 2013, 9, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Talbott, E.O.; Malek, A.M.; Lacomis, D. The epidemiology of amyotrophic lateral sclerosis. Handb. Clin. Neurol. 2016, 138, 225–238. [Google Scholar] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Vucic, S.; Rothstein, J.D.; Kiernan, M.C. Advances in treating amyotrophic lateral sclerosis: Insights from pathophysiological studies. Trends Neurosci. 2014, 37, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Gurney, A.L.; Carver-Moore, K.; de Sauvage, F.J.; Moore, M.W. Thrombocytopenia in c-mpl-deficient mice. Science 1994, 265, 1445–1447. [Google Scholar] [CrossRef] [PubMed]
- Turner, B.J.; Talbot, K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog. Neurobiol. 2008, 85, 94–134. [Google Scholar] [CrossRef] [PubMed]
- Babin, P.J.; Goizet, C.; Raldua, D. Zebrafish models of human motor neuron diseases: Advantages and limitations. Prog. Neurobiol. 2014, 118, 36–58. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.R.; Lagow, R.D.; Xu, K.; Zhang, B.; Bonini, N.M. A drosophila model for amyotrophic lateral sclerosis reveals motor neuron damage by human SOD1. J. Biol. Chem. 2008, 283, 24972–24981. [Google Scholar] [CrossRef] [PubMed]
- Bahadorani, S.; Mukai, S.T.; Rabie, J.; Beckman, J.S.; Phillips, J.P.; Hilliker, A.J. Expression of zinc-deficient human superoxide dismutase in Drosophila neurons produces a locomotor defect linked to mitochondrial dysfunction. Neurobiol. Aging 2013, 34, 2322–2330. [Google Scholar] [CrossRef] [PubMed]
- Islam, R.; Kumimoto, E.L.; Bao, H.; Zhang, B. ALS-linked SOD1 in glial cells enhances ss-N-Methylamino l-Alanine (BMAA)-induced toxicity in Drosophila. F1000Research 2012, 1, 47. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, T.; Zhang, X.; Tang, Y.; Yang, J.; Le, W. Human superoxide dismutase 1 overexpression in motor neurons of Caenorhabditis elegans causes axon guidance defect and neurodegeneration. Neurobiol. Aging 2014, 35, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Oeda, T.; Shimohama, S.; Kitagawa, N.; Kohno, R.; Imura, T.; Shibasaki, H.; Ishii, N. Oxidative stress causes abnormal accumulation of familial amyotrophic lateral sclerosis-related mutant SOD1 in transgenic Caenorhabditis elegans. Hum. Mol. Genet. 2001, 10, 2013–2023. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Farr, G.W.; Hall, D.H.; Li, F.; Furtak, K.; Dreier, L.; Horwich, A.L. An ALS-linked mutant SOD1 produces a locomotor defect associated with aggregation and synaptic dysfunction when expressed in neurons of Caenorhabditis elegans. PLoS Genet. 2009, 5, e1000350. [Google Scholar] [CrossRef] [PubMed]
- Philips, T.; Rothstein, J.D. Glial cells in amyotrophic lateral sclerosis. Exp. Neurol. 2014, 262 Pt B, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Arbour, D.; Vande Velde, C.; Robitaille, R. New perspectives on amyotrophic lateral sclerosis: The role of glial cells at the neuromuscular junction. J. Physiol. 2017, 595, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, J.P.; Picchiarelli, G.; Dupuis, L.; Gonzalez de Aguilar, J.L. The role of skeletal muscle in amyotrophic lateral sclerosis. Brain Pathol. 2016, 26, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Brettschneider, J.; Ludolph, A.C.; Lee, V.M.; Trojanowski, J.Q.; del Tredici, K. Amyotrophic lateral sclerosis—A model of corticofugal axonal spread. Nat. Rev. Neurol. 2013, 9, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Moloney, E.B.; de Winter, F.; Verhaagen, J. ALS as a distal axonopathy: Molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease. Front. Neurosci. 2014, 8, 252. [Google Scholar] [CrossRef] [PubMed]
- Ilieva, H.; Polymenidou, M.; Cleveland, D.W. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 2009, 187, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Pramatarova, A.; Laganiere, J.; Roussel, J.; Brisebois, K.; Rouleau, G.A. Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, 3369–3374. [Google Scholar]
- Lino, M.M.; Schneider, C.; Caroni, P. Accumulation of SOD1 mutants in postnatal motoneurons does not cause motoneuron pathology or motoneuron disease. J. Neurosci. Off. J. Soc. Neurosci. 2002, 22, 4825–4832. [Google Scholar]
- Clement, A.M.; Nguyen, M.D.; Roberts, E.A.; Garcia, M.L.; Boillee, S.; Rule, M.; McMahon, A.P.; Doucette, W.; Siwek, D.; Ferrante, R.J.; et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 2003, 302, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Jaarsma, D.; Teuling, E.; Haasdijk, E.D.; de Zeeuw, C.I.; Hoogenraad, C.C. Neuron-specific expression of mutant superoxide dismutase is sufficient to induce amyotrophic lateral sclerosis in transgenic mice. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 2075–2088. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.A.; Southam, K.A.; Blizzard, C.A.; King, A.E.; Dickson, T.C. Axonal degeneration, distal collateral branching and neuromuscular junction architecture alterations occur prior to symptom onset in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. J. Chem. Neuroanat. 2016, 76, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Pollari, E.; Goldsteins, G.; Bart, G.; Koistinaho, J.; Giniatullin, R. The role of oxidative stress in degeneration of the neuromuscular junction in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2014, 8, 131. [Google Scholar] [CrossRef] [PubMed]
- Pun, S.; Santos, A.F.; Saxena, S.; Xu, L.; Caroni, P. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat. Neurosci. 2006, 9, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Xia, R.; Liu, Y.; Yang, L.; Gal, J.; Zhu, H.; Jia, J. Motor neuron apoptosis and neuromuscular junction perturbation are prominent features in a Drosophila model of Fus-mediated ALS. Mol. Neurodegener. 2012, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Chai, A.; Withers, J.; Koh, Y.H.; Parry, K.; Bao, H.; Zhang, B.; Budnik, V.; Pennetta, G. hVAPB, the causative gene of a heterogeneous group of motor neuron diseases in humans, is functionally interchangeable with its Drosophila homologue DVAP-33A at the neuromuscular junction. Hum. Mol. Genet. 2008, 17, 266–280. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, L.; Ghilardi, A.; Rottoli, E.; de Maglie, M.; Prosperi, L.; Perego, C.; Baruscotti, M.; Bucchi, A.; del Giacco, L.; Francolini, M. INaP selective inhibition reverts precocious inter- and motorneurons hyperexcitability in the Sod1-G93R zebrafish ALS model. Sci. Rep. 2016, 6, 24515. [Google Scholar] [CrossRef] [PubMed]
- Dzieciolowska, S.; Drapeau, P.; Armstrong, G.A.B. Augmented quantal release of acetylcholine at the vertebrate neuromuscular junction following tdp-43 depletion. PLoS ONE 2017, 12, e0177005. [Google Scholar] [CrossRef] [PubMed]
- Pansarasa, O.; Rossi, D.; Berardinelli, A.; Cereda, C. Amyotrophic lateral sclerosis and skeletal muscle: An update. Mol. Neurobiol. 2014, 49, 984–990. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Yi, J.; Ma, C.; Xiao, Y.; Yi, F.; Yu, T.; Zhou, J. Defective mitochondrial dynamics is an early event in skeletal muscle of an amyotrophic lateral sclerosis mouse model. PLoS ONE 2013, 8, e82112. [Google Scholar] [CrossRef] [PubMed]
- Echaniz-Laguna, A.; Degos, B.; Mohr, M.; Kessler, R.; Urban-Kraemer, E.; Tranchant, C. A study of three patients with amyotrophic lateral sclerosis and a polyneuropathy resembling CIDP. Muscle Nerve 2006, 33, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Da Cruz, S.; Parone, P.A.; Lopes, V.S.; Lillo, C.; McAlonis-Downes, M.; Lee, S.K.; Vetto, A.P.; Petrosyan, S.; Marsala, M.; Murphy, A.N.; et al. Elevated PGC-1α activity sustains mitochondrial biogenesis and muscle function without extending survival in a mouse model of inherited ALS. Cell Metab. 2012, 15, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolny, G.; Aucello, M.; Molinaro, M.; Musaro, A. Local expression of mIgf-1 modulates ubiquitin, caspase and CDK5 expression in skeletal muscle of an ALS mouse model. Neurol. Res. 2008, 30, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; Martin, L.J. Skeletal muscle-restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Hum. Mol. Genet. 2010, 19, 2284–2302. [Google Scholar] [CrossRef] [PubMed]
- Marcuzzo, S.; Zucca, I.; Mastropietro, A.; de Rosbo, N.K.; Cavalcante, P.; Tartari, S.; Bonanno, S.; Preite, L.; Mantegazza, R.; Bernasconi, P. Hind limb muscle atrophy precedes cerebral neuronal degeneration in G93A-SOD1 mouse model of amyotrophic lateral sclerosis: A longitudinal MRI study. Exp. Neurol. 2011, 231, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolny, G.; Giacinti, C.; Pelosi, L.; Nicoletti, C.; Winn, N.; Barberi, L.; Molinaro, M.; Rosenthal, N.; Musaro, A. Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model. J. Cell Biol. 2005, 168, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, B.K.; Frost, L.M.; Christian, L.; Umapathi, P.; Gage, F.H. Synergy of insulin-like growth factor-1 and exercise in amyotrophic lateral sclerosis. Ann. Neurol. 2005, 57, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Azzouz, M.; Ralph, G.S.; Storkebaum, E.; Walmsley, L.E.; Mitrophanous, K.A.; Kingsman, S.M.; Carmeliet, P.; Mazarakis, N.D. VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature 2004, 429, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Brakefield, D.; Pan, Y.; Hunter, D.; Myckatyn, T.M.; Parsadanian, A. Muscle-derived but not centrally derived transgene GDNF is neuroprotective in G93A-SOD1 mouse model of ALS. Exp. Neurol. 2007, 203, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Skold, M.K.; Li, J.; Nennesmo, I.; Fadeel, B.; Henter, J.I. VEGF reduces astrogliosis and preserves neuromuscular junctions in ALS transgenic mice. Biochem. Biophys. Res. Commun. 2007, 363, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Rocha, M.C.; Pousinha, P.A.; Correia, A.M.; Sebastiao, A.M.; Ribeiro, J.A. Early changes of neuromuscular transmission in the SOD1(G93A) mice model of ALS start long before motor symptoms onset. PLoS ONE 2013, 8, e73846. [Google Scholar] [CrossRef] [PubMed]
- Campanari, M.L.; Garcia-Ayllon, M.S.; Ciura, S.; Saez-Valero, J.; Kabashi, E. Neuromuscular Junction Impairment in Amyotrophic Lateral Sclerosis: Reassessing the Role of Acetylcholinesterase. Front. Mol. Neurosci. 2016, 9, 160. [Google Scholar] [CrossRef] [PubMed]
- Palma, E.; Inghilleri, M.; Conti, L.; Deflorio, C.; Frasca, V.; Manteca, A.; Pichiorri, F.; Roseti, C.; Torchia, G.; Limatola, C.; et al. Physiological characterization of human muscle acetylcholine receptors from ALS patients. Proc. Natl. Acad. Sci. USA 2011, 108, 20184–20188. [Google Scholar] [CrossRef] [PubMed]
- Palma, E.; Reyes-Ruiz, J.M.; Lopergolo, D.; Roseti, C.; Bertollini, C.; Ruffolo, G.; Cifelli, P.; Onesti, E.; Limatola, C.; Miledi, R.; et al. Acetylcholine receptors from human muscle as pharmacological targets for ALS therapy. Proc. Natl. Acad. Sci. USA 2016, 113, 3060–3065. [Google Scholar] [CrossRef] [PubMed]
- Tsujihata, M.; Hazama, R.; Yoshimura, T.; Satoh, A.; Mori, M.; Nagataki, S. The motor end-plate fine structure and ultrastructural localization of acetylcholine receptors in amyotrophic lateral sclerosis. Muscle Nerve 1984, 7, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Bjornskov, E.K.; Norris, F.H., Jr.; Mower-Kuby, J. Quantitative axon terminal and end-plate morphology in amyotrophic lateral sclerosis. Arch. Neurol. 1984, 41, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, T.; Ishii, T.; Iwata, M.; Nomoto, M. Ultrastructural and histochemical study of the motor end plates of the intrinsic laryngeal muscles in amyotrophic lateral sclerosis. Ultrastruct. Pathol. 1998, 22, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, L.; Baranzini, M.; Berardinelli, M.G.; Lattanzi, W.; Monforte, M.; Tasca, G.; Conte, A.; Logroscino, G.; Michetti, F.; Ricci, E.; et al. Potential therapeutic targets for ALS: MIR206, MIR208b and MIR499 are modulated during disease progression in the skeletal muscle of patients. Sci. Rep. 2017, 7, 9538. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.H.; Parsadanian, A.S.; Andreeva, A.; Snider, W.D.; Elliott, J.L. Restricted expression of G86R Cu/Zn superoxide dismutase in astrocytes results in astrocytosis but does not cause motoneuron degeneration. J. Neurosci. Off. J. Soc. Neurosci. 2000, 20, 660–665. [Google Scholar]
- Yamanaka, K.; Boillee, S.; Roberts, E.A.; Garcia, M.L.; McAlonis-Downes, M.; Mikse, O.R.; Cleveland, D.W.; Goldstein, L.S. Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proc. Natl. Acad. Sci. USA 2008, 105, 7594–7599. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Chun, S.J.; Boillee, S.; Fujimori-Tonou, N.; Yamashita, H.; Gutmann, D.H.; Takahashi, R.; Misawa, H.; Cleveland, D.W. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 2008, 11, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Qian, K.; Chen, W.; Hu, B.; Blackbourn, L.W.; Du, Z.; Ma, L.; Liu, H.; Knobel, K.M.; Ayala, M.; et al. Human-derived neural progenitors functionally replace astrocytes in adult mice. J. Clin. Investig. 2015, 125, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Qian, K.; Huang, H.; Peterson, A.; Hu, B.; Maragakis, N.J.; Ming, G.L.; Chen, H.; Zhang, S.C. Sporadic ALS Astrocytes Induce Neuronal Degeneration In Vivo. Stem Cell Rep. 2017, 8, 843–855. [Google Scholar] [CrossRef] [PubMed]
- Boillee, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.; Kassiotis, G.; Kollias, G.; Cleveland, D.W. Onset and progression in inherited ALS determined by motor neurons and microglia. Science 2006, 312, 1389–1392. [Google Scholar] [CrossRef] [PubMed]
- Valori, C.F.; Brambilla, L.; Martorana, F.; Rossi, D. The multifaceted role of glial cells in amyotrophic lateral sclerosis. Cell. Mol. Life Sci. CMLS 2014, 71, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Perrie, W.T.; Lee, G.T.; Curtis, E.M.; Sparke, J.; Buller, J.R.; Rossi, M.L. Changes in the myelinated axons of femoral nerve in amyotrophic lateral sclerosis. J. Neural Transm. Suppl. 1993, 39, 223–233. [Google Scholar] [PubMed]
- Keller, A.F.; Gravel, M.; Kriz, J. Live imaging of amyotrophic lateral sclerosis pathogenesis: Disease onset is characterized by marked induction of GFAP in Schwann cells. Glia 2009, 57, 1130–1142. [Google Scholar] [CrossRef] [PubMed]
- Turner, B.J.; Ackerley, S.; Davies, K.E.; Talbot, K. Dismutase-competent SOD1 mutant accumulation in myelinating Schwann cells is not detrimental to normal or transgenic ALS model mice. Hum. Mol. Genet. 2010, 19, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Lobsiger, C.S.; Boillee, S.; McAlonis-Downes, M.; Khan, A.M.; Feltri, M.L.; Yamanaka, K.; Cleveland, D.W. Schwann cells expressing dismutase active mutant SOD1 unexpectedly slow disease progression in ALS mice. Proc. Natl. Acad. Sci. USA 2009, 106, 4465–4470. [Google Scholar] [CrossRef] [PubMed]
- Duregotti, E.; Negro, S.; Scorzeto, M.; Zornetta, I.; Dickinson, B.C.; Chang, C.J.; Montecucco, C.; Rigoni, M. Mitochondrial alarmins released by degenerating motor axon terminals activate perisynaptic Schwann cells. Proc. Natl. Acad. Sci. USA 2015, 112, E497–E505. [Google Scholar] [CrossRef] [PubMed]
- Muyderman, H.; Chen, T. Mitochondrial dysfunction in amyotrophic lateral sclerosis—A valid pharmacological target? Br. J. Pharmacol. 2014, 171, 2191–2205. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Lichtman, J.W. Motor axon regeneration and muscle reinnervation in young adult and aged animals. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 19480–19491. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Tian, L.; Mikesh, M.; Lichtman, J.W.; Thompson, W.J. Terminal Schwann cells participate in neuromuscular synapse remodeling during reinnervation following nerve injury. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 6323–6333. [Google Scholar] [CrossRef] [PubMed]
- Arbour, D.; Tremblay, E.; Martineau, E.; Julien, J.P.; Robitaille, R. Early and persistent abnormal decoding by glial cells at the neuromuscular junction in an ALS model. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 688–706. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, H.; Ng, S.K.; Diaz, N.; Burstein, S.; Morel, L.; Osgood, A.; Sider, B.; Higashimori, H.; Haydon, P.G.; Manfredi, G.; et al. Abnormal intracellular calcium signaling and SNARE-dependent exocytosis contributes to SOD1G93A astrocyte-mediated toxicity in amyotrophic lateral sclerosis. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 2331–2348. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, D.I.; Seburn, K.L.; Pinter, M.J. Altered terminal Schwann cell morphology precedes denervation in SOD1 mice. Exp. Neurol. 2016, 275, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, D.I.; Bahr, B.A.; Seburn, K.L.; Pinter, M.J. Abnormal response of distal Schwann cells to denervation in a mouse model of motor neuron disease. Exp. Neurol. 2016, 278, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Lasiene, J.; Komine, O.; Fujimori-Tonou, N.; Powers, B.; Endo, F.; Watanabe, S.; Shijie, J.; Ravits, J.; Horner, P.; Misawa, H.; et al. Neuregulin 1 confers neuroprotection in SOD1-linked amyotrophic lateral sclerosis mice via restoration of C-boutons of spinal motor neurons. Acta Neuropathol. Commun. 2016, 4, 15. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Fukuda, Y.; Yoshimura, J.; Toyoda, A.; Kurppa, K.; Moritoyo, H.; Belzil, V.V.; Dion, P.A.; Higasa, K.; Doi, K.; et al. ERBB4 mutations that disrupt the neuregulin-ErbB4 pathway cause amyotrophic lateral sclerosis type 19. Am. J. Hum. Genet. 2013, 93, 900–905. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cappello, V.; Francolini, M. Neuromuscular Junction Dismantling in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2017, 18, 2092. https://doi.org/10.3390/ijms18102092
Cappello V, Francolini M. Neuromuscular Junction Dismantling in Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences. 2017; 18(10):2092. https://doi.org/10.3390/ijms18102092
Chicago/Turabian StyleCappello, Valentina, and Maura Francolini. 2017. "Neuromuscular Junction Dismantling in Amyotrophic Lateral Sclerosis" International Journal of Molecular Sciences 18, no. 10: 2092. https://doi.org/10.3390/ijms18102092
APA StyleCappello, V., & Francolini, M. (2017). Neuromuscular Junction Dismantling in Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences, 18(10), 2092. https://doi.org/10.3390/ijms18102092