A Combination of Immune Checkpoint Inhibition with Metronomic Chemotherapy as a Way of Targeting Therapy-Resistant Cancer Cells

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
Tumors Engage and Modify Their Microenvironment
2. Alternative to Maximal Tolerated Dose (MTD): Metronomic Chemotherapy
2.1. Decreased Angiogenesis
2.2. Decrease Therapeutic Resistance
2.3. Promote Anti-Tumor Immunity
2.4. Targeting Cancer Stem Cells
3. Checkpoint Inhibitors
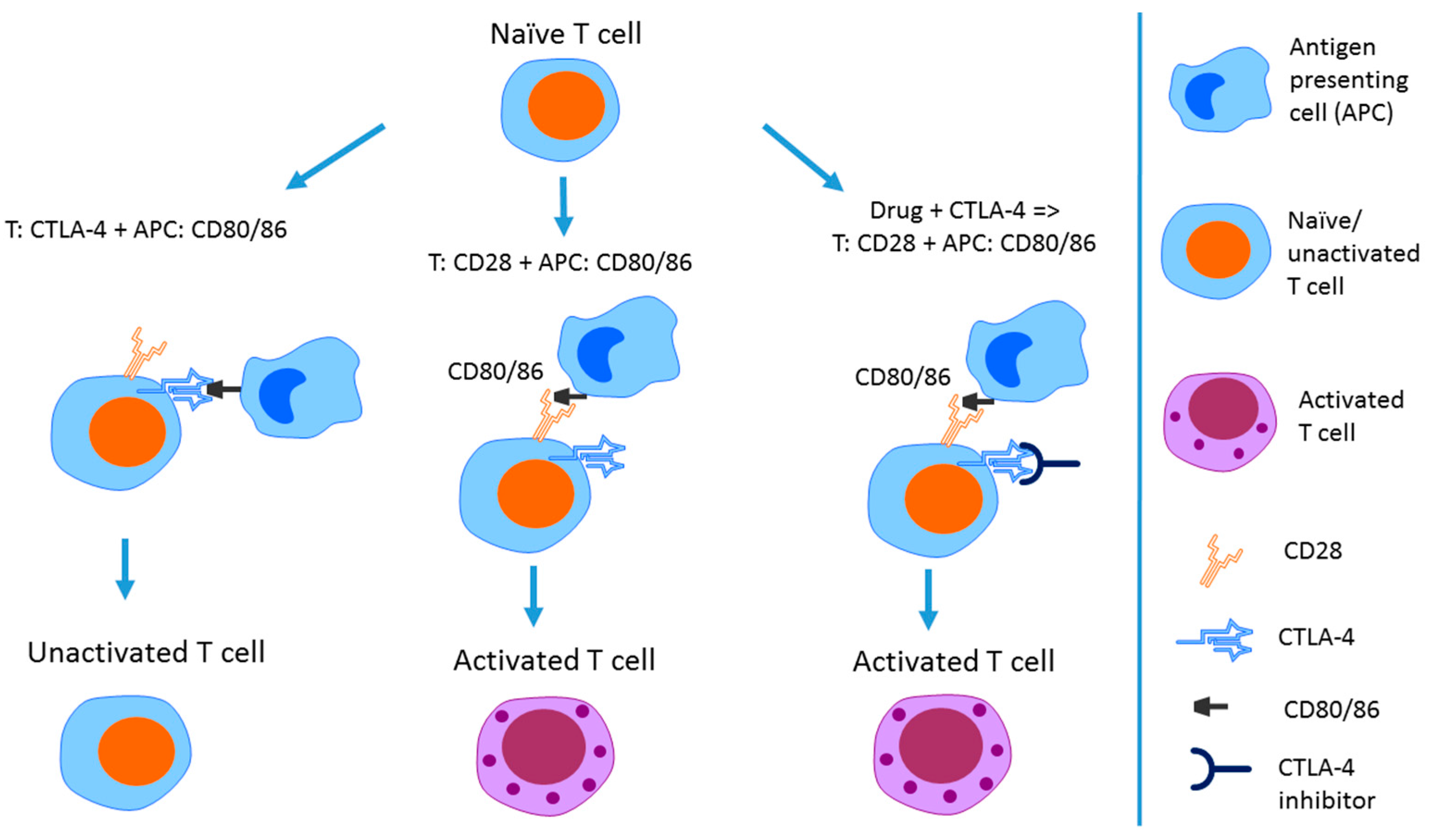
3.1. CTLA-4 Inhibition
3.2. PD-1, PD-L1 and PD-L2 Inhibition
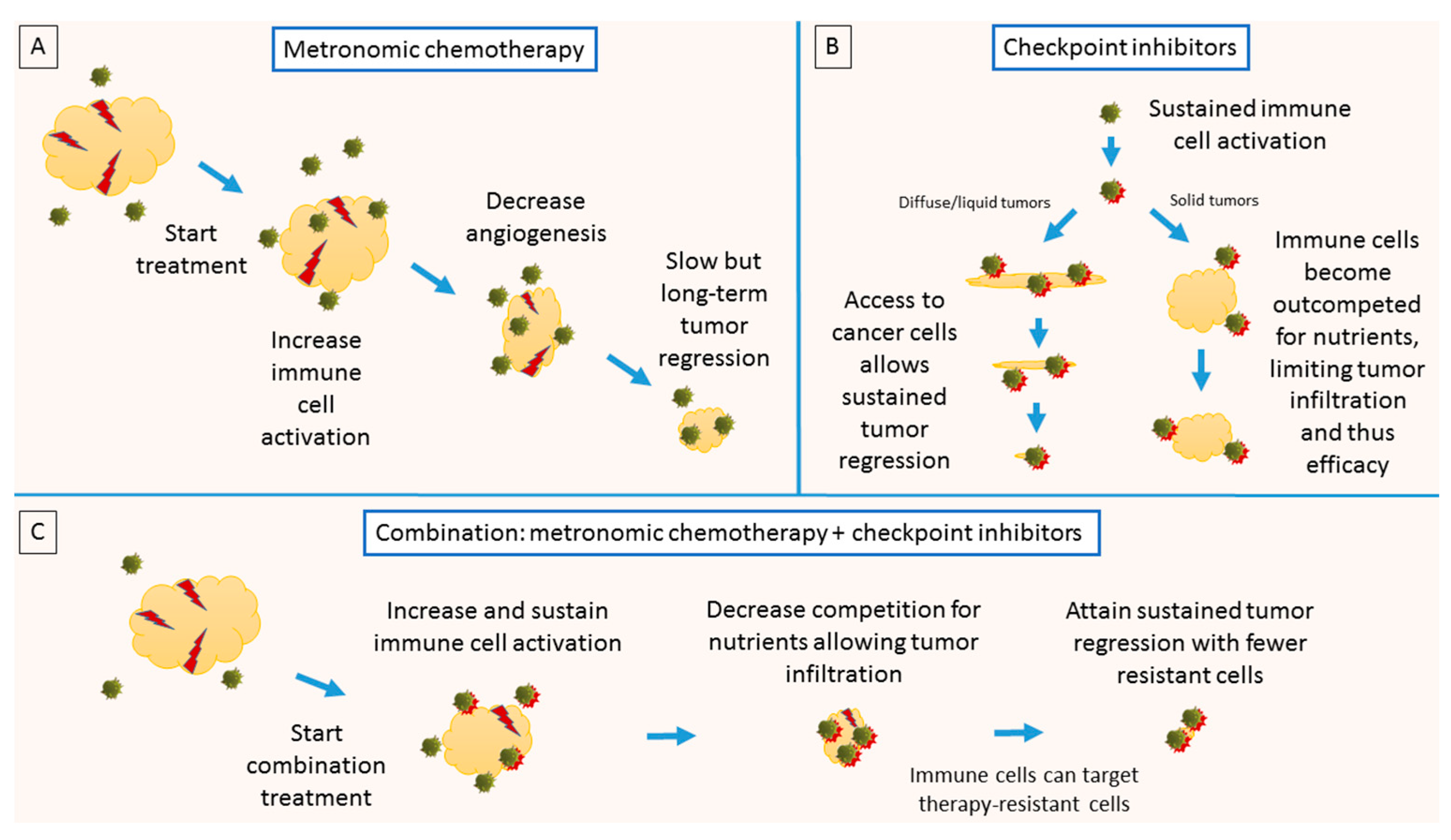
4. Combination: Metronomic Chemotherapy and Checkpoint Inhibitors
- (1)
- Both immune checkpoint inhibitors and metronomic chemotherapy increase immune cell activation. While metronomic chemotherapy can promote tumor-specific immune activation, concurrent administration of immune checkpoint inhibitors would maintain the activated state of T cells.
- (2)
- Administration of metronomic chemotherapy would allow competition for nutrients between tumor and immune cells to be reduced via gradual removal of tumor cells. This would facilitate tumor infiltration by cytotoxic immune cells, which has been associated with improved clinical outcome.
- (3)
- Experimental evidence [6] has shown that blocking PD-L1 directly on tumors dampens glycolysis, giving cytotoxic lymphocytes additional competitive advantage.
Acknowledgments
Conflicts of Interest
References
- DeVita, V.T.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S. The Emperor of All Maladies: A Biography of Cancer; Simon and Schuster: New York, NY, USA, 2010. [Google Scholar]
- Klement, G.L. Eco-evolution of cancer resistance. Sci. Transl. Med. 2016, 8, 327fs5. [Google Scholar] [CrossRef] [PubMed]
- Kareva, I. What can ecology teach us about cancer? Transl. Oncol. 2011, 4, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Kareva, I.; Hahnfeldt, P. The emerging “hallmarks” of metabolic reprogramming and immune evasion: Distinct or linked? Cancer Res. 2013, 73, 2737–2742. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.W.; et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Kareva, I. Cancer ecology: Niche construction, keystone species, ecological succession, and ergodic theory. Biol. Theory 2015, 10, 283–288. [Google Scholar] [CrossRef]
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Vahdat, L.T.; Wong, S.; Chang, J.C.; Mittal, V. Microenvironmental regulation of epithelial—Mesenchymal transitions in cancer. Cancer Res. 2012, 72, 4883–4889. [Google Scholar] [CrossRef] [PubMed]
- Stout, R.D.; Jiang, C.; Matta, B.; Tietzel, I.; Watkins, S.K.; Suttles, J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J. Immunol. 2005, 175, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Gillies, R.J. A microenvironmental model of carcinogenesis. Nat. Rev. Cancer 2008, 8, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Gillies, R.J.; Gatenby, R.A. Metabolism and its sequelae in cancer evolution and therapy. Cancer J. 2015, 21, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, V.; Thangaraju, M.; Prasad, P.D. Nutrient transporters in cancer: Relevance to Warburg hypothesis and beyond. Pharmacol. Ther. 2009, 121, 29–40. [Google Scholar] [CrossRef] [PubMed]
- MacIver, N.J.; Jacobs, S.R.; Wieman, H.L.; Wofford, J.A.; Coloff, J.L.; Rathmell, J.C. Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. J. Leukoc. Biol. 2008, 84, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.J.; Hammerman, P.S.; Thompson, C.B. Fuel feeds function: Energy metabolism and the T-cell response. Nat. Rev. Cancer 2005, 5, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Kareva, I.; Berezovskaya, F. Cancer immunoediting: A process driven by metabolic competition as a predator-prey-shared resource type model. J. Theor. Biol. 2015, 380, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Elser, J.J.; Kyle, M.M.; Smith, M.S.; Nagy, J.D. Biological stoichiometry in human cancer. PLoS ONE 2007, 2, e1028. [Google Scholar] [CrossRef] [PubMed]
- Elser, J.J.; Nagy, J.D.; Kuang, Y. Biological stoichiometry: An ecological perspective on tumor dynamics. BioScience 2003, 53, 1112–1120. [Google Scholar] [CrossRef]
- Kareva, I. Prisoner’s dilemma in cancer metabolism. PLoS ONE 2011, 6, e28576. [Google Scholar] [CrossRef] [PubMed]
- Kareva, I. Biological stoichiometry in tumor micro-environments. PLoS ONE 2013, 8, e51844. [Google Scholar] [CrossRef] [PubMed]
- Kareva, I.; Waxman, D.J.; Klement, G.L. Metronomic chemotherapy: An attractive alternative to maximum tolerated dose therapy that can activate anti-tumor immunity and minimize therapeutic resistance. Cancer Lett. 2015, 358, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Kerbel, R.S.; Kamen, B.A. The anti-angiogenic basis of metronomic chemotherapy. Nat. Rev. Cancer 2004, 4, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, E.; Kavallaris, M.; André, N. Metronomic chemotherapy: New rationale for new directions. Nat. Rev. Clin. Oncol. 2010, 7, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Kerbel, R.; Klement, G.; Pritchard, K.; Kamen, B. Continuous low-dose anti-angiogenic/metronomic chemotherapy: From the research laboratory into the oncology clinic. Ann. Oncol. 2002, 13, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, G. Metronomic scheduling: The future of chemotherapy? Lancet Oncol. 2001, 2, 733–740. [Google Scholar] [CrossRef]
- Kareva, I. Understanding Cancer from a Systems Biology Point of View; Academic Press: San Diego, CA, USA, 2018. [Google Scholar]
- Naumov, G.N.; Akslen, L.A.; Folkman, J. Role of angiogenesis in human tumor dormancy: Animal models of the angiogenic switch. Cell Cycle 2006, 5, 1779–1787. [Google Scholar] [CrossRef] [PubMed]
- Naumov, G.N.; Folkman, J.; Straume, O. Tumor dormancy due to failure of angiogenesis: Role of the microenvironment. Clin. Exp. Metastasis 2009, 26, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Italiano, J.; Shivdasani, R. Megakaryocytes and beyond: The birth of platelets. J. Thromb. Haemost. 2003, 1, 1174–1182. [Google Scholar] [CrossRef] [PubMed]
- Hellström, M.; Phng, L.-K.; Hofmann, J.J.; Wallgard, E.; Coultas, L.; Lindblom, P.; Alva, J.; Nilsson, A.K.; Karlsson, L.; Gaiano, N.; et al. DLL4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 2007, 445, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Blanco, R.; Gerhardt, H. VEGF and Notch in tip and stalk cell selection. Cold Spring Harb. Perspect. Med. 2013, 3, a006569. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, H. VEGF and endothelial guidance in angiogenic sprouting. In VEGF in Development; Springer: New York, NY, USA, 2008; pp. 68–78. [Google Scholar]
- Xie, L.; Duncan, M.B.; Pahler, J.; Sugimoto, H.; Martino, M.; Lively, J.; Mundel, T.; Soubasakos, M.; Rubin, K.; Takeda, T.; et al. Counterbalancing angiogenic regulatory factors control the rate of cancer progression and survival in a stage-specific manner. Proc. Natl. Acad Sci. USA 2011, 108, 9939–9944. [Google Scholar] [CrossRef] [PubMed]
- Pravinkumar, E.; Webster, N. HIT/HITT and alternative anticoagulation: Current concepts. Br. J. Anaesth. 2003, 90, 676–685. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.S.; Holmgren, L.; Chen, C.; Folkman, J. Angiostatin induces and sustains dormancy of human primary tumors in mice. Nat. Med. 1996, 2, 689–692. [Google Scholar] [CrossRef] [PubMed]
- Grandi, F.; Sandal, M.; Guarguaglini, G.; Capriotti, E.; Casadio, R.; Samorì, B. Hierarchical mechanochemical switches in angiostatin. Chembiochem 2006, 7, 1774–1782. [Google Scholar] [CrossRef] [PubMed]
- Vlodavsky, I.; Eldor, A.; Bar-Ner, M.; Fridman, R.; Cohen, I.R.; Klagsbrun, M. Heparan sulfate degradation in tumor cell invasion and angiogenesis. Adv. Exp. Med. Biol. 1988, 233, 201–210. [Google Scholar] [PubMed]
- Vlodavsky, I.; Miao, H.; Atzmon, R.; Levi, E.; Zimmermann, J.; Bar-Shavit, R.; Peretz, T.; Ben-Sasson, S.A. Control of cell proliferation by heparan sulfate and heparin-binding growth factors. Thromb. Haemost. 1995, 74, 534. [Google Scholar] [PubMed]
- Kareva, I.; Abou-Slaybi, A.; Dodd, O.; Dashevsky, O.; Klement, G.L. Normal wound healing and tumor angiogenesis as a game of competitive inhibition. PLoS ONE 2016, 11, e0166655. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef] [PubMed]
- Klement, G.; Baruchel, S.; Rak, J.; Man, S.; Clark, K.; Hicklin, D.J.; Bohlen, P.; Kerbel, R.S. Continuous low-dose therapy with vinblastine and VEGF receptor-2 antibody induces sustained tumor regression without overt toxicity. J. Clin. Investig. 2000, 105, R15–R24. [Google Scholar] [CrossRef] [PubMed]
- Browder, T.; Butterfield, C.E.; Kräling, B.M.; Shi, B.; Marshall, B.; O’Reilly, M.S.; Folkman, J. Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res. 2000, 60, 1878–1886. [Google Scholar] [PubMed]
- Folkins, C.; Man, S.; Xu, P.; Shaked, Y.; Hicklin, D.J.; Kerbel, R.S. Anticancer therapies combining antiangiogenic and tumor cell cytotoxic effects reduce the tumor stem-like cell fraction in glioma xenograft tumors. Cancer Res. 2007, 67, 3560–3564. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Waxman, D.J. Combination of antiangiogenesis with chemotherapy for more effective cancer treatment. Mol. Cancer Ther. 2008, 7, 3670–3684. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Waxman, D.J. Dominant effect of antiangiogenesis in combination therapy involving cyclophosphamide and axitinib. Clin. Cancer Res. 2009, 15, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Banissi, C.; Ghiringhelli, F.; Chen, L.; Carpentier, A.F. Treg depletion with a low-dose metronomic temozolomide regimen in a rat glioma model. Cancer Immunol. Immunother. 2009, 58, 1627–1634. [Google Scholar] [CrossRef] [PubMed]
- Generali, D.; Bates, G.; Berruti, A.; Brizzi, M.P.; Campo, L.; Bonardi, S.; Bersiga, A.; Allevi, G.; Milani, M.; Aguggini, S.; et al. Immunomodulation of FOXP3+ regulatory T cells by the aromatase inhibitor letrozole in breast cancer patients. Clin. Cancer Res. 2009, 15, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Menard, C.; Puig, P.E.; Ladoire, S.; Roux, S.; Martin, F.; Solary, E.; Le Cesne, A.; Zitvogel, L.; Chauffert, B. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol. Immunother. 2007, 56, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Matsushima, H.; Mizumoto, N.; Takashima, A. Classification of chemotherapeutic agents based on their differential in vitro effects on dendritic cells. Cancer Res. 2009, 69, 6978–6986. [Google Scholar] [CrossRef] [PubMed]
- Nars, M.S.; Kaneno, R. Immunomodulatory effects of low dose chemotherapy and perspectives of its combination with immunotherapy. Int. J. Cancer 2013, 132, 2471–2478. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Waxman, D.J. Metronomic cyclophosphamide eradicates large implanted GL261 gliomas by activating antitumor CD8+ T-cell responses and immune memory. Oncoimmunology 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Waxman, D.J. Metronomic cyclophosphamide schedule-dependence of innate immune cell recruitment and tumor regression in an implanted glioma model. Cancer Lett. 2014, 353, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Doloff, J.C.; Waxman, D.J. Vegf receptor inhibitors block the ability of metronomically dosed cyclophosphamide to activate innate immunity—Induced tumor regression. Cancer Res. 2012, 72, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived-suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Highfill, S.L.; Cui, Y.; Giles, A.J.; Smith, J.P.; Zhang, H.; Morse, E.; Kaplan, R.N.; Mackall, C.L. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci. Transl. Med. 2014, 6, 237ra67. [Google Scholar] [CrossRef] [PubMed]
- Tu, N.; Le Trinh, T.; Zhou, J.-M.; Gilvary, D.L.; Coppola, D.; Wei, S.; Djeu, J.Y. Chemotherapeutic sensitivity of myeloid-derived suppressor cells during cancer therapy is dictated by selective expression of clusterin. Cancer Res. 2017, 77. [Google Scholar] [CrossRef]
- Suzuki, E.; Kapoor, V.; Jassar, A.S.; Kaiser, L.R.; Albelda, S.M. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin. Cancer Res. 2005, 11, 6713–6721. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.-J.; Kim, Y.-J.; Kim, Y.-S.; Chang, W.-S.; Ko, S.-Y.; Chang, S.-Y.; Sakaguchi, S.; Kang, C.Y. A combination of chemoimmunotherapies can efficiently break self-tolerance and induce antitumor immunity in a tolerogenic murine tumor model. Cancer Res. 2007, 67, 7477–7486. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Bronte, V. Immune suppressive mechanisms in the tumor microenvironment. Curr. Opin. Immunol. 2016, 39, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.-S.; Hsu, C.-C.; Pai, V.C.; Liao, W.-Y.; Huang, S.-S.; Tan, K.-T.; Yen, C.J.; Hsu, S.C.; Chen, W.Y.; Shan, Y.S.; et al. Metronomic chemotherapy prevents therapy-induced stromal activation and induction of tumor-initiating cells. J. Exp. Med. 2016, 213, 2967–2988. [Google Scholar] [CrossRef] [PubMed]
- Relation, T.; Dominici, M.; Horwitz, E.M. Concise review: An (Im) penetrable shield: How the tumor microenvironment protects cancer stem cells. Stem Cells 2017, 35, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Farnie, G.; Sotgia, F.; Lisanti, M.P. High mitochondrial mass identifies a sub-population of stem-like cancer cells that are chemo-resistant. Oncotarget 2015, 6, 30472–30486. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Seymour, T.; Nowak, A.; Kakulas, F. Targeting aggressive cancer stem cells in glioblastoma. Front. Oncol. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Eggermont, A.M.; Maio, M.; Robert, C. Immune checkpoint inhibitors in melanoma provide the cornerstones for curative therapies. Semin. Oncol. 2015, 42, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 pathways: Similarities, differences, and implications of their inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.; Weaver, C. Janeway’s Immunobiology; Garland Science: New York, NY, USA, 2016. [Google Scholar]
- Lim, T.S.; Goh, J.K.H.; Mortellaro, A.; Lim, C.T.; Hämmerling, G.J.; Ricciardi-Castagnoli, P. CD80 and CD86 differentially regulate mechanical interactions of T-cells with antigen-presenting dendritic cells and B-cells. PLoS ONE 2012, 7, e45185. [Google Scholar] [CrossRef] [PubMed]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Fife, B.T.; Bluestone, J.A. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol. Rev. 2008, 224, 166–182. [Google Scholar] [CrossRef] [PubMed]
- Cameron, F.; Whiteside, G.; Perry, C. Ipilimumab. Drugs 2011, 71, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Fife, B.T.; Pauken, K.E.; Eagar, T.N.; Obu, T.; Wu, J.; Tang, Q.; Azuma, M.; Krummel, M.F.; Bluestone, J.A. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR—Induced stop signal. Nat. Immunol. 2009, 10, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Tagami, T.; Yamazaki, S.; Uede, T.; Shimizu, J.; Sakaguchi, N.; Mak, T.W.; Sakaguchi, S. Immunologic self-tolerance maintained by CD25+ CD4+ regulatory T cells constitutively expressing cytotoxic T lymphocyte—Associated antigen 4. J. Exp. Med. 2000, 192, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.; Hoffmann, R.; Muskens, F.; Voehringer, D. Alternatively activated macrophages inhibit T-cell proliferation by Stat6-dependent expression of PD-L2. Blood 2010, 116, 3311–3320. [Google Scholar] [CrossRef] [PubMed]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Hino, R.; Kabashima, K.; Kato, Y.; Yagi, H.; Nakamura, M.; Honjo, T.; Okazaki, T.; Tokura, Y. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer 2010, 116, 1757–1766. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kareva, I. A Combination of Immune Checkpoint Inhibition with Metronomic Chemotherapy as a Way of Targeting Therapy-Resistant Cancer Cells. Int. J. Mol. Sci. 2017, 18, 2134. https://doi.org/10.3390/ijms18102134
Kareva I. A Combination of Immune Checkpoint Inhibition with Metronomic Chemotherapy as a Way of Targeting Therapy-Resistant Cancer Cells. International Journal of Molecular Sciences. 2017; 18(10):2134. https://doi.org/10.3390/ijms18102134
Chicago/Turabian StyleKareva, Irina. 2017. "A Combination of Immune Checkpoint Inhibition with Metronomic Chemotherapy as a Way of Targeting Therapy-Resistant Cancer Cells" International Journal of Molecular Sciences 18, no. 10: 2134. https://doi.org/10.3390/ijms18102134
APA StyleKareva, I. (2017). A Combination of Immune Checkpoint Inhibition with Metronomic Chemotherapy as a Way of Targeting Therapy-Resistant Cancer Cells. International Journal of Molecular Sciences, 18(10), 2134. https://doi.org/10.3390/ijms18102134