A Mini-Review of Reactive Oxygen Species in Urological Cancer: Correlation with NADPH Oxidases, Angiogenesis, and Apoptosis


 , ,
, ,
Abstract
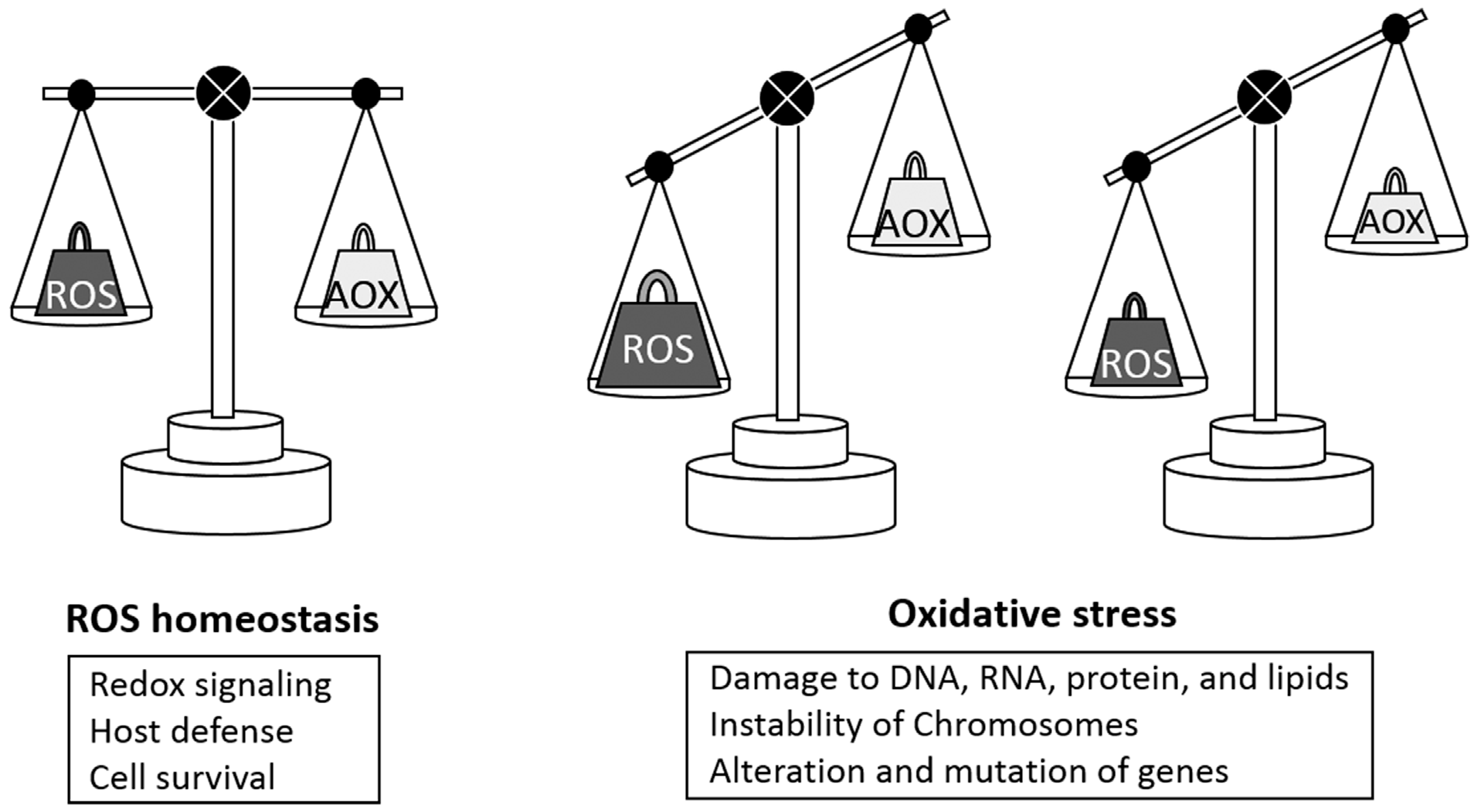
:1. Introduction
2. NOX and Urological Cancer
2.1. Pathological Significance of NOX1–5 in Malignancies
2.2. Pathological Significance of DUOX1 and 2 in Malignancies
3. Pathological Significance of the NOX Family in Urological Cancers
3.1. NOX 1–5 in Prostate Cancer
3.2. DUOX1 and 2 in Prostate Cancer
3.3. NOXs and DUOXs in RCC
3.4. NOXs and DUOXs in Urothelial Cancer
4. Angiogenesis and ROS
4.1. Angiogenesis and ROS in Prostate Cancer
4.2. Angiogenesis and ROS in RCC and Urothelial Cancer
5. Apoptosis and ROS
5.1. ROS and Apoptosis in Prostate Cancer
5.2. Apoptosis and ROS in RCC
5.3. Apoptosis and ROS in Urothelial Cancer
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sies, H. Oxidative stress: Oxidants and antioxidants. Exp. Physiol. 1997, 82, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M.; Bostick, R.M.; Dash, C.; Terry, P.; Flanders, W.D.; Mandel, J. A summary measure of pro- and anti-oxidant exposures and risk of incident, sporadic, colorectal adenomas. Cancer Causes Control 2008, 19, 1051–1064. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Andrisic, L.; Dudzik, D.; Barbas, C.; Milkovic, L.; Grune, T.; Zarkovic, N. Short overview on metabolomics approach to study pathophysiology of oxidative stress in cancer. Redox Biol. 2017, 14, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Carini, F.; Mazzola, M.; Rappa, F.; Jurjus, A.; Geagea, A.G.; Al Kattar, S.; Bou-Assi, T.; Jurjus, R.; Damiani, P.; Leone, A.; et al. Colorectal carcinogenesis: Role of oxidative stress and antioxidants. Anticancer Res. 2017, 37, 4759–4766. [Google Scholar] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- D’Autréaux, B.; Toledano, M.B. ROS as signaling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. The antioxidant paradox: Less paradoxical now? Br. J. Clin. Pharmacol. 2013, 75, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.I.; Griendling, K.K. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ. Res. 2015, 116, 531–549. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Du, M.Q.; Carmichael, P.I.; Phillips, D.H. Induction of activating mutations in the human c-Ha-ras-a proto-oncogene by oxygen free radicals. Mol. Carcinog. 1994, 11, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Ushijima, T. detection and interpretation of altered methylation patterns in cancer cells. Nat. Rev. Cancer. 2005, 5, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Toyokuni, S. Molecular mechanisms of oxidative stress-induced carcinogenesis: From epidemiology to oxygenomics. IUBMB Life 2008, 60, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Kim, S.W.; Kim, J.R. Reactive oxygen species regulate urokinase plasminogen activator expression and cell invasion via mitogen-activated protein kinase pathways after treatment with hepatocyte growth factor in stomach cancer cells. J. Exp. Clin. Cancer Res. 2009, 28, 73. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Engelhardt, J.F. Interleukin-1β induction of NFκB is partially regulated by H2O2-mediated activation of NFκB-inducing kinase. J. Biol. Chem. 2006, 281, 1495–1505. [Google Scholar] [CrossRef] [PubMed]
- Fresno Vara, J.A.; Casado, E.; de Castro, J.; Cejas, P.; Belda-Iniesta, C.; González-Barón, M. PI3K/Akt signaling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Lee, S.R.; Yang, K.S.; Ahn, Y.; Kim, Y.J.; Stadtman, E.R.; Rhee, S.G. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factor. Proc. Natl. Acad. Sci. USA 2004, 101, 16419–16424. [Google Scholar] [CrossRef] [PubMed]
- Saybasilli, H.; Yülsel, M.; Haklar, G.; Yalçin, A.S. Effect of mitochondrial electron transport chain inhibitors on superoxide radical generation in rat hippocampal and striatal slices. Antioxid. Redox Signal. 2001, 3, 1099–1104. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Segal, A.W.; Shatwell, K.P. The NAPDH oxidase of phagocytic leukemia. Ann. N. Y. Acad. Sci. 1997, 832, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Kröller-Schön, S.; Steven, S.; Kossmann, S.; Scholz, A.; Daub, S.; Oelze, M.; Xia, N.; Hausding, M.; Mikhed, Y.; Zinssius, E.; et al. Molecular mechanisms of the crosstalk between mitochondria and NADPH oxidase through reactive oxygen species-studies in white blood cells and in animal models. Antioxid. Redox Signal. 2015, 20, 247–266. [Google Scholar] [CrossRef] [PubMed]
- Korbecki, J.; Baranowska-Bosiacka, I.; Gutowska, I.; Chlubek, D. The effect of reactive oxygen species on the synthesis of prostanoids from arachidonic acid. J. Physiol. Pharmacol. 2013, 64, 409–421. [Google Scholar] [PubMed]
- Nelson, K.K.; Melendez, J.A. Mitochondrial redox control of matrix metalloproteinases. Free Radic. Biol. Med. 2004, 37, 768–784. [Google Scholar] [CrossRef] [PubMed]
- Siwik, D.A.; Colucci, W.S. Regulation of matrix metalloproteinases by cytokines and reactive oxygen/nitrogen species in the myocardium. Heart Fail. Rev. 2004, 9, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Fiaschi, T.; Cozzi, G.; Raugei, G.; Formigli, L.; Ramponi, G.; Chiarugi, P. Redox regulation of β-actin during integrin-mediated cell adhesion. J. Biol. Chem. 2006, 281, 22983–22991. [Google Scholar] [CrossRef] [PubMed]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef] [PubMed]
- Leufkens, A.M.; van Duijnhoven, F.J.; Woudt, S.H.; Siersema, P.D.; Jenab, M.; Jansen, E.H.; Pischon, T.; Tjønneland, A.; Olsen, A.; Overvad, K.; et al. Biomarkers of oxidative stress and risk of developing colorectal cancer: A cohort-nested case-control study in the European Prospective Investigation Into Cancer and Nutrition. Am. J. Epidemiol. 2012, 175, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Tochhawng, L.; Deng, S.; Pervaiz, S.; Yap, C.T. Redox regulation of cancer cell migration and invasion. Mitochondrion 2013, 13, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Gupta, S.C.; Tyagi, A.K. Reactive oxygen species (ROS) and cancer: Role of antioxidative nutraceuticals. Cancer Lett. 2017, 387, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Toyokuni, S.; Okamoto, K.; Yodoi, J.; Hiai, H. Persistent oxidative stress in cancer. FEBS Lett. 1995, 358, 1–3. [Google Scholar] [CrossRef]
- Schumacker, P.T. Reactive oxygen species in cancer: A dance with the devil. Cancer Cell. 2015, 27, 156–157. [Google Scholar] [CrossRef] [PubMed]
- Morry, J.; Ngamcherdtrakul, W.; Yantasee, W. Oxidative stress in cancer and fibrosis: Opportunity for therapeutic intervention with antioxidant compounds, enzymes, and nanoparticles. Redox Biol. 2017, 11, 240–253. [Google Scholar] [CrossRef] [PubMed]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar] [PubMed]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D.; Kawahara, T.; Diebold, B. Regulation of Nox and Duox enzymatic activity and expression. Free Radic. Biol. Med. 2007, 43, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Zhang, Y.; Dusting, G.J. NADPH oxidase-mediated redox signaling: Roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol. Rev. 2011, 63, 218–242. [Google Scholar] [CrossRef] [PubMed]
- Gào, X.; Schöttker, B. Reduction-oxidation pathways involved in cancer development: A systematic review of literature reviews. Oncotarget 2017, 8, 51888–51906. [Google Scholar] [CrossRef] [PubMed]
- Block, K.; Gorin, Y. Aiding and abetting roles of NOX oxidases in cellular transformation. Nat. Rev. Cancer 2012, 12, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.A.; Arnold, R.S.; Lassegue, B.; Shi, J.; Xu, X.; Sorescu, D.; Chung, A.B.; Griendling, K.K.; Lambeth, J.D. Cell transformation by the superoxide-generating oxidase MOX1. Nature 1999, 13, 16–22. [Google Scholar]
- Arnold, R.S.; Shi, J.; Murad, E.; Whalen, A.M.; Sun, C.Q.; Polavarapu, R.; Parthasarathy, S.; Petros, J.A.; Lambeth, J.D. Hydrogen peroxide mediates the cell growth and transformation caused by the mitogenic oxidase Nox1. Proc. Natl. Acad. Sci. USA 2001, 98, 5550–5555. [Google Scholar] [CrossRef] [PubMed]
- Böhm, B.; Heinzelmann, S.; Motz, M.; Bauer, G. Extracellular localization of catalase is associated with the transformed state of malignant cells. Biol. Chem. 2015, 396, 1339–1356. [Google Scholar] [CrossRef] [PubMed]
- Heinzelmann, S.; Bauer, G. Multiple protective functions of catalase against intercellular apoptosis inducing ROS signaling of human tumor cells. Biol. Chem. 2010, 391, 675–693. [Google Scholar] [CrossRef] [PubMed]
- Bauer, G. Increasing the endogenous NO level causes catalase inactivation and reactivation of intercellular apoptosis signaling specifically in tumor cells. Redox Biol. 2015, 6, 353–371. [Google Scholar] [CrossRef] [PubMed]
- Bánfi, B.; Muturana, A.; Jaconi, S.; Arnaudeau, S.; Laforge, T.; Sinha, B.; Ligeti, E.; Demaurex, N.; Krause, K.H. A mammalian H+ channel generated trough alternative splicing of the NADPH oxidase homolog NOH-1. Science 2000, 287, 138–142. [Google Scholar] [PubMed]
- Kobayashi, S.; Nojima, Y.; Shibuya, M.; Maru, Y. Nox1 regulates apoptosis and potentially stimulates branching morphogenesis in sinusoidal endothelial cells. Exp. Cell Res. 2004, 300, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Shinihara, M.; Shang, W.H.; Kubodera, M.; Harada, S.; Mitsushita, J.; Kato, M.; Miyazaki, H.; Sumimoto, H.; Kamata, T. Nox1 redox signaling mediates oncogenic Ras-induced disruption of stress fibers and focal adhesion by down-regulating Rho. J. Biol. Chem. 2007, 282, 17640–17648. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Nakamura, M.; Anai, S.; De Velasco, M.; Tanaka, M.; Tsujikawa, K.; Ouji, Y.; Konishi, N. A novel human AlkB homologue, ALKBH8, contributes to human bladder cancer progression. Cancer Res. 2009, 69, 3157–3164. [Google Scholar] [CrossRef] [PubMed]
- De Carvalbo, D.D.; Sadok, A.; Bourgarel-Rey, V.; Gattacceca, F.; Penel, C.; Lehmann, M.; Kovacic, H. Nox1 downstream of 12-lipoxygenase controls cell proliferation but not cell spreading of colon cancer. Int. J. Cancer 2008, 122, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Raad, H.; Serrano-Sanchez, M.; Harfouche, G.; Mahfouf, W.; Bortolotto, D.; Bergeron, V.; Kasraian, Z.; Dousset, L.; Hosseini, M.; Taieb, A.; et al. NADPH oxidase-1 plays a key role in keratinocyte responses to UV radiation and UVB-induced skin carcinogenesis. J. Investig. Dermatol. 2017, 137, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, V.; Gokulan, R.C.; Horvat, A.; Yermalitskaya, L.; Korolkova, O.; Washington, K.M.; El-Rifai, W.; Dikalov, S.I.; Zaika, A.I. Activation of NADPH oxidases leads to DNA damage in esophageal cells. Sci. Rep. 2017, 7, 9956. [Google Scholar] [CrossRef] [PubMed]
- Aydin, E.; Johansson, J.; Nazir, F.H.; Hellstrand, K.; Martner, A. Role of NOX2-derived reactive oxygen species in NK cell-mediated control of murine melanoma metastasis. Cancer Immunol. Res. 2017, 5, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Aurelius, J.; Hallner, A.; Werlenius, O.; Riise, R.; Möllgård, L.; Brune, M.; Hansson, M.; Martner, A.; Thorén, F.B.; Hellstrand, K. NOX2-dependent immunosuppression in chronic myelomonocytic leukemia. J. Leukoc. Biol. 2017, 102, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Montalvo-Javé, E.E.; Olguín-Martínez, M.; Hernández-Espinosa, D.R.; Sánchez-Sevilla, L.; Mendieta-Condado, E.; Contreras-Zentella, M.L.; Oñate-Ocaña, L.F.; Escalante-Tatersfield, T.; Echegaray-Donde, A.; Ruiz-Molina, J.M.; et al. Role of NADPH oxidases in inducing a selective increase of oxidant stress and cyclin D1 and checkpoint 1 over-expression during progression to human gastric adenocarcinoma. Eur. J. Cancer. 2016, 57, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Bánfi, B.; Malgrange, B.; Knisz, J.; Steger, K.; Steger, K.; Dubois-Dauphin, M.; Krause, K.H. NOX3, a superoxide-generating NADPH oxidase of the inner ear. J. Biol. Chem. 2004, 279, 46065–46072. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Q.; Zhao, W.; Zhou, X.; Miao, G.; Sun, C.; Zhang, H. NADPH oxidase activation contributes to heavy ion irradiation-induced cell death. Dose Response 2017, 15, 1559325817699697. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.L.; Fan, X.X.; Wong, M.P.; Jiang, Z.H.; Liu, Z.Q.; Yao, X.J.; Lu, L.L.; Zhou, Y.L.; Yau, L.F.; Tin, V.P.; et al. Targeting tyrosine kinase inhibitor-resistant non-small cell lung cancer by inducing epidermal growth factor receptor degradation via methionine 790 oxidation. Antioxid. Redox Signal. 2016, 24, 263–279. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Cao, Z.; Xu, X.; van Meir, E.G.; Lambeth, J.D. Homologs of gp91phox: Cloning and tissue expression of Nox3, Nox4, and Nox5. Gene 2001, 269, 131–140. [Google Scholar] [CrossRef]
- Lee, J.K.; Edderkaoui, M.; Truong, P.; Ohno, I.; Jang, K.T.; Berti, A.; Pandol, S.J.; Gukovskaya, A.S. NADPH oxidase promotes pancreatic cancer cell survival via inhibiting JAK2 dephosphorylation by tyrosine phosphatases. Gastroenterology 2007, 133, 1637–1648. [Google Scholar] [CrossRef] [PubMed]
- Maloney, E.; Sweet, I.R.; Hockenbery, D.M.; Pharm, M.; Rizzo, N.O.; Tateya, S.; Handa, P.; Schwartz, M.W.; Kim, F. Activation of NF-κB by palmitate in endothelial cells, a key role for NADPH oxidase-derived superoxide in response to TLR4. Activation 2009, 29, 1370–1375. [Google Scholar]
- Liu, R.-M.; Choi, J.; Wu, J.-H.; Gaston Pravia, K.A.; Lewis, K.M.; Brand, J.D.; Mochel, N.S.; Krzywanski, D.M.; Lambeth, J.D.; Hagood, J.S.; et al. Oxidative modification of nuclear mitogen-activated protein kinase phosphatase 1 is involved in transforming growth factor β1-induced expression of plasminogen activator inhibitor 1 in fibroblasts. J. Biol. Chem. 2010, 285, 16239–16247. [Google Scholar] [CrossRef] [PubMed]
- Brar, S.S.; Kennedy, T.P.; Whorton, A.R.; Sturrock, A.B.; Huecksteadt, T.P.; Ghio, A.J.; Hoidal, J.R. Reactive oxygen species from NAD(P)H: Quinone oxidoreductase constitutively activate NF-κB in malignant melanoma cells. Am. J. Physiol. 2001, 280, C659–C676. [Google Scholar]
- Vaquero, E.C.; Edderkaoui, M.; Pandol, S.; Gukovsky, I.; Gukovskaya, A.S. Reactive oxygen species produced by NAD(P)H oxidase inhibit apoptosis in pancreatic cancer cells. J. Biol. Chem. 2004, 279, 34643–34654. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Meng, Q.; Liu, L.Z.; Rojanasakul, Y.; Wang, X.R.; Jiang, B.H. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res. 2007, 67, 10823–10830. [Google Scholar] [CrossRef] [PubMed]
- Datla, S.R.; Peshavariya, H.; Dusting, G.J.; Mahadev, K.; Goldstein, B.J.; Jiang, F. Important role of Nox4 type NADPH oxidase in angiogenic responses in human microvascular endothelial cells in vitro. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2319–2324. [Google Scholar] [CrossRef] [PubMed]
- Vallet, P.; Charnay, Y.; Steger, K.; Ogier-Denis, E.; Kovari, E.; Herrmann, F.; Michel, J.P.; Szanto, I. Neuronal expression of the NADPH oxidase NOX4, and its regulation in mouse experimental brain ischemia. Neuroscience 2005, 132, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Juhasz, A.; Ge, Y.; Markel, S.; Chiu, A.; Matsumoto, L.; Van, B.J.; Roy, K.; Doroshow, J.H. Expression of NADPH oxidase homologues and accessory genes in human cancer cell lines, tumours and adjacent normal tissues. Free Radic. Res. 2009, 43, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Si, J.; Fu, X.; Behar, J.; Wands, J.; Beer, D.G.; Souza, R.F.; Spechler, S.J.; Lambeth, D.; Cao, W. NADPH oxidase NOX5-S mediates acid-induced cyclooxygenase-2 expression via activation of NF-κB in Barrett’s esophageal adenocarcinoma cells. J. Biol. Chem. 2007, 282, 16244–16255. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M.; Nakamura, Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008, 266, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Luxen, S.; Belinsky, S.A.; Knaus, U.G. Silencing of DUOX NADPH oxidases by promoter hypermethylation in lung cancer. Cancer Res. 2008, 68, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Ling, Q.; Yu, K.; Huang, C.; Li, N.; Zheng, J.; Bao, S.; Cheng, Q.; Zhu, M.; Chen, M. Dual oxidase 1: A predictive tool for the prognosis of hepatocellular carcinoma patients. Oncol. Rep. 2016, 35, 3198–3208. [Google Scholar] [CrossRef] [PubMed]
- Little, A.C.; Sulovari, A.; Danyal, K.; Heppner, D.E.; Seward, D.J.; van der Vliet, A. Paradoxical roles of dual oxidases in cancer biology. Free Radic. Biol. Med. 2017, 110, 117–132. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.S.; Yu, K.K.; Ling, Q.X.; Huang, C.; Li, N.; Zheng, J.M.; Bao, S.X.; Cheng, Q.; Zhu, M.Q.; Chen, M.Q. The combination of three molecular markers can be a valuable predictive tool for the prognosis of hepatocellular carcinoma patients. Sci. Rep. 2016, 6, 24582. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Antony, S.; Hewitt, S.M.; Jiang, G.; Yang, S.X.; Meitzler, J.L.; Juhasz, A.; Lu, J.; Liu, H.; Doroshow, J.H.; Roy, K. Functional activity and tumor-specific expression of dual oxidase 2 in pancreatic cancer cells and human malignancies characterized with a novel monoclonal antibody. Int. J. Oncol. 2013, 42, 1229–1238. [Google Scholar] [CrossRef] [PubMed]
- Qi, R.; Zhou, Y.; Li, X.; Guo, H.; Gao, L.; Wu, L.; Wang, Y.; Gao, Q. DUOX2 expression is increased in Barrett esophagus and cancerous tissues of stomach and colon. Gastroenterol. Res. Pract. 2016, 2016, 1835684. [Google Scholar] [CrossRef] [PubMed]
- Brar, S.S.; Corbib, Z.; Kennedy, T.P.; Hemendinger, R.; Thornton, L.; Bommarius, B.; Arnold, R.S.; Whorton, A.R.; Sturrock, A.B.; Huecksteadt, T.P.; et al. NOX5 NAD(P)H oxidase regulates growth and apoptosis in DU145 prostate cancer cells. Am. J. Physiol. Cell Physiol. 2003, 285, C353–C369. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Koul, S.; Khandrika, L.; Meacham, R.B.; Koul, H.K. Oxidative stress in inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res. 2008, 68, 1777–1785. [Google Scholar] [CrossRef] [PubMed]
- Höll, M.; Koziel, R.; Schäfer, G.; Pircher, H.; Pauck, A.; Hermann, M.; Klocker, H.; Jansen-Dürr, P.; Sampson, N. ROS signaling by NADPH oxidase 5 modulates the proliferation and survival of prostate carcinoma cells. Mol. Carcinog. 2016, 55, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Arbiser, J.L.; Petros, J.; Klafter, R.; Govindajaran, B.; McLaughlin, E.R.; Brown, L.F.; Cohen, C.; Moses, M.; Kilroy, S.; Arnold, R.S.; et al. Reactive oxygen generated by Nox1 triggers the angiogenic switch. Proc. Natl. Acad. Sci. USA 2001, 99, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.D.; Sun, C.Q.; Lambeth, J.D.; Marshall, F.; Amin, M.; Chung, L.; Petros, J.A.; Arnold, R.S. Increased Nox1 and hydrogen peroxide in prostate cancer. Prostate 2005, 62, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Deep, G.; Kumar, R.; Jain, A.K.; Dhar, D.; Panigrahi, G.K.; Hussain, A.; Agarwal, C.; El-Elimat, T.; Sica, V.P.; Oberlies, N.H.; et al. Graviola inhibits hypoxia-induced NADPH oxidase activity in prostate cancer cells reducing their proliferation and clonogenicity. Sci. Rep. 2016, 6, 23135. [Google Scholar] [CrossRef] [PubMed]
- Tamura, R.E.; Hunger, A.; Fernandes, D.C.; Laurindo, F.R.; Costanzi-Strauss, E.; Strauss, B.E. Induction of oxidants distinguishes susceptibility of prostate carcinoma cell lines to p53 gene transfer mediated by an improved adenoviral vector. Hum. Gene Ther. 2017, 28, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Arnold, R.S.; He, J.; Remo, A.; Ritsick, D.; Yin-Goen, Q.; Lambeth, J.D.; Datta, M.W.; Young, A.N.; Petros, J.A. Nox1 expression determines cellular reactive oxygen and modulates c-fos-induced growth factor, interleukin-8 and cav-1. Am. J. Pahol. 2007, 171, 2021–2032. [Google Scholar] [CrossRef] [PubMed]
- Pettigrew, C.A.; Clerkin, J.S.; Cotter, T.G. DUOX enzyme activity promotes AKT signalling in prostate cancer cells. Anticancer Res. 2012, 32, 5175–5181. [Google Scholar] [PubMed]
- Tam, N.; Gao, Y.; Leung, Y.; Ho, S.M. Androgenic regulation of oxidative stress in the rat prostate: Involvement of NAD(P)H oxidase and antioxidant defense machinery during prostatic involution and regrowth. Am. J. Pathol. 2003, 163, 2513–2522. [Google Scholar] [CrossRef]
- Lu, J.P.; Monardo, L.; Bryskin, I.; Hou, Z.F.; Trachtenberg, J.; Wilson, B.C.; Pinthus, J.H. Androgens induce oxidative stress and radiation resistance in prostate cancer cells though NADPH oxidase. Prostate Cancer Prostatic Dis. 2010, 13, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.P.; Hou, Z.F.; Duivenvoorden, W.C.; Whelan, K.; Honig, A.; Pinthus, J.H. Adiponectin inhibits oxidative stress in human prostate carcinoma cells. Prostate Cancer Prostatic Dis. 2012, 15, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Antony, S.; Wu, Y.; Hewitt, S.M.; Anver, M.R.; Butcher, D.; Jiang, G.; Meitzler, J.L.; Liu, H.; Juhasz, A.; Lu, J.; et al. Characterization of NADPH oxidase 5 expression in human tumors and tumor cell lines with a novel mouse monoclonal antibody. Free Radic. Biol. Med. 2013, 65, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Li, X.; Liu, J.; Lin, J.; Chung, L.W. Activation of androgen receptor, lipogenesis, and oxidative stress converged by SREBP-1 is responsible for regulating growth and progression of prostate cancer cells. Mol. Cancer Res. 2012, 10, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Block, K.; Gorin, Y.; Hoover, P.; Williams, P.; Chelmicki, T.; Clark, R.A.; Yoneda, T.; Abboud, H.E. NAD(P)H oxidases regulate HIF-2α protein expression. J. Biol. Chem. 2007, 282, 8019–8026. [Google Scholar] [CrossRef] [PubMed]
- Gregg, J.L.; Turner, R.M., 2nd; Chang, G.; Joshi, D.; Zhan, Y.; Chen, L.; Maranchie, J.K. NADPH oxidase NOX4 supports renal tumorigenesis by promoting the expression and nuclear accumulation of HIF2α. Cancer Res. 2014, 74, 3501–3511. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.; Chen, L.; Lin, H.M.; Lin, Y.; Maranchie, J.K. Nox4 inhibition enhances the cytotoxicity of cisplatin in human renal cancer cells. J. Exp. Ther. Oncol. 2012, 10, 9–18. [Google Scholar] [PubMed]
- Shanmugasundaram, K.; Block, K. Renal carcinogenesis, tumor heterogeneity, and reactive oxygen species: Tactics evolved. Antioxid. Redox Signal. 2016, 25, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Tsujikawa, K.; Koike, K.; Kitae, K.; Shinkawa, A.; Arima, H.; Suzuki, T.; Tsuchiya, M.; Makino, Y.; Furukawa, T.; Konishi, N.; et al. Expression and sub-cellular localization of human ABH family molecules. J. Cell. Mol. Med. 2007, 11, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Rathore, K.; Wang, H.C. Differential induction of reactive oxygen species through Erk1/1 and Nox-1 by FK228 for selective apoptosis of oncogenic H-Ras-expressing human urinary bladder cancer J82 cells. J. Cancer Res. Clin. Oncol. 2011, 137, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Fujii, T.; Tsujikawa, K.; Anai, S.; Fujimoto, K.; Konishi, N. ALKBH3 contributes to survival and angiogenesis of human urothelial carcinoma cells through NADPH oxidase and Tweak/Fn14/VEGF signals. Clin. Cancer Res. 2012, 18, 5247–5255. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Fujii, T.; Anai, S. ROS generation via NOX4 and its utility in the cytological diagnosis of urothelial carcinoma of the urinary bladder. BMC Cancer 2011, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Seo, J.M.; Kim, C.; Lee, J.E.; Lee, K.M.; Kim, J.H. BLT2 promotes the invasion and metastasis of aggressive bladder cancer cells through a reactive oxygen species-linked pathway. Free Radic. Biol. Med. 2010, 49, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, T.; Miyata, Y.; Asai, A.; Sagara, Y.; Furusato, B.; Fukuoka, J.; Sakai, H. Green tea polyphenol induces changes in cancer-related factors in an animal model of bladder cancer. PLoS ONE 2017, 12, e0171091. [Google Scholar] [CrossRef] [PubMed]
- Miyata, Y.; Mitsunari, K.; Asai, A.; Takehara, K.; Mochizuki, Y.; Sakai, H. Pathological significance and prognostic role of microvessel density, evaluated using CD31, CD34, and CD105 in prostate cancer patients after radical prostatectomy with neoadjuvant therapy. Prostate 2015, 75, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Abid, M.R.; Spokes, S.C.; Shih, W.C.; Aird, W.C. NAPDH oxidase activity selectively modulates vascular endothelial growth factor signaling pathway. J. Biol. Chem. 2007, 282, 35373–35385. [Google Scholar] [CrossRef] [PubMed]
- Kilmova, T.; Chandel, N.S. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 2008, 15, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Liu, L.Z.; Jiang, Y.; Zhu, Y.; Guo, N.L.; Barnett, J.; Rojanasakul, Y.; Agani, F.; Jiang, B.H. Cadmium increases HIF-1 and VEGF expression through ROS, ERK, and Akt signaling pathways and induces malignant transformation of human bronchial epithelial cells. Toxicol. Sci. 2012, 125, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Ding, M.; Zheng, J.Z.; Zhang, Z.; Leonard, S.S.; Liu, K.J.; Shi, X.; Jiang, B.H. Vanadate-induced expression of hypoxia-inducible factor 1 alpha and vascular endothelial growth factor through phosphatidylinositol 3-kinase/Akt pathway and reactive oxygen species. J. Biol. Chem. 2002, 277, 31963–31971. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, S.; Desrosiers, R.R.; Béliveau, R. HIF-1α mRNA and protein upregulation involves Rho GTPase expression during hypoxia in renal cell carcinoma. J. Cell Sci. 2003, 116, 2247–2260. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.H.; Dier, U.; Melendez, J.A.; Hempel, N. Regulation of MMP-1 expression in response to hypoxia is dependent on the intracellular redox status of metastatic bladder cancer cells. Biochim. Biophys. Acta 2015, 1852, 2593–2602. [Google Scholar] [CrossRef] [PubMed]
- Coso, S.; Harrison, I.; Harrison, C.B.; Vinh, A.; Sobey, C.G.; Drummond, G.R.; Williams, E.D.; Selemidis, S. NADPH oxidases as regulators of tumor angiogenesis: Current and emerging concepts. Antioxid. Redox Signal. 2012, 16, 1229–1247. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Bermejo, R.; Hernández-Hernández, A. The Importance of NADPH Oxidases and Redox Signaling in Angiogenesis. Antioxidants (Basel) 2017, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Shen, L.; Zhang, Z.; Leonard, S.S.; He, H.; Zhang, X.G.; Shi, X.; Jiang, B.H. Arsenite induces HIF-1α and VEGF through PI3K, Akt and reactive oxygen species in DU145 human prostate carcinoma cells. Mol. Cell. Biochem. 2004, 255, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Kou, X.; Fan, J.; Chen, N. Potential molecular targets of ampelopsin in prevention and treatment of cancers. Anticancer Agents Med. Chem. 2017. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, C.; Zhang, Z.; Yang, S.; Zhang, B.; Yin, L.; Swarts, S.; Vidyasagar, S.; Zhang, L.; Okunieff, P. A new flavonoid regulates angiogenesis and reactive oxygen species production. Adv. Exp. Med. Biol. 2014, 812, 149–155. [Google Scholar] [PubMed]
- Delle, M.S.; Sanità, P.; Calgani, A.; Schenone, S.; Botta, L.; Angelucci, A. Src inhibition potentiates antitumoral effect of paclitaxel by blocking tumor-induced angiogenesis. Exp. Cell Res. 2014, 328, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.T.; Chen, M.F.; Chen, W.C.; Hsieh, C.C. The role of IL-6 in the radiation response of prostate cancer. Radiat. Oncol. 2013, 8, 159. [Google Scholar] [CrossRef] [PubMed]
- Alcayaga-Miranda, F.; González, P.; Lopez-Verrilli, A.; Varas-Godoy, M.; Aguila-Diaz, C.; Contreras, L.; Khoury, M. Prostate tumor-induced angiogenesis is blocked by exosomes derived from menstrual stem cells through the inhibition of reactive oxygen species. Oncotarget 2016, 7, 44462–44477. [Google Scholar] [CrossRef] [PubMed]
- Golovine, K.; Makhov, P.; Naito, S.; Raiyani, H.; Tomaszewski, J.; Mehrazin, R.; Tulin, A.; Kutikov, A.; Uzzo, R.G.; Kolenko, V.M. Piperlongumine and its analogs down-regulate expression of c-Met in renal cell carcinoma. Cancer Biol. Ther. 2015, 16, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, M.; Xu, Y.F.; Feng, Y.; Che, J.P.; Wang, G.C.; Zheng, J.H. Combination of quercetin and hyperoside has anticancer effects on renal cancer cells through inhibition of oncogenic microRNA-27a. Oncol. Rep. 2014, 31, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Miyajima, A.; Kosaka, T.; Shirotake, S.; Hasegawa, M.; Kikuchi, E.; Oya, M. Cis-dichlorodiammineplatinum upregulates angiotensin II type 1 receptors through reactive oxygen species generation and enhances VEGF production in bladder cancer. Mol. Cancer Ther. 2010, 9, 2982–2992. [Google Scholar] [CrossRef] [PubMed]
- Shirotake, S.; Miyajima, A.; Kosaka, T.; Tanaka, N.; Maeda, T.; Kikuchi, E.; Oya, M. Angiotensin II type 1 receptor expression and microvessel density in human bladder cancer. Urology 2011, 77, e19–e25. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Urta, E.; Kimura, T.; Yamamoto, Y.; Osaki, T. Reactive oxygen species (ROS) control the expression of Bcl-2 family proteins by regulating their phosphorylation and ubiquitination. Cancer Sci. 2004, 95, 644–650. [Google Scholar] [CrossRef] [PubMed]
- Scheit, K.; Bauer, G. Direct and indirect inactivation of tumor cell protective catalase by salicylic acid and anthocyanidins reactivates intercellular ROS signaling and allows for synergistic effects. Carcinogenesis 2015, 36, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Bauer, G. Central signaling elements of intercellular reactive oxygen/nitrogen species-dependent induction of apoptosis in malignant cells. Anticancer Res. 2017, 37, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Luanpitpong, S.; Chanvorachote, P.; Stehlik, C.; Tse, W.; Callery, P.S.; Wang, L.; Rojanasakul, Y. Regulation of apoptosis by Bcl-2 cysteine oxidation in human lung epithelial cells. Mol. Biol. Cell 2013, 24, 858–869. [Google Scholar] [CrossRef] [PubMed]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Hu, X.; Liu, Y.; Dong, S.; Wen, Z.; He, W.; Zhang, S.; Huang, Q.; Shi, M. ROS signaling under metabolic stress: Cross-talk between AMPK and AKT pathway. Mol. Cancer 2017, 16, 79. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Ren, K.; Dong, H.; Song, F.; Chen, J.; Guo, Y.; Liu, Y.; Tao, W.; Zhang, Y. Flavonoids from persimmon (Diospyros kaki L.) leaves inhibit proliferation and induce apoptosis in PC-3 cells by activation of oxidative stress and mitochondrial apoptosis. Chem. Biol. Interact. 2017, 275, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Elkady, A.I. Anethole inhibits the proliferation of human prostate cancer cells via induction of cell cycle arrest and apoptosis. Anticancer Agents Med. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Li, Y.; Yang, F.; Zeng, A.; Yang, S.; Luo, Y.; Zhang, Y.; Xie, Y.; Ye, T.; Xia, Y.; et al. The extract from Punica granatum (pomegranate) peel induces apoptosis and impairs metastasis in prostate cancer cells. Biomed. Pharmacother. 2017, 93, 976–984. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Sahabjada; Akhtar, J.; Hussain, A.; Badaruddeen; Arshad, M.; Mishra, A. Development of a new rutin nanoemulsion and its application on prostate carcinoma PC3 cell line. EXCLI J. 2017, 16, 810–823. [Google Scholar] [PubMed]
- Lin, J.F.; Tsai, T.F.; Yang, S.C.; Lin, Y.C.; Chen, H.E.; Chou, K.Y.; Hwang, T.I. Benzyl isothiocyanate induces reactive oxygen species-initiated autophagy and apoptosis in human prostate cancer cells. Oncotarget 2017, 8, 20220–20234. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Garcia, A.; Hevia, D.; Mayo, J.C.; Gonzalez-Menendez, P.; Coppo, L.; Lu, J.; Holmgren, A.; Sainz, R.M. Thioredoxin 1 modulates apoptosis induced by bioactive compounds in prostate cancer cells. Redox Biol. 2017, 12, 634–647. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.B.; Tian, F.J.; Liu, L.Q. Chikusetsu (CHI) triggers mitochondria-regulated apoptosis in human prostate cancer via reactive oxygen species (ROS) production. Biomed. Pharmacother. 2017, 90, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.; Lim, W.; Bazer, F.W.; Song, G. Chrysin induces death of prostate cancer cells by inducing ROS and ER stress. J. Cell. Physiol. 2017, 232, 3786–3797. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.Y.; Kim, S.H.; Yu, S.N.; Park, S.G.; Kim, Y.W.; Nam, H.W.; An, H.H.; Yu, H.S.; Kim, Y.W.; Ji, J.H.; et al. Lasalocid induces cytotoxic apoptosis and cytoprotective autophagy through reactive oxygen species in human prostate cancer PC-3 cells. Biomed. Pharmacother. 2017, 8, 1016–1024. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.; Park, S.; Bazer, F.W.; Song, G. Naringenin-induced apoptotic cell death in prostate cancer cells Is mediated via the PI3K/AKT and MAPK signaling pathways. J. Cell. Biochem. 2017, 118, 1118–1131. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.; Jeong, M.; Bazer, F.W.; Song, G. Coumestrol inhibits proliferation and migration of prostate cancer cells by regulating AKT, ERK1/2, and JNK MAPK cell signaling cascades. J. Cell. Physiol. 2017, 232, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhou, B.; Zhong, P.; Rajamanickam, V.; Dai, X.; Karvannan, K.; Zhou, H.; Zhang, X.; Liang, G. Increased intracellular reactive oxygen species mediates the anti-cancer effects of WZ35 via activating mitochondrial apoptosis pathway in prostate cancer cells. Prostate 2017, 77, 489–504. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.Y.; Park, K.I.; Kim, S.H.; Yu, S.N.; Park, S.G.; Kim, Y.W.; Seo, Y.K.; Ma, J.Y.; Ahn, S.C. Inhibition of autophagy promotes salinomycin-induced apoptosis via reactive oxygen species-mediated PI3K/AKT/mTOR and ERK/p38 MAPK-dependent signaling in human prostate cancer. Int. J. Mol. Sci. 2017, 18, 1088. [Google Scholar] [CrossRef] [PubMed]
- Park, S.G.; Kim, S.H.; Kim, K.Y.; Yu, S.N.; Choi, H.D.; Kim, Y.W.; Nam, H.W.; Seo, Y.K.; Ahn, S.C. Toyocamycin induces apoptosis via the crosstalk between reactive oxygen species and p38/ERK MAPKs signaling pathway in human prostate cancer PC-3 cells. Pharmacol. Rep. 2017, 69, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.; Chen, L.; Tan, G.; Ke, L.; Zhang, S.; Chen, H.; Liu, J. JS-K promotes apoptosis by inducing ROS production in human prostate cancer cells. Oncol. Lett. 2017, 13, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Thamilselvan, V.; Menon, M.; Stein, G.S.; Valeriote, F.; Thamilselvan, S. Combination of carmustine and selenite inhibits EGFR mediated growth signaling in androgen-independent prostate cancer cells. J. Cell. Biochem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Thamilselvan, V.; Menon, M.; Thamilselvan, S. Combination of carmustine and selenite effectively inhibits tumor growth by targeting androgen receptor, androgen receptor-variants, and Akt in preclinical models: New hope for patients with castration resistant prostate cancer. Int. J. Cancer 2016, 139, 1632–1647. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.; Iyer, A.K.V.; Kaushik, V.; Azad, N. Anti-tumorigenic potential of a novel orlistat-AICAR combination in prostate cancer cells. J. Cell. Biochem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Wang, H.; Chen, F.; Fang, J.; Xu, A.; Xi, W.; Zhang, S.; Wu, G.; Wang, Z. Galangin inhibits cell invasion by suppressing the epithelial-mesenchymal transition and inducing apoptosis in renal cell carcinoma. Mol. Med. Rep. 2016, 13, 4238–4244. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.F.; Wang, X.H.; Pan, B.; Li, F.; Kuang, L.; Su, Z.X. Eupatilin induces human renal cancer cell apoptosis via ROS-mediated MAPK and PI3K/AKT signaling pathways. Oncol. Lett. 2016, 12, 2894–2899. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Park, B.; Chae, I.G.; Kim, D.H.; Kundu, J.; Kundu, J.K.; Chun, K.S. Carnosic acid induces apoptosis through inactivation of Src/STAT3 signaling pathway in human renal carcinoma Caki cells. Oncol. Rep. 2016, 35, 2723–2732. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zheng, W.; Rong, L.; Xing, Y.; Hu, D. Bicyclol exerts an anti-tumor effect via ROS-mediated endoplasmic reticulum stress in human renal cell carcinoma cells. Biomed. Pharmacother. 2017, 91, 1184–1192. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; He, Q.; Gong, Z.; Chen, S.; Cui, L. Niclosamide suppresses renal cell carcinoma by inhibiting Wnt/β-catenin and inducing mitochondrial dysfunctions. Springerplus 2016, 5, 1436. [Google Scholar] [CrossRef] [PubMed]
- Yin, P.; Jia, J.; Li, J.; Song, Y.; Zhang, Y.; Chen, F. ABT-737, a Bcl-2 selective inhibitor, and chloroquine synergistically kill renal cancer cells. Oncol. Res. 2016, 24, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Gillissen, B.; Richter, A.; Richter, A.; Preissner, R.; Schulze-Osthoff, K.; Essmann, F.; Daniel, P.T. Bax/Bak-independent mitochondrial depolarization and reactive oxygen species induction by sorafenib overcome resistance to apoptosis in renal cell carcinoma. J. Biol. Chem. 2017, 292, 6478–6492. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Azad, N.; Kongkaneramit, L.; Chen, F.; Lu, Y.; Jiang, B.H.; Rojanasakul, Y. The Fas death signaling pathway connecting reactive oxygen species generation and FLICE inhibitory protein down-regulation. J. Immunol. 2008, 180, 3072–3080. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Noh, H.J.; Sung, E.G.; Song, I.H.; Kim, J.Y.; Kwon, T.K.; Lee, T.J. Berberine sensitizes TRAIL-induced apoptosis through proteasome-mediated downregulation of c-FLIP and Mcl-1 proteins. Int. J. Oncol. 2011, 38, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Han, M.A.; Woo, S.M.; Min, K.J.; Kim, S.; Park, J.W.; Kim, D.E.; Kim, S.H.; Choi, Y.H.; Kwon, T.K. 6-Shogaol enhances renal carcinoma Caki cells to TRAIL-induced apoptosis through reactive oxygen species-mediated cytochrome c release and down-regulation of c-FLIP(L) expression. Chem. Biol. Interact. 2015, 228, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Chauhan, A.K.; Min, K.J.; Park, D.C.; Kwon, T.K. Thymoquinone induces apoptosis through downregulation of c-FLIP and Bcl-2 in renal carcinoma Caki cells. Oncol. Rep. 2016, 36, 2261–2267. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.C.; Shen, C.H.; Chang, C.B.; Hsieh, H.Y.; Wu, J.D.; Tseng, L.H.; Hwang, D.W.; Chen, S.Y.; Wu, S.F.; Chan, M.W.; et al. Guizhi Fuling Wan as a novel agent for intravesical treatment for bladder cancer in mouse model. Mol. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Romanov, V.; Whyard, T.C.; Waltzer, W.C.; Grollman, A.P.; Rosenquist, T. Aristolochic acid-induced apoptosis and G2 cell cycle arrest depends on ROS generation and MAP kinases activation. Arch. Toxicol. 2015, 89, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wen, J.M.; Du, C.J.; Hu, S.M.; Chen, J.X.; Zhang, S.G.; Zhang, N.; Gao, F.; Li, S.J.; Mao, X.W.; et al. Thymol inhibits bladder cancer cell proliferation via inducing cell cycle arrest and apoptosis. Biochem. Biophys. Res. Commun. 2017. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.; Chen, L.; Tan, G.; Ke, L.; Zhang, S.; Chen, H.; Liu, J. A reactive oxygen species activation mechanism contributes to JS-K-induced apoptosis in human bladder cancer cells. Sci. Rep. 2015, 5, 15104. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.Q.; Dou, Z.L.; Jia, Z.H. 5-bromo-3-(3-hydroxyprop-1-ynyl)-2H-pyran-2-one induces apoptosis in T24 human bladder cancer cells through mitochondria-dependent signaling pathways. Mol. Med. Rep. 2017, 15, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Saleem, A.; Dvorzhinski, D.; Santanam, U.; Mathew, R.; Bray, K.; Stein, M.; White, E.; DiPaola, R.S. Effect of dual inhibition of apoptosis and autophagy in prostate cancer. Prostate 2012, 72, 1374–1381. [Google Scholar] [CrossRef] [PubMed]
- Hsin, I.L.; Wang, S.C.; Li, J.R.; Ciou, T.C.; Wu, C.H.; Wu, H.M.; Ko, J.L. Immunomodulatory proteins FIP-gts and chloroquine induce caspase-independent cell death via autophagy for resensitizing cisplatin-resistant urothelial cancer cells. Phytomedicine 2016, 23, 1566–1573. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Characteristics as Malignant Cells | Types | NOX | DUOX | References | |||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 1 | 2 | |||
| Overexpression in cancer cells | PCa | Y/N | Y/N | N | Y/N | Y/N | N | N | [68,77,78,79,80,81,82,84,85,89] |
| RCC | N | N | N | Y/N | N | N | N | [68,91] | |
| UC | – | Y | – | Y | – | – | – | [97,98] | |
| Tumorigenesis | PCa | Y | – | – | – | – | – | – | [80,81] |
| RCC | Y | – | – | Y | – | – | – | [91,92] | |
| UC | – | – | – | – | – | – | – | – | |
| Cell death/Apoptosis | PCa | Y | – | – | – | Y | Y | Y | [77,79,83,85] |
| RCC | – | – | – | Y | – | – | – | [93] | |
| UC | Y | – | – | N | – | – | – | [49,96] | |
| Angiogenesis/Angiogenesis-related factors | PCa | Y | – | – | Y | – | – | – | [65,80] |
| RCC | – | – | – | – | – | – | – | – | |
| UC | – | N | – | – | – | – | – | [97] | |
| Gleason score/Grade | PCa | N | – | – | – | – | – | – | [84] |
| RCC | – | – | – | – | – | – | – | – | |
| UC | Y | N | – | N | – | – | – | [49,97,98] | |
| T stage/Tumor growth/Invasion | PCa | Y/N | Y | – | Y | Y | – | – | [65,77,78,79,80,82,84] |
| RCC | – | – | – | Y | – | – | – | [92] | |
| UC | Y | Y/N | – | Y/N | – | – | – | [49,97,98,99] | |
| N stage/M stage/Metastasis | PCa | Y/N | – | – | – | – | – | – | [81,84] |
| RCC | – | – | – | – | – | – | – | – | |
| UC | Y | – | – | Y | – | – | – | [99] | |
| Outcome/Survival | PCa | N | – | – | – | – | – | – | [84] |
| RCC | – | – | – | – | – | – | – | – | |
| UC | – | – | – | – | – | – | – | ||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miyata, Y.; Matsuo, T.; Sagara, Y.; Ohba, K.; Ohyama, K.; Sakai, H. A Mini-Review of Reactive Oxygen Species in Urological Cancer: Correlation with NADPH Oxidases, Angiogenesis, and Apoptosis. Int. J. Mol. Sci. 2017, 18, 2214. https://doi.org/10.3390/ijms18102214
Miyata Y, Matsuo T, Sagara Y, Ohba K, Ohyama K, Sakai H. A Mini-Review of Reactive Oxygen Species in Urological Cancer: Correlation with NADPH Oxidases, Angiogenesis, and Apoptosis. International Journal of Molecular Sciences. 2017; 18(10):2214. https://doi.org/10.3390/ijms18102214
Chicago/Turabian StyleMiyata, Yasuyoshi, Tomohiro Matsuo, Yuji Sagara, Kojiro Ohba, Kaname Ohyama, and Hideki Sakai. 2017. "A Mini-Review of Reactive Oxygen Species in Urological Cancer: Correlation with NADPH Oxidases, Angiogenesis, and Apoptosis" International Journal of Molecular Sciences 18, no. 10: 2214. https://doi.org/10.3390/ijms18102214
APA StyleMiyata, Y., Matsuo, T., Sagara, Y., Ohba, K., Ohyama, K., & Sakai, H. (2017). A Mini-Review of Reactive Oxygen Species in Urological Cancer: Correlation with NADPH Oxidases, Angiogenesis, and Apoptosis. International Journal of Molecular Sciences, 18(10), 2214. https://doi.org/10.3390/ijms18102214