The Emerging Role of the Major Histocompatibility Complex Class I in Amyotrophic Lateral Sclerosis


 ,
, 
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Amyotrophic Lateral Sclerosis (ALS): The Role of the Major Histocompatibility Complex Class I in Motoneurons
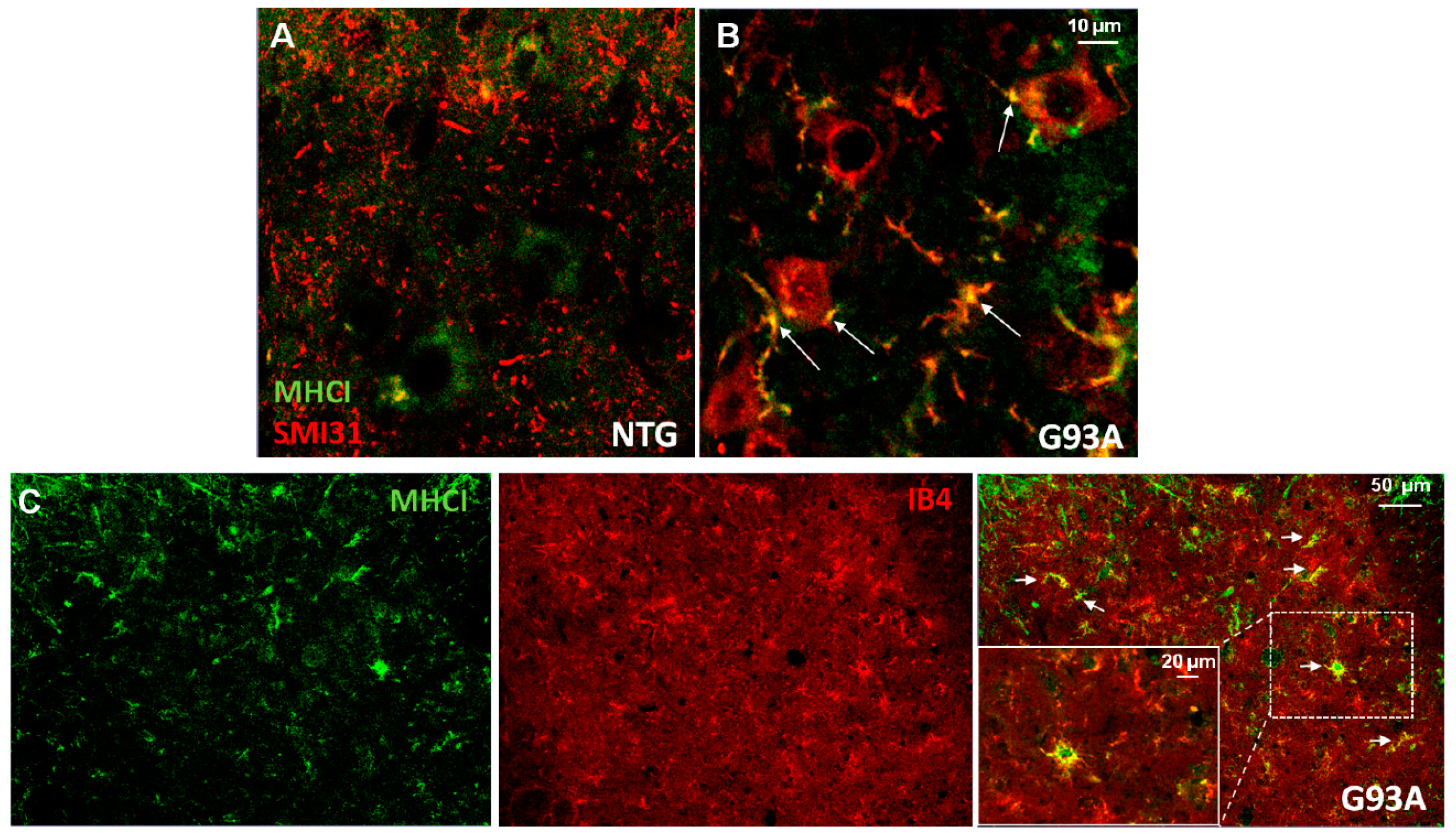
3. The Expression of Major Histocompatibility Complex Class I (MHCI) by Non-Neuronal Cells
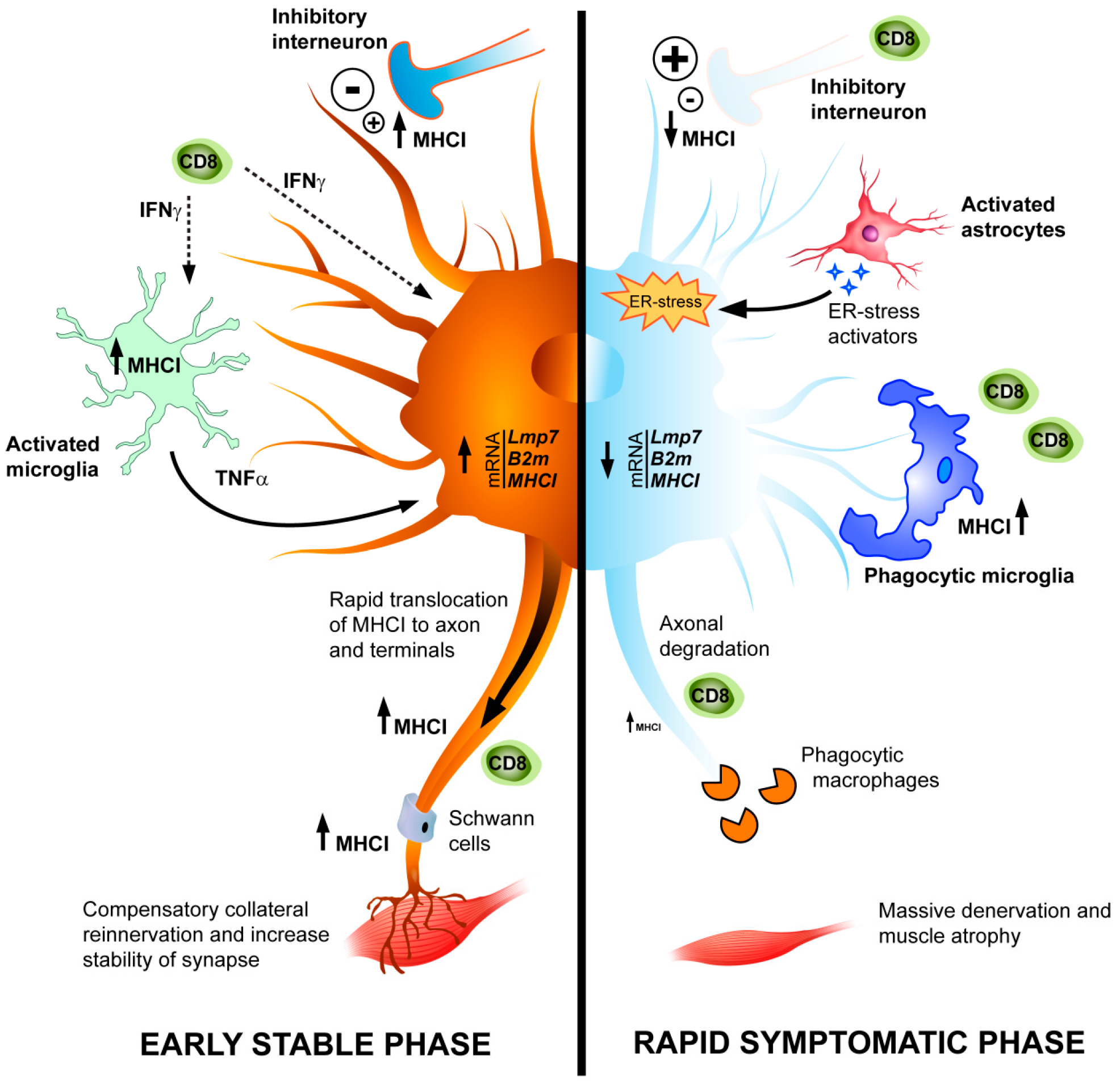
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hardiman, O.; Van Den Berg, L.H.; Kiernan, M.C. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Logroscino, G.; Traynor, B.J.; Collins, J.; Simeone, J.C.; Goldstein, L.A.; White, L.A. Global epidemiology of amyotrophic lateral sclerosis: A systematic review of the published literature. Neuroepidemiology 2013, 41, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Pupillo, E.; Messina, P.; Logroscino, G.; Beghi, E. Long-term survival in amyotrophic lateral sclerosis: A population-based study. Ann. Neurol. 2014, 75, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Logroscino, G.; Hardiman, O.; Swingler, R.; Mitchell, D.; Beghi, E.; Traynor, B.G.; Eurals Consortium. Prognostic factors in ALS: A critical review. Amyotroph. Lateral Scler. 2009, 10, 310–323. [Google Scholar] [CrossRef] [PubMed]
- Byrne, S.; Walsh, C.; Lynch, C.; Bede, P.; Elamin, M.; Kenna, K.; McLaughlin, R.; Hardiman, O. Rate of familial amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2011, 82, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; Jones, A.; Troakes, C.; King, A.; Al-Sarraj, S.; van den Berg, L.H. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol. 2012, 124, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Gurney, M.E. Transgenic-mouse model of amyotrophic lateral sclerosis. N. Engl. J. Med. 1994, 331, 1721–1722. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Chiò, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Velde, C.V.; Bouchard, J.P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F.; et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008, 40, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, T.J.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [PubMed]
- Baloh, R.H. How do the RNA-binding proteins TDP-43 and FUS relate to amyotrophic lateral sclerosis and frontotemporal degeneration, and to each other? Curr. Opin. Neurol. 2012, 25, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Ciura, S.; Lattante, S.; Le Ber, I.; Latouche, M.; Tostivint, H.; Brice, A.; Kabashi, E. Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann. Neurol. 2013, 74, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Beers, D.R.; Appel, S.H. Immune-mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis. J. Neuroimmune Pharmacol. 2013, 8, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Ilieva, E.V.; Ayala, V.; Jové, M.; Dalfó, E.; Cacabelos, D.; Povedano, M.; Bellmunt, M.J.; Ferrer, I.; Pamplona, R.; Portero-Otín, M. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain 2007, 130, 3111–3123. [Google Scholar] [CrossRef] [PubMed]
- Ayala, V.; Granado-Serrano, A.B.; Cacabelos, D.; Naudí, A.; Ilieva, E.V.; Boada, J.; Caraballo-Miralles, V.; Lladó, J.; Ferrer, I.; Pamplona, R.; et al. Cell stress induces TDP-43 pathological changes associated with ERK1/2 dysfunction: Implications in ALS. Acta Neuropathol. 2011, 122, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, M.; Ferri, A.; Valle, C.; Carrì, M.T. Mitochondria and ALS: Implications from novel genes and pathways. Mol. Cell. Neurosci. 2013, 55, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D. Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann. Neurol. 2009, 65, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, M.; Pesaresi, M.G.; Gerbino, V.; Grosskreutz, J.; Carri, M.T. Amyotrophic lateral sclerosis: New insights into underlying molecular mechanisms and opportunities for therapeutic intervention. Antioxid. Redox Signal. 2012, 17, 1277–1330. [Google Scholar] [CrossRef] [PubMed]
- Guareschi, S.; Cova, E.; Cereda, C.; Ceroni, M.; Donetti, E.; Bosco, D.A.; Trotti, D.; Pasinelli, P. An over-oxidized form of superoxide dismutase found in sporadic amyotrophic lateral sclerosis with bulbar onset shares a toxic mechanism with mutant SOD1. Proc. Natl. Acad. Sci. USA 2012, 109, 5074–5079. [Google Scholar] [CrossRef] [PubMed]
- Magrane, J.; Manfredi, G. Mitochondrial function, morphology, and axonal transport in amyotrophic lateral sclerosis. Antioxid. Redox Signal. 2009, 11, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Iwata, M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2007, 66, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Pasinelli, P.; Houseweart, M.K.; Brown, R.H.; Cleveland, D.W. Caspase-1 and -3 are sequentially activated in motor neuron death in Cu, Zn superoxide dismutase-mediated familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2000, 97, 13901–13906. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Ruegsegger, C.; Saxena, S. Proteostasis impairment in ALS. Brain Res. 2016, 1648, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Parakh, S.; Atkin, J.D. Protein folding alterations in amyotrophic lateral sclerosis. Brain Res. 2016, 1648, 633–649. [Google Scholar] [CrossRef] [PubMed]
- Atkin, J.D.; Farg, M.A.; Walker, A.K.; McLean, C.; Tomas, D.; Horne, M.K. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 2008, 30, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Thielen, P.; Matus, S.; Nassif, M.; Kiffin, R.; Martinez, G.; Cuervo, A.M.; Brown, R.H.; Glimcher, L.H. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009, 23, 2294–2306. [Google Scholar] [CrossRef] [PubMed]
- Boillée, S.; Velde, C.V.; Cleveland, D.W. ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron 2006, 52, 39–59. [Google Scholar] [CrossRef] [PubMed]
- Appel, E.A.; Biedermann, F.; Rauwald, U.; Jones, S.T.; Zayed, J.M.; Scherman, O.A. Supramolecular cross-linked networks via host-guest complexation with cucurbit [8] uril. J. Am. Chem. Soc. 2010, 132, 14251–14260. [Google Scholar] [CrossRef] [PubMed]
- Robberecht, W.; Philips, T. The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 2013, 14, 248–264. [Google Scholar] [CrossRef] [PubMed]
- Lino, M.M.; Schneider, C.; Caroni, P. Accumulation of SOD1 mutants in postnatal motoneurons does not cause motoneuron pathology or motoneuron disease. J. Neurosci. 2002, 22, 4825–4832. [Google Scholar] [PubMed]
- Nagai, M.; Re, D.B.; Nagata, T.; Chalazonitis, A.; Jessell, T.M.; Wichterle, H.; Przedborski, S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 2007, 10, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Brites, D.; Vaz, A.R. Microglia centered pathogenesis in ALS: Insights in cell interconnectivity. Front. Cell. Neurosci. 2014, 8, 117. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Miranda, C.J.; Braun, L.; Meyer, K.; Frakes, A.E.; Ferraiuolo, L.; Likhite, S.; Bevan, A.K.; Foust, K.D.; McConnell, M.J.; et al. Major histocompatibility complex class I molecules protect motor neurons from astrocyte-induced toxicity in amyotrophic lateral sclerosis. Nat. Med. 2016, 22, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, F.P.; Boulting, G.L.; Bobrowicz, S.; Eggan, K.C. Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell 2008, 3, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, F.P.; Carrasco, M.A.; Siao, M.C.; Maniatis, T.; Eggan, K. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat. Neurosci. 2007, 10, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Siddiqui, S.; Gabriely, G.; Lanser, A.J.; Dake, B.; Murugaiyan, G.; Doykan, C.E.; Wu, P.M.; Gali, R.R.; Iyer, L.K.; et al. Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J. Clin. Investig. 2012, 122, 3063–3087. [Google Scholar] [CrossRef] [PubMed]
- Chiu, I.M.; Chen, A.; Zheng, Y.; Kosaras, B.; Tsiftsoglou, S.A.; Vartanian, T.K.; Brown, R.H.; Carroll, M.C. T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proc. Natl. Acad. Sci. USA 2008, 105, 17913–17918. [Google Scholar] [CrossRef] [PubMed]
- Beers, D.R.; Henkel, J.S.; Zhao, W.; Wang, J.; Appel, S.H. CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc. Natl. Acad. Sci. USA 2008, 105, 15558–15563. [Google Scholar] [CrossRef] [PubMed]
- Rojas, F.; Cortes, N.; Abarzua, S.; Dyrda, A.; van Zundert, B. Astrocytes expressing mutant SOD1 and TDP43 trigger motoneuron death that is mediated via sodium channels and nitroxidative stress. Front. Cell. Neurosci. 2014, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Bendotti, C.; Marino, M.; Cheroni, C.; Fontana, E.; Crippa, V.; Poletti, A.; de Biasi, S. Dysfunction of constitutive and inducible ubiquitin-proteasome system in amyotrophic lateral sclerosis: Implication for protein aggregation and immune response. Prog. Neurobiol. 2012, 97, 101–126. [Google Scholar] [CrossRef] [PubMed]
- Cheroni, C.; Marino, M.; Tortarolo, M.; Veglianese, P.; De Biasi, S.; Fontana, E.; Zuccarello, L.V.; Maynard, C.J.; Dantuma, N.P.; Bendotti, C. Functional alterations of the ubiquitin-proteasome system in motor neurons of a mouse model of familial amyotrophic lateral sclerosis. Hum. Mol. Genet. 2009, 18, 82–96. [Google Scholar] [CrossRef] [PubMed]
- Nardo, G.; Iennaco, R.; Fusi, N.; Heath, P.R.; Marino, M.; Trolese, M.C.; Ferraiuolo, L.; Lawrence, N.; Shaw, P.J.; Bendotti, C. Transcriptomic indices of fast and slow disease progression in two mouse models of amyotrophic lateral sclerosis. Brain 2013, 136, 3305–3332. [Google Scholar] [CrossRef] [PubMed]
- Radisky, D.C.; Stallings-Mann, M.; Hirai, Y.; Bissell, M.J. Single proteins might have dual but related functions in intracellular and extracellular microenvironments. Nat. Rev. Mol. Cell Biol. 2009, 10, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Huh, G.S.; Boulanger, L.M.; Du, H.; Riquelme, P.A.; Brotz, T.M.; Shatz, C.J. Functional requirement for class I MHC in CNS development and plasticity. Science 2000, 290, 2155–2159. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, L.M. Immune proteins in brain development and synaptic plasticity. Neuron 2009, 64, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Elmer, B.M.; McAllister, A.K. Major histocompatibility complex class I proteins in brain development and plasticity. Trends Neurosci. 2012, 35, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.L.; Thams, S.; Lidman, O.; Piehl, F.; Hökfelt, T.; Kärre, K.L.A.S.; Lindå, H.; Cullheim, S. A role for MHC class I molecules in synaptic plasticity and regeneration of neurons after axotomy. Proc. Natl. Acad. Sci. USA 2004, 101, 17843–17848. [Google Scholar] [CrossRef] [PubMed]
- McConnell, M.J.; Huang, Y.H.; Datwani, A.; Shatz, C.J. H2-K(b) and H2-D(b) regulate cerebellar long-term depression and limit motor learning. Proc. Natl. Acad. Sci. USA 2009, 106, 6784–6789. [Google Scholar] [CrossRef] [PubMed]
- Lindå, H.; Olsson, T.; Piehl, F. Expression of MHC class I and β2-microglobulin in rat spinal motoneurons: Regulatory influences by IFN-gamma and axotomy. Exp. Neurol. 1998, 150, 282–295. [Google Scholar] [CrossRef] [PubMed]
- Corriveau, R.A.; Huh, G.S.; Shatz, C.J. Regulation of class I MHC gene expression in the developing and mature CNS by neural activity. Neuron 1998, 21, 505–520. [Google Scholar] [CrossRef]
- Garay, P.A.; McAllister, A.K. Novel roles for immune molecules in neural development: Implications for neurodevelopmental disorders. Front. Synaptic Neurosci. 2010, 2, 136. [Google Scholar] [CrossRef] [PubMed]
- Shatz, C.J. MHC class I: An unexpected role in neuronal plasticity. Neuron 2009, 64, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Stephan, A.H.; Barres, B.A.; Stevens, B. The complement system: An unexpected role in synaptic pruning during development and disease. Annu. Rev. Neurosci. 2012, 35, 369–389. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Brott, B.K.; Kirkby, L.A.; Adelson, J.D.; Cheng, S.; Feller, M.B.; Datwani, A.; Shatz, C.J. Synapse elimination and learning rules co-regulated by MHC class I H2-Db. Nature 2014, 509, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Bilousova, T.; Dang, H.; Xu, W.; Gustafson, S.; Jin, Y.; Wickramasinghe, L.; Won, T.; Bobarnac, G.; Middleton, B.; Tian, J.; et al. Major histocompatibility complex class I molecules modulate embryonic neuritogenesis and neuronal polarization. J. Neuroimmunol. 2012, 247, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sabha, M., Jr.; Emirandetti, A.; Cullheim, S.; De Oliveira, A.L.R. MHC I expression and synaptic plasticity in different mice strains after axotomy. Synapse 2008, 62, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Joseph, M.S.; Bilousova, T.; Zdunowski, S.; Wu, Z.P.; Middleton, B.; Boudzinskaia, M.; Wong, B.; Ali, N.; Zhong, H.; Yong, J.; et al. Transgenic mice with enhanced neuronal major histocompatibility complex class I expression recover locomotor function better after spinal cord injury. J. Neurosci. Res. 2011, 89, 365–372. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Thams, S.; Brodin, P.; Plantman, S.; Saxelin, R.; Kärre, K.; Cullheim, S. Classical major histocompatibility complex class I molecules in motoneurons: New actors at the neuromuscular junction. J. Neurosci. 2009, 29, 13503–13515. [Google Scholar] [CrossRef] [PubMed]
- Nardo, G.; Trolese, M.C.; de Vito, G.; Cecchi, R.; Riva, N.; Dina, G.; Heath, P.R.; Quattrini, A.; Shaw, P.J.; Piazza, V.; et al. Immune response in peripheral axons delays disease progression in SOD1(G93A) mice. J. Neuroinflamm. 2016, 13, 261. [Google Scholar] [CrossRef] [PubMed]
- Staats, K.A.; Schönefeldt, S.; Van Rillaer, M.; Van Hoecke, A.; van Damme, P.; Robberecht, W.; Liston, A.; Van Den Bosch, L. Beta-2 microglobulin is important for disease progression in a murine model for amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2013, 7, 249. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, A.D.; Popovich, P.G.; Ramer, M.S. Wallerian degeneration: Gaining perspective on inflammatory events after peripheral nerve injury. J. Neuroinflamm. 2011, 8, 110. [Google Scholar] [CrossRef] [PubMed]
- Moalem, G.; Leibowitz-Amit, R.; Yoles, E.; Mor, F.; Cohen, I.R.; Schwartz, M. Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat. Med. 1999, 5, 49–55. [Google Scholar] [PubMed]
- Drouin-Ouellet, J.; Cicchetti, F. Inflammation and neurodegeneration: The story ‘retolled’. Trends Pharmacol. Sci. 2012, 33, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Lo Coco, D.; Veglianese, P.; Allievi, E.; Bendotti, C. Distribution and cellular localization of high mobility group box protein 1 (HMGB1) in the spinal cord of a transgenic mouse model of ALS. Neurosci. Lett. 2007, 412, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Tobisawa, S.; Hozumi, Y.; Arawaka, S.; Koyama, S.; Wada, M.; Nagai, M.; Aoki, M.; Itoyama, Y.; Goto, K.; Kato, T. Mutant SOD1 linked to familial amyotrophic lateral sclerosis, but not wild-type SOD1, induces ER stress in COS7 cells and transgenic mice. Biochem. Biophys. Res. Commun. 2003, 303, 496–503. [Google Scholar] [CrossRef]
- Atkin, J.D.; Farg, M.A.; Turner, B.J.; Tomas, D.; Lysaght, J.A.; Nunan, J.; Rembach, A.; Nagley, P.; Beart, P.M.; Cheema, S.S.; et al. Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J. Biol. Chem. 2006, 281, 30152–30165. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, H.; Ozaki, T.; Furuya, K.; Hanamoto, T.; Nakanishi, M.; Yamamoto, H.; Yoshida, K.; Todo, S.; Nakagawara, A. NF-kappaB regulates the stability and activity of p73 by inducing its proteolytic degradation through a ubiquitin-dependent proteasome pathway. Oncogene 2006, 25, 7608–7617. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.; Cozzolino, M.; Carrì, M.T. Old versus New Mechanisms in the Pathogenesis of ALS. Brain Pathol. 2016, 26, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Mollereau, B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 2014, 15, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Shi, G.; Sha, H.; Ji, Y.; Han, X.; Shu, X.; Ma, H.; Inoue, T.; Gao, B.; Kim, H.; et al. IRE1α is an endogenous substrate of endoplasmic-reticulum-associated degradation. Nat. Cell Biol. 2015, 17, 1546–1555. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [PubMed]
- Lilley, B.N.; Ploegh, H.L. Viral modulation of antigen presentation: Manipulation of cellular targets in the ER and beyond. Immunol. Rev. 2005, 207, 126–144. [Google Scholar] [CrossRef] [PubMed]
- Nishitoh, H.; Kadowaki, H.; Nagai, A.; Maruyama, T.; Yokota, T.; Fukutomi, H.; Noguchi, T.; Matsuzawa, A.; Takeda, K.; Ichijo, H. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 2008, 22, 1451–1464. [Google Scholar] [CrossRef] [PubMed]
- Bombeiro, A.L.; Hell, R.C.; Simões, G.F.; de Castro, M.V.; de Oliveira, A.L.R. Importance of major histocompatibility complex of class I (MHC-I) expression for astroglial reactivity and stability of neural circuits in vitro. Neurosci. Lett. 2017, 647, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.A.; Quan, N.; Stern, E.L.; Kristensson, K.; Herkenham, M. Induced neuronal expression of class I major histocompatibility complex mRNA in acute and chronic inflammation models. J. Neuroimmunol. 2002, 131, 83–91. [Google Scholar] [CrossRef]
- Redwine, J.M.; Buchmeier, M.J.; Evans, C.F. In vivo expression of major histocompatibility complex molecules on oligodendrocytes and neurons during viral infection. Am. J. Pathol. 2001, 159, 1219–1224. [Google Scholar] [CrossRef]
- Bombeiro, A.L.; Thomé, R.; Nunes, S.L.; Moreira, B.M.; Verinaud, L.; de Oliveira, A.L.R. Correction: MHC-I and PirB Upregulation in the Central and Peripheral Nervous System Following Sciatic Nerve Injury. PLoS ONE 2016, 11, e0165185. [Google Scholar] [CrossRef] [PubMed]
- Van Guilder Starkey, H.D.; Van Kirk, C.A.; Bixler, G.V.; Imperio, C.G.; Kale, V.P.; Serfass, J.M.; Farley, J.A.; Yan, H.; Warrington, J.P.; Han, S.; et al. Neuroglial expression of the MHCI pathway and PirB receptor is upregulated in the hippocampus with advanced aging. J. Mol. Neurosci. 2012, 48, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Gerber, Y.N.; Sabourin, J.C.; Rabano, M.; Vivanco, Md.; Perrin, F.E. Early functional deficit and microglial disturbances in a mouse model of amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e36000. [Google Scholar] [CrossRef] [PubMed]
- Weydt, P.; Yuen, E.C.; Ransom, B.R.; Möller, T. Increased cytotoxic potential of microglia from ALS-transgenic mice. Glia 2004, 48, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Beauvillain, C.; Donnou, S.; Jarry, U.; Scotet, M.; Gascan, H.; Delneste, Y.; Guermonprez, P.; Jeannin, P.; Couez, D. Neonatal and adult microglia cross-present exogenous antigens. Glia 2008, 56, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Jarry, U.; Jeannin, P.; Pineau, L.; Donnou, S.; Delneste, Y.; Couez, D. Efficiently stimulated adult microglia cross-prime naive CD8+ T cells injected in the brain. Eur. J. Immunol. 2013, 43, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Nardo, G.; Trolese, M.C.; Bendotti, C. Major Histocompatibility Complex I Expression by Motor Neurons and Its Implication in Amyotrophic Lateral Sclerosis. Front. Neurol. 2016, 7, 89. [Google Scholar] [CrossRef] [PubMed]
- Bombeiro, A.L.; Santini, J.C.; Thomé, R.; Ferreira, E.R.; Nunes, S.L.; Moreira, B.M.; Bonet, I.J.; Sartori, C.R.; Verinaud, L.; Oliveira, A.L. Enhanced Immune Response in Immunodeficient Mice Improves Peripheral Nerve Regeneration Following Axotomy. Front. Cell. Neurosci. 2016, 10, 151. [Google Scholar] [CrossRef] [PubMed]
- Turner, B.J.; Talbot, K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog. Neurobiol. 2008, 85, 94–134. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, M.J.; Klenowski, P.M.; Lee, J.D.; Drieberg-Thompson, J.R.; Bartlett, S.E.; Ngo, S.T.; Hilliard, M.A.; Bellingham, M.C.; Noakes, P.G. Cortical synaptic and dendritic spine abnormalities in a presymptomatic TDP-43 model of amyotrophic lateral sclerosis. Sci. Rep. 2016, 6, 37968. [Google Scholar] [CrossRef] [PubMed]
- McCombe, P.A.; Henderson, R.D. The Role of immune and inflammatory mechanisms in ALS. Curr. Mol. Med. 2011, 11, 246–254. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiarotto, G.B.; Nardo, G.; Trolese, M.C.; França Jr., M.C.; Bendotti, C.; Rodrigues de Oliveira, A.L. The Emerging Role of the Major Histocompatibility Complex Class I in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2017, 18, 2298. https://doi.org/10.3390/ijms18112298
Chiarotto GB, Nardo G, Trolese MC, França Jr. MC, Bendotti C, Rodrigues de Oliveira AL. The Emerging Role of the Major Histocompatibility Complex Class I in Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences. 2017; 18(11):2298. https://doi.org/10.3390/ijms18112298
Chicago/Turabian StyleChiarotto, Gabriela Bortolança, Giovanni Nardo, Maria Chiara Trolese, Marcondes Cavalcante França Jr., Caterina Bendotti, and Alexandre Leite Rodrigues de Oliveira. 2017. "The Emerging Role of the Major Histocompatibility Complex Class I in Amyotrophic Lateral Sclerosis" International Journal of Molecular Sciences 18, no. 11: 2298. https://doi.org/10.3390/ijms18112298
APA StyleChiarotto, G. B., Nardo, G., Trolese, M. C., França Jr., M. C., Bendotti, C., & Rodrigues de Oliveira, A. L. (2017). The Emerging Role of the Major Histocompatibility Complex Class I in Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences, 18(11), 2298. https://doi.org/10.3390/ijms18112298