1. Introduction
Animal models are limited in their ability to mimic extremely complex carcinogenic and physiological processes in humans and more than 90% of drugs that pass animal trials fail in the clinical trials [
1,
2]. Various researchers have developed biomimetic models that can produce results similar to actual responses in humans, and three-dimensionally (3D) cultured cells have been found to better represent human tissue than two-dimensionally (2D) cultured cells [
3,
4,
5,
6,
7,
8,
9]. Most cells in tissues are surrounded by an extracellular matrix (ECM), which provides a structural chemical environment for cells and interacts with cells/ECM to regulate diverse functions including proliferation, differentiation, and migration [
10]. Therefore, we employed an electrospun nanofibrous membrane (NF) whose structure is similar to that of the extracellular matrix. The cell-laden NF has various morphologies, in contrast to two-dimensionally cultured cells, and the cells interact more actively based on the surface characteristics of this membrane.
In the drug discovery process, multiple tests are required to verify various factors such as efficacy, safety, and dose of the drug [
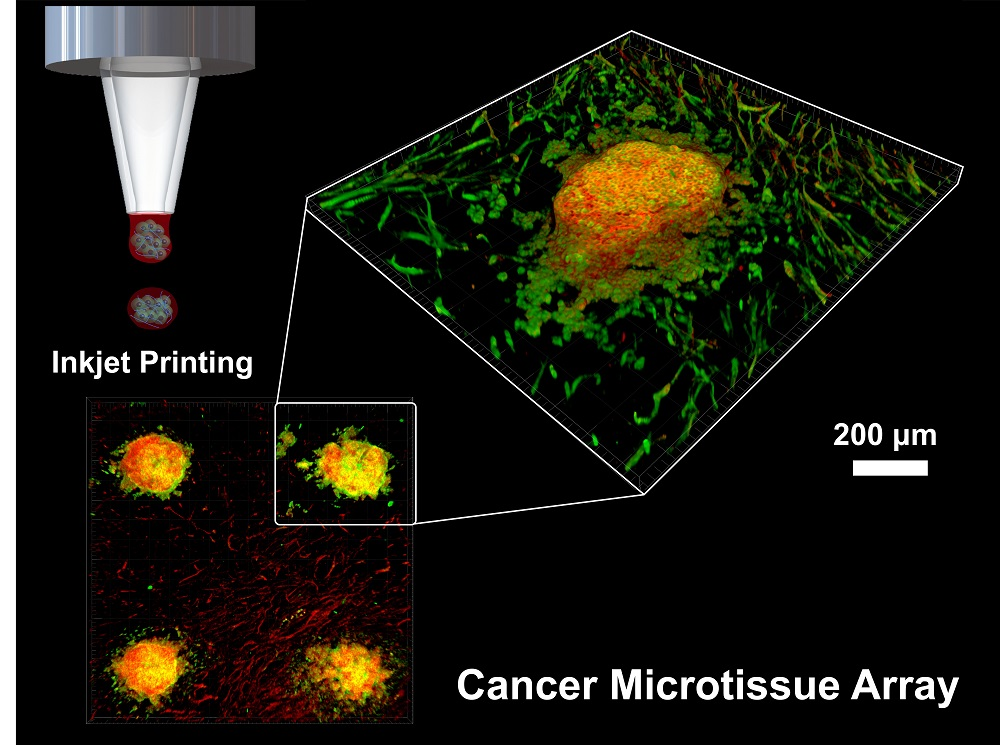
11]. Current 3D culture models have various difficulties such as limited control over cell positioning and multiply arrayed cells and batch-to-batch variability. To this end, this study developed and evaluated a patterned cancer microtissue array with controlled size and shape as well as batch-to-batch structures using an inkjet printer. Multiple tests can be simultaneously performed on the developed microtissue array (
Figure 1). The inkjet printer enabled precise adjustment of the spatial placement of the tissue formed on the NF with microscale resolution, and quantitative cell seeding was possible, thus improving the repeatability and reproducibility of the model.
The tumor microenvironment is constantly evolving owing to tissue remodeling and changes in the recruitment of stromal cells including a diversity of immune cells with angiogenesis. As the tumor becomes larger, the phenotype of cancer cells changes to aggressive, and in the tumor microenvironment, the extracellular matrix also changes [
12]. In addition, fibroblasts, which are cancer stromal cells, play an important role in tumor progression, growth, and metastasis [
13]. Therefore, developing functional and gradational cancer models that mimic the tumor microenvironment, including stromal cells, are required for testing drugs and investigating mechanisms of tumor progression as well as cancer metastasis.
This study used cervical cancer cells, which are invasive and aggressive and closely associated with matrix metalloproteinases (MMPs). In particular, MMP2 and MMP9 have been found to be the most important in the degradation of the basement membrane and extracellular matrix needed for cells to acquire invasive capabilities [
14]. In this study, MMP2 and MMP9 levels were measured through enzyme-linked immunosorbent assay (ELISA) to verify the effect of co-culturing with fibroblasts. The 3D structure of the fabricated cancer microtissue was examined using histological analysis, scanning electron microscopy (SEM), and confocal microscopy by staining for human papillomavirus 18 (HPV18), which is specifically found in cervical cancer cells. Finally, we treated the model with an anticancer drug for assessment of the drug response.
3. Discussion
Environmental factors have significant biochemical influences on cells [
15]. A two-dimensional culture is advantageous in terms of manipulation to observe cells and extract proteins and mRNA. However, most of these cells are flat, in contrast to their actual shapes in the human body, and in many cases, the experimental results are different from the actual in vivo situations. In contrast, a three-dimensional culture model is more complex and difficult to manipulate and evaluate than a two-dimensional culture model, but it can accurately mimic the 3D structures of cells in the human body, and their behaviors and the expression of proteins and mRNA in actual in vivo situations [
16]. Thus, in this study, fibroblasts were cultured with NF to provide a sufficient niche for cancer cells, and the co-cultured microtissue showed a different tissue-like structure and drug resistance compared to the monoculture.
The complex test processes and high throughput of each cell are the greatest obstacles to quantitative and in-depth molecular analysis of single cells or cell populations. The distribution of cells can be quantified by patterning cells to aid in the study of cell processes, such as reactions between individual tissues and cells. The advantage of patterning technology is the exposure of tissues to the same conditions owing to the arrangement of tissues. This study exploited inkjet printing to arrange cancer microtissues on nanofibrous membranes and analyzed the viability, size changes, and other characteristics of the patterned microtissues. The analysis showed that the viability of the cells was close to 100% when they were printed on a nanofibrous membrane using an inkjet printer, and the cancer microtissues produced and patterned using inkjet printing maintained their initial patterns even after culturing for one week. Such patterned cancer microtissues can be multi-screened, thus enabling simultaneous verification of the changes in the size of multiple cancer microtissues and observation of various tissues.
The rapid evaporation of droplets is one of the greatest drawbacks of the inkjet cell printer. The easy evaporation of droplets in micro units makes it difficult to culture cells, and many researchers have addressed this problem by printing hydrogels in wells containing a crosslinking agent [
17,
18,
19]. In contrast, in this study, a nanofibrous membrane soaked in medium was used as the substrate, and printing on this wetted nanofibrous membrane prevented the rapid evaporation of droplets. The nanofibrous membrane provided an appropriate niche for inkjet printing without the use of a crosslinking agent when culturing the printed cells and also provided structural and biochemical elements of the extracellular matrix when culturing the cells together with fibroblasts. We also showed that inkjet printing on nanofibrous membrane is advantageous in terms of shaping and maintaining cell aggregates.
In future studies, the immunity network based on the interaction between immune cells and the fabricated cancer microtissues will be simulated, and drug tests with a sample with such an immunity network are expected to produce results that are closer to the results obtained from the human body. The significance of this study is the simulation of in vitro cancer tissues in drug tests and the production of highly reliable samples. The failure rates of clinical trials are expected to decrease as the reactions to drugs would better replicate those in the human body, and this will have positive effects on pharmaceutical discovery processes with low success rates.
4. Materials and Methods
4.1. Fabrication of NFs by Electrospinning
NF was fabricated by a previously reported electrospinning method [
20,
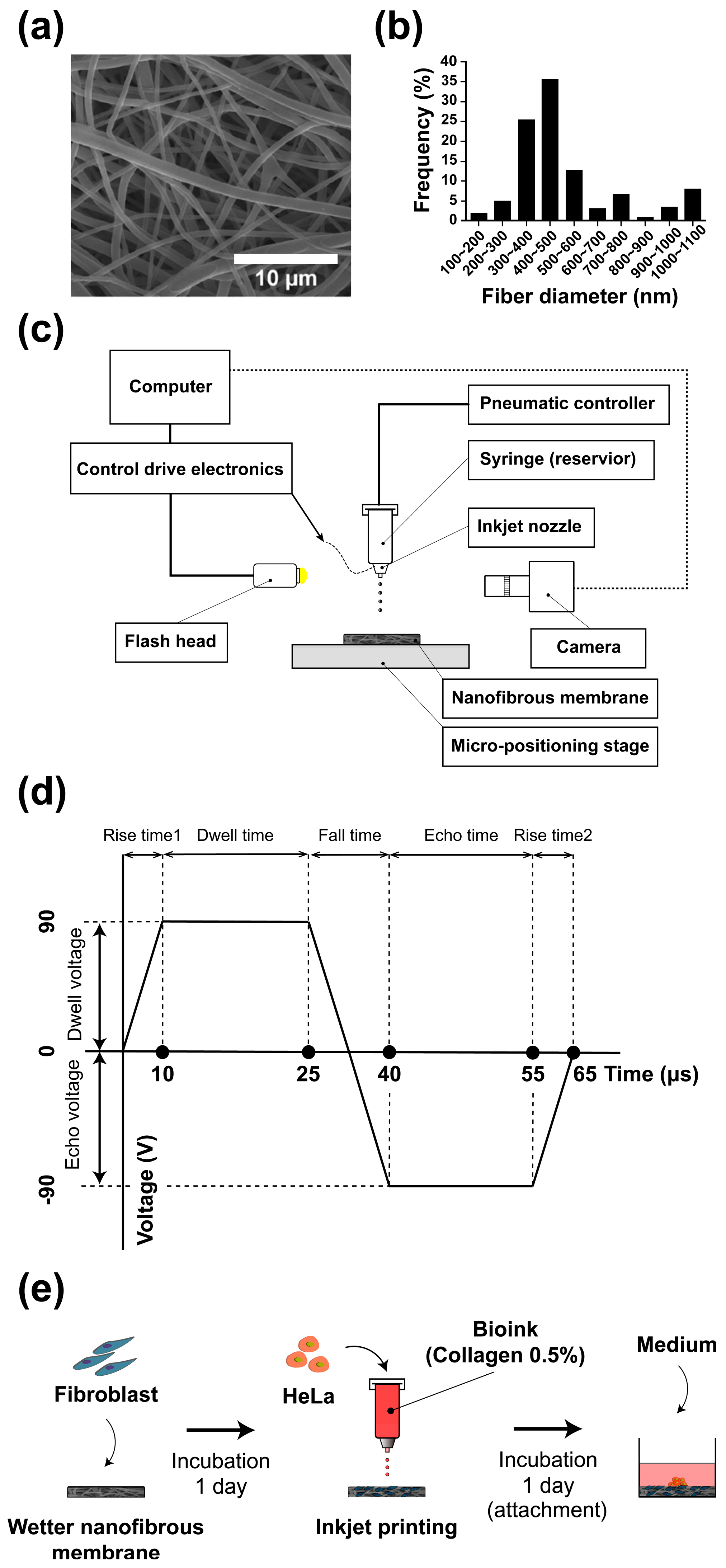
21]. Briefly, nanofibers were produced by diluting high molecular weight (80,000 MW) polycaprolactone (Sigma-Aldrich, St. Louis, MO, USA) at a concentration of 17% in a 1:1 mixture of chloroform (99.5%, Samchun Pure Chemical Co., Ltd., Seoul, Korea) and dimethylformamide (Sigma-Aldrich). During electrospinning, the temperature and humidity were maintained at 20–21 °C and 50–55%, respectively. The voltage, tip-to-collector distance, and flow rate of the solution were maintained at 15 kV, 18 cm, and 0.5 mL/h, respectively. The fabricated NF was dried for 24 h to evaporate the remaining solvent, and the dried membrane was then baked in an oven at 60 °C for 10 s to improve the interconnectivity of interstitial fibers before they were cut to a diameter of 10 mm. The fabricated NF was soaked in 70% ethanol and sterilized under an ultraviolet lamp overnight. Then, the sample was washed out three times with phosphate-buffered saline (PBS; pH 7.4, Gibco, Rockvile, MD, USA) and soaked in Dulbecco’s modified Eagle’s medium (DMEM, GenDEPOT) containing 10% fetal bovine serum (FBS, GenDEPOT) and 1% penicillin-streptomycin (Pen Strep, Gibco). The diameter distribution of the nanofibers characterized by an SEM image (
Figure 2a) is shown in
Figure 2b; the diameter of the nanofibers was estimated to be 400–500 nm.
4.2. Cell and Bioink Preparation
Fibroblasts (CCD-1112SK, ATCC, Manassas, VA, USA) and HPV18-positive cervical cancer cells (HeLa; KCLB, Seoul, Korea) were cultured with a medium containing 10% FBS and 1% Pen Strep in a 100-mm Petri dish (Nunclon Delta Surface, Thermo Fisher Scientific, Waltham, MA, USA). When cell confluency reached 80%, the cells were washed three times with PBS and treated with 2 mL of trypsin-EDTA (0.25%; Gibco) to produce single-cell units before culturing in 5% CO2 at 37 °C for 2 min. After culturing, the detached cells were collected in a 15-mL conical tube into which culture medium was added, diluted 10 times to neutralize the trypsin, and centrifuged for 3 min at 1200 rpm. The cells obtained after removal of the supernatant were mixed with the culture medium and then used in the experiment.
The fibroblasts ( cells/mL) were seeded on NF with a diameter of 10 mm. After culturing in an incubator overnight, fibroblast-layered NF or bare NF was used as a substrate for inkjet cell printing.
For the cell-laden bioink, cell suspensions ( cells/mL) were produced containing 0.5% collagen (collagen type I, Thermo Fisher Scientific) or cell suspension alone.
4.3. Inkjet Cell Printing
Inkjet printing technology that improves cell aggregation is essential for fabricating cancer microtissues. Therefore, the formation of the printed droplets on different substrates (glass and NF) and bioink was verified. To examine the effects of the bioink on the droplets, the culture medium with or without 0.5% collagen was mixed with HeLa cells ( cells/mL) stained with Hoechst 33342 (Thermo Fisher Scientific), and this mixture was then used as the bioink for printing. To examine the effect of the substrate, the droplets were printed on a glass and the NF wetted in the medium.
To fabricate cancer microtissue samples, droplets of the 0.5% collagen bioink containing cancer cells were dropped 150 times using an inkjet printer on a NF as shown in
Figure 2e. Three different conditions of NF were tested: the first was bare membrane, and the others ware fibroblast-layered membranes (
cells/mL). An array of a maximum of four microtissues was patterned on the membrane (
10 mm). Immediately afterwards, the microtissues were cultured for 2 h in 5% CO
2 at 37 °C to enable the cancer cells to positively adhere. A sufficient amount of culture medium was added after adhesion of the cancer cells to the sample. Furthermore, for the co-cultured sample, fibroblasts were seeded on the NF and then soaked in the medium. The cells were then cultured in 5% CO
2 at 37 °C for one day before the cancer cells were printed as described above (
Figure 2d).
4.4. Cell Viability
To verify the viability of the printed cells, a Live/Dead kit (Invitrogen) was used in this experiment according to the manufacturer’s recommended procedure. In brief, 20 μL of 2 mM EthD-1 stock solution enclosed in the product was added to 10 mL of PBS and mixed by vortexing. Then, 5 μL of 4 mM calcein AM stock solution was added to the mixed solution, which was vortexed again to produce a live/dead solution. The samples were washed out three times with PBS and treated with the live/dead solution on the NF. Then, the specimen was cultured at room temperature, i.e., 20 °C, for 40 min.
Specifically, to compare the viability of printed cells on dried glass and wetted nanofibrous membrane (
Figure 3), the printed cells were gently subjected to the live/dead assay immediately after the printing. Thereafter, the sample was cultured in situ at 25 °C for 30 min.
4.5. Immunocytochemistry/Immunofluorescence
The sample was fixed by treatment with 4% paraformaldehyde for 20 min at room temperature and then washed three times with PBS to remove the paraformaldehyde. The washed sample was treated with 0.1% Triton X-100 for 15 min at room temperature to ensure permeability and then washed out with PBS three times. BSA buffer (1%) was used as the blocking buffer, and after treatment for 1 h at room temperature, the sample was washed with PBS three times. Then, the specific antibodies (MMP-2, MMP-9, and HPV18) were diluted in PBS at a ratio of 1:500. The primary and secondary antibodies were applied to the sample for 1 h at room temperature without light exposure.
4.6. Enzyme-Linked Immunosorbent Assay (ELISA)
Human MMP2 (ab100606, Abcam, Cambridge, UK) and MMP9 (ab100610, Abcam, Cambridge, UK) ELISA kits were used to measure the level of MMP2 and MMP9. The procedure was followed the manufacturer’s instructions. To summarize, 100 μL of the standard sample, which was the benchmark, and 100 μL of the specific experimental sample to be measured in this experiment were added to the well plates and cultured for 150 min at room temperature. Then, the solution was removed and the plates were washed with distilled water and PBS-Triton. Then, 100 μL of biotinylated (MMP2, MMP9) detection antibody was added to the well plates and mixed carefully for 1 h at room temperature. The solution was removed and the well plates were washed again with distilled water and PBS-Triton. Then, 100 μL of HRP–streptavidin solution was added to each well plate and carefully mixed for 45 min at room temperature. The well plates were washed with distilled water and PBS-Triton again, and 100 μL of TMB one-step substrate reagent was added to each well plate and carefully mixed for 30 min at room temperature without light exposure. Finally, 50 μL of the stop solution was added to each well plate and the absorbance at 450 nm was measured using a microreader. The amount of MMP in the cancer microtissue sample was compared with that in the standard sample.
4.7. Drug Testing
Doxorubicin (1 mg, Sigma Aldrich) was used to verify the drug resistance of the fabricated samples. The doxorubicin was diluted to 0.1, 1.0, and 2.0 μM in cell culture medium and added to the day-7 co-cultured samples, after which further culture was performed in 5% CO2 at 37 °C. Then, the amounts of MMP2 and MMP9 expressed after one and two days of administration were measured with ELISA, and a Live/Dead assay was used to examine the drug resistance of the fabricated cancer tissues.
4.8. Statistical Analysis
All of the data were presented as the mean ± standard deviation (SD). Statistical significance was defined as * p < 0.01, ** p < 0.005, *** p < 0.001.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







