Autophagy Roles in the Modulation of DNA Repair Pathways


Abstract
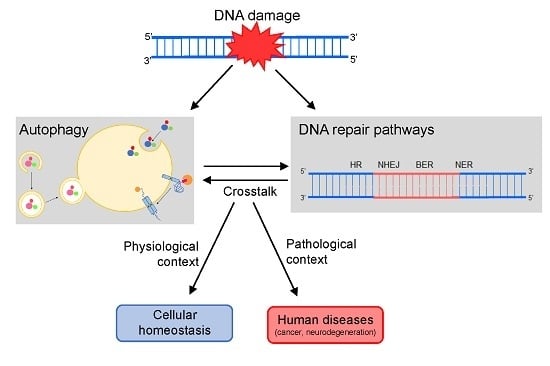
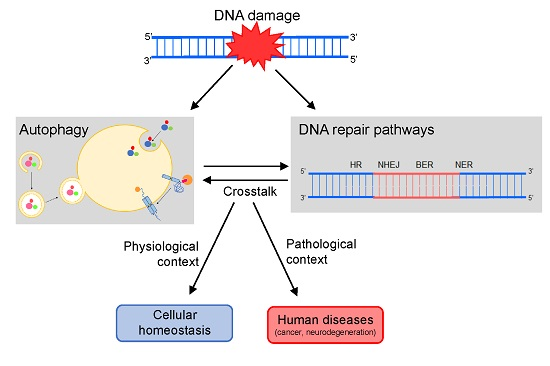
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Autophagy and p53: How the Guardians of Proteome and Genome Relate to Each Other
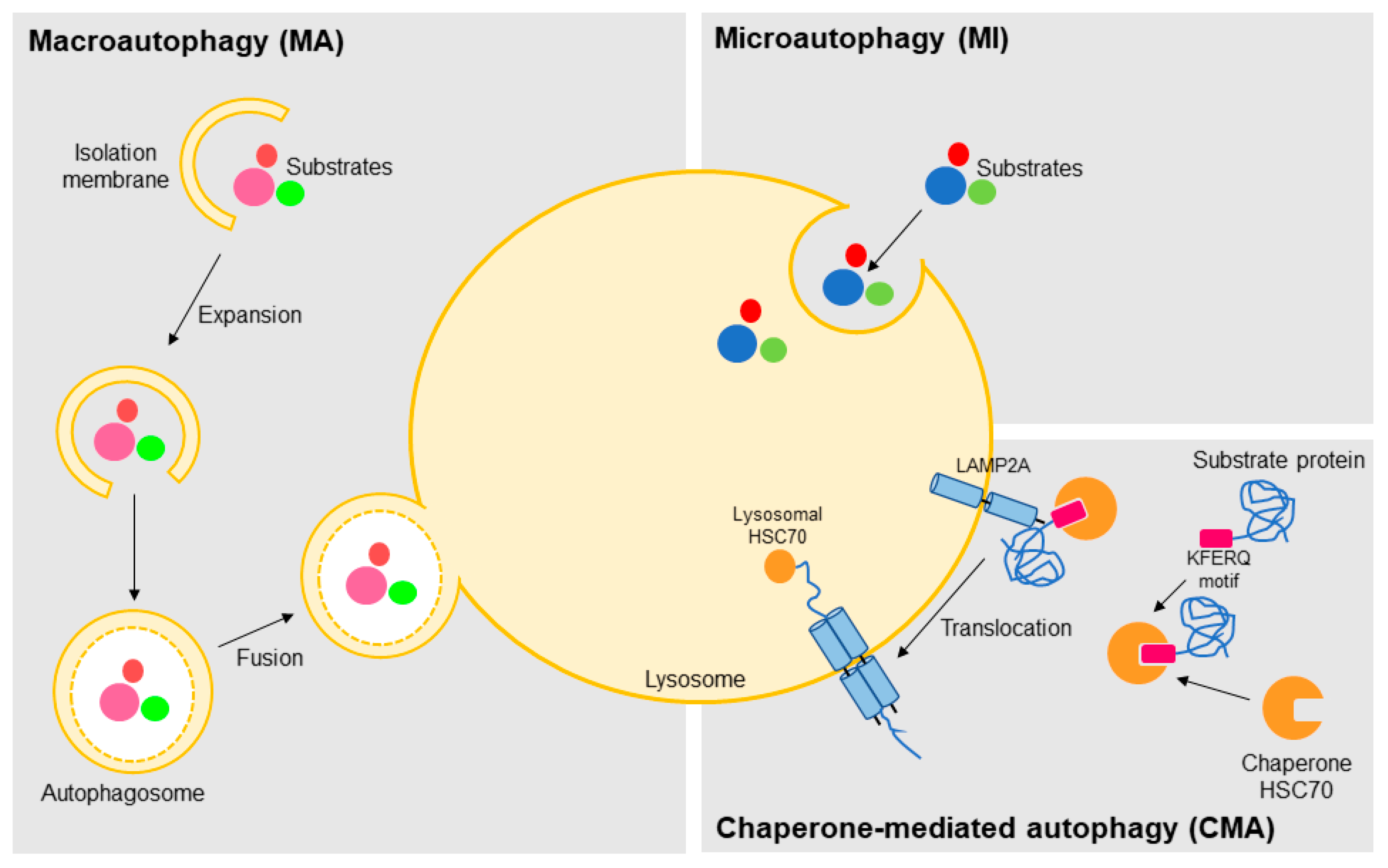
1.2. Autophagy: Mechanisms and Functions
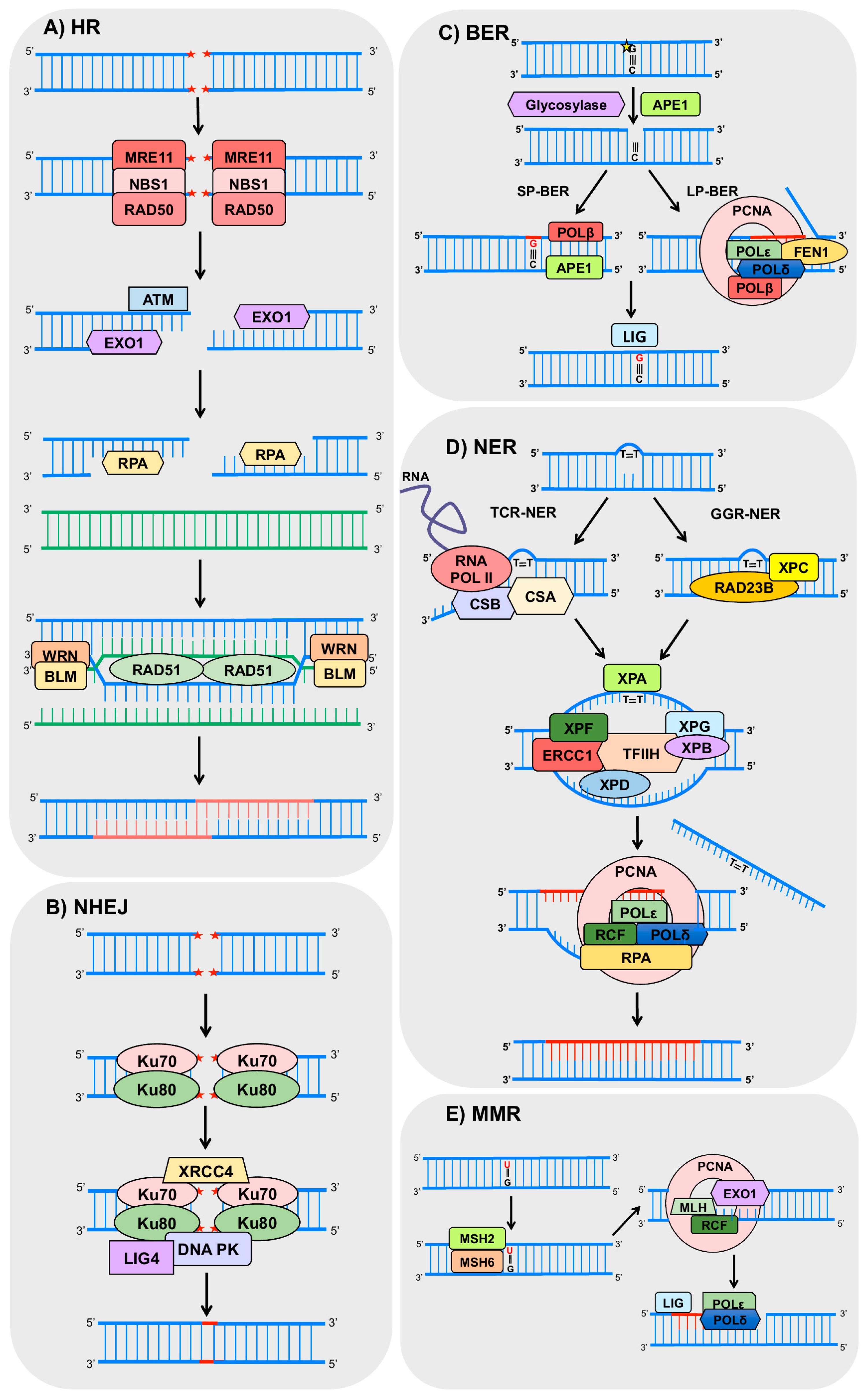
1.3. DNA Repair Machinery
2. Modulation of DNA Repair Pathways by Autophagy
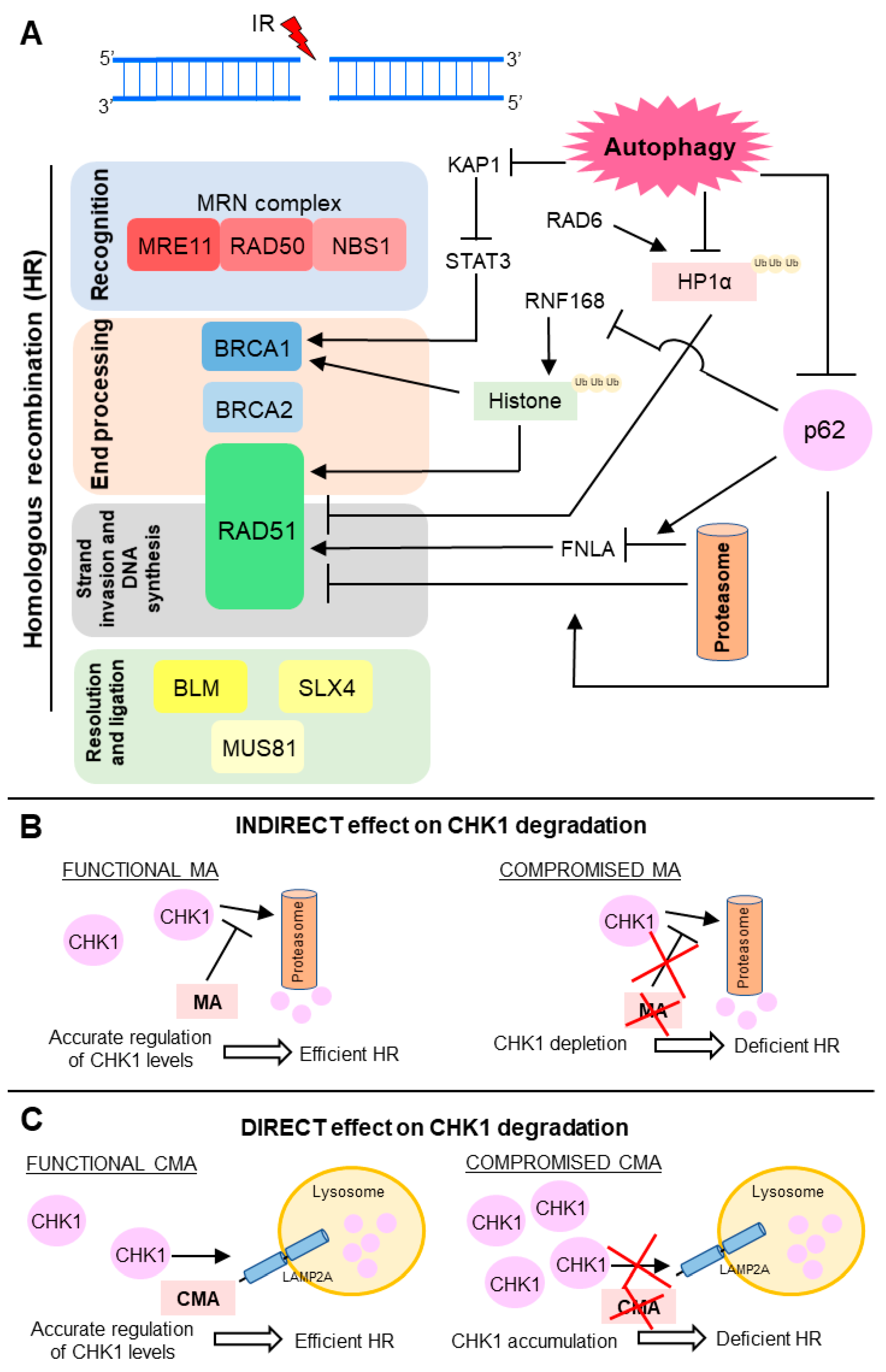
2.1. HR and NHEJ
2.2. BER
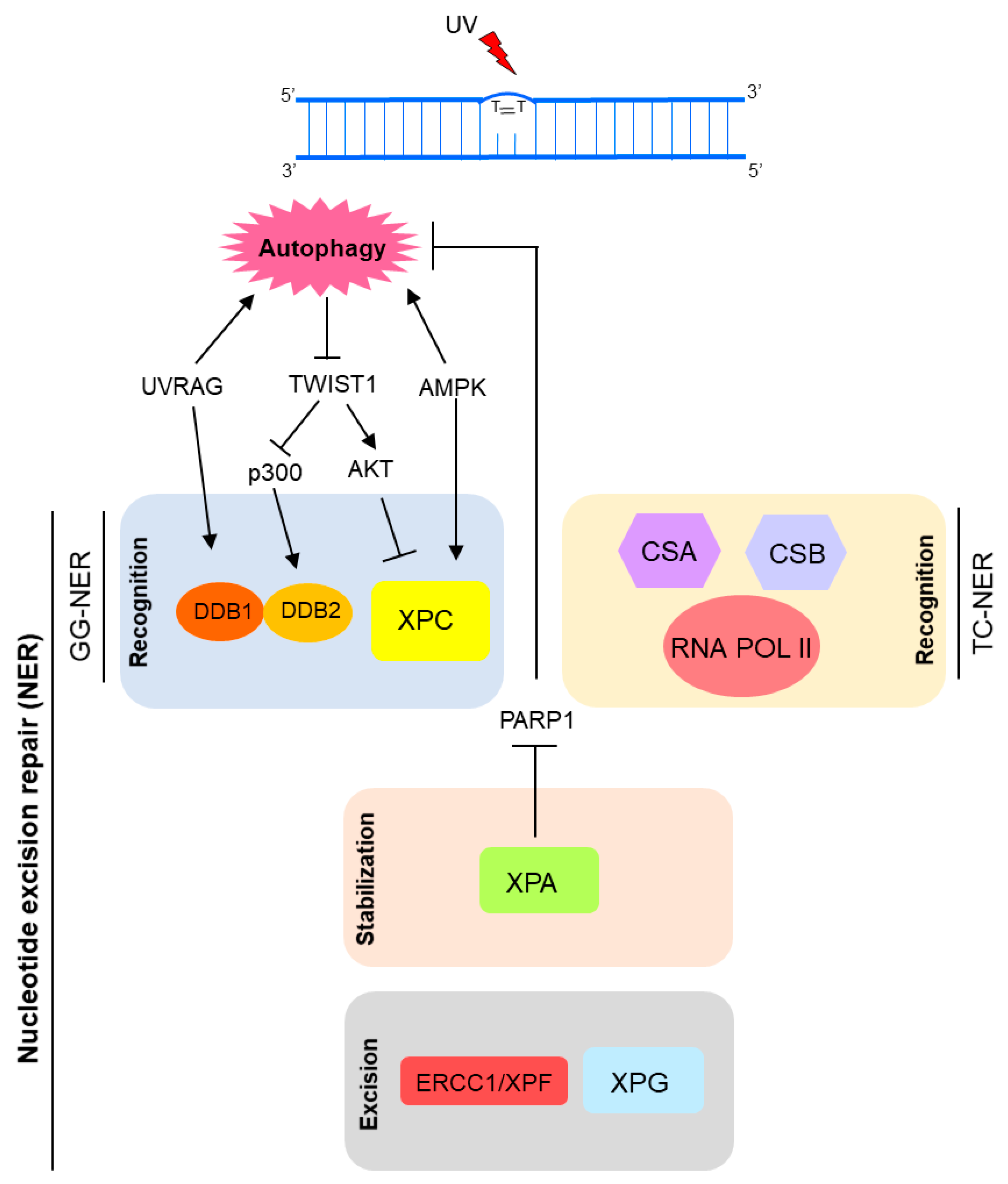
2.3. NER
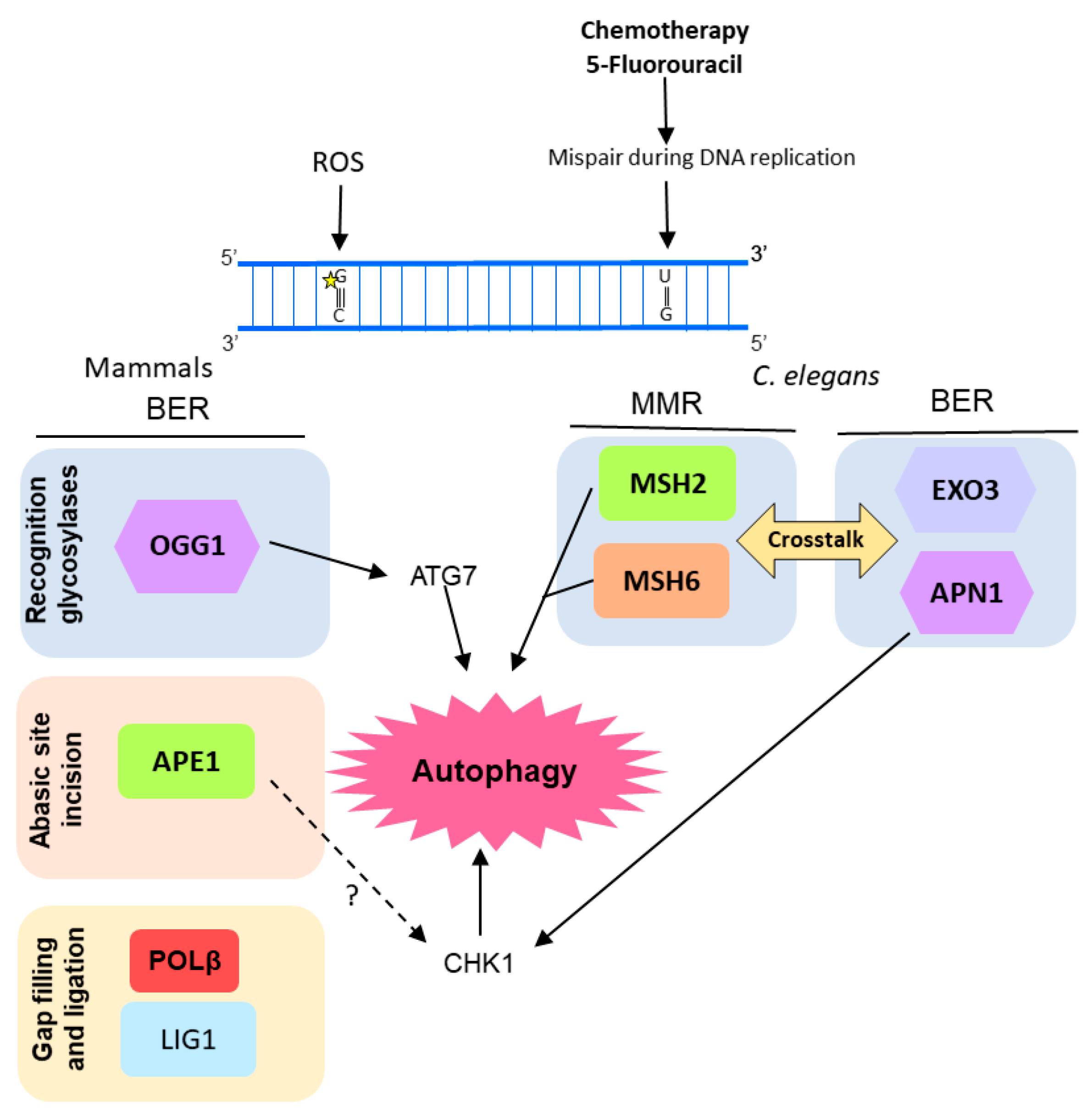
2.4. MMR
2.5. Other DNA Repair-Related Pathways
3. Concluding Remarks
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPK | AMP-activated protein kinase |
| AP | Abasic |
| APE1 | Apurinic/apyrimidinic endonuclease 1 |
| ATG | Autophagy-related |
| ATM | Ataxia telangiectasia mutated |
| BER | Base excision repair |
| BLM | Bloom syndrome REcQ like helicase |
| BNIP3 | Bcl-2 nineteen-kilodaltoni protein 3 |
| BRCA1 and 2 | Breast cancer 1 and 2, respectively |
| CHK1 | Checkpoint kinase 1 |
| CMA | Chaperone-mediated autophagy |
| CPD | Cyclobutane pyrimidine dimers |
| CS | Cockayne syndrome |
| CSA and CSB | Cockayne syndrome A and B, respectively |
| DDB1 and 2 | Damage-DNA binding protein 1 and 2, respectively |
| DDR | DNA damage Response |
| DNA-PK | DNA-dependent protein kinase |
| DRAM | Damage-regulated autophagy modulator |
| DSB | DNA double-strand break |
| ERCC1 | Excision repair cross-complementation group 1 |
| E2F4-RBL2 | E2F transcription factor 4-Retinoblastoma-like protein 2 |
| EXO 1 | Exonuclease 1 |
| FA | Fanconi anemia |
| FEN1 | Flap endonuclease 1 |
| FLA | Filamin A |
| GGR | Global genome repair |
| HP1α | Heterochromatin protein 1 Alfa |
| HR | Homologous recombination |
| HSC70 | heat shock cognate protein of 70-kDa |
| ICL | Interstrand DNA crosslink |
| IDL | Insertion/deletions loops |
| KAP1 | Corepressor for the Kruppel-associated box-domain-containing zinc-finger proteins, for degradation |
| IDL | Insertion/deletion loops |
| KAP1 | KRAB (Kruppel-Associated Box Domain)-Associated Protein 1 |
| LAMP2A | Lysosome-associated membrane protein type 2A |
| LC3B | Microtubule associated protein 1 light chain 3 beta |
| LIG1 | DNA ligase 1 |
| LIG3 | DNA ligase 3 |
| LIG4 | DNA ligase 4 |
| MLH | MutL homolog |
| MMR | Mismatch repair |
| MRE11 | Double strand break repair nuclease |
| MSH2, 3 and 6 | MutS homolog 2, 3 and 6, respectively |
| NBS1 | Nijmegen breakage syndrome 1 |
| NER | Nucleotide excision repair |
| NHEJ | Non-homologous end joining |
| OGG1 | 8-oxoguanine glycosylase 1 |
| PARP 1 | Poly (ADP-Ribose) polymerase 1 |
| PCNA | Proliferating cell nuclear antigen |
| POLβ, ε and δ | DNA polymerase beta, epsilon and delta, respectively |
| RAD23B | Human homolog B of S. cerevisiae RAD23 |
| RAD50, 51 and 52 | Human homolog of S. cerevisiae RAD50, RAD51 and RAD52, respectively |
| RFC | Replication factor C |
| RNA POL II | RNA polymerase II |
| ROS | Reactive oxygen species |
| RPA | Replication Protein A |
| SIRT1 | Sirtuin 1 |
| SQSTM1 | Sequestosome 1 |
| SSB | DNA single-strand breaks |
| STAT3 | Signal transducer and activator of transcription 3 |
| TCR | Transcription coupled repair |
| TP53BP1 | Tumor protein p53 binding protein 1 |
| TTD | Trichothiodystrophy |
| TFIIH | Transcription factor IIH |
| UPS | Ubiquitin proteasome system |
| UV | Ultraviolet |
| UVRAG | UV radiation resistance-associated gene |
| XPA, B, C, D, F and G | Xeroderma Pigmentosum complementation group A, B, C, D, F and G, respectively |
| XRCC1 and 4 | X-ray repair cross complementing 1 and 4, respectively |
| WRN | Werner syndrome RecQ like helicase |
| 5-FU | 5-fluoracil |
| 6-4PP | 6-4 pyrimidine pyrimidone |
| 6-TG | 6-thioguanine |
References
- Eliopoulos, A.G.; Havaki, S.; Gorgoulis, V.G. DNA damage response and autophagy: A meaningful partnership. Front. Genet. 2016, 7, 204. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Quach, C.; Liang, C. Autophagy modulator plays a part in UV protection. Autophagy 2016, 12, 1677–1678. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Zhao, B.; Shah, P.; Sample, A.; Yang, S.; He, Y.Y. Autophagy positively regulates DNA damage recognition by nucleotide excision repair. Autophagy 2016, 12, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, G.; Korolchuk, V.I. Repair, reuse, recycle: The expanding role of autophagy in genome maintenance. Trends Cell Biol. 2017, 27, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, D.A.; Ryan, K.M. Autophagy is critically required for DNA repair by homologous recombination. Mol. Cell. Oncol. 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhu, W.G.; Zhao, Y. Autophagy substrate SQSTM1/p62 regulates chromatin ubiquitination during the DNS damage response. Autophagy 2017, 13, 212–213. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, G.; Carroll, B.; Sarallah, R.; Correia-Melo, C.; Ogrodnik, M.; Nelson, G.; Otten, E.G.; Manni, D.; Antrobus, R.; Morgan, B.A.; et al. SQSTM1/p62 mediates crosstalk between autophagy and the ups in DNA repair. Autophagy 2016, 12, 1917–1930. [Google Scholar] [CrossRef] [PubMed]
- Czarny, P.; Pawlowska, E.; Bialkowska-Warzecha, J.; Kaarniranta, K.; Blasiak, J. Autophagy in DNA damage response. Int. J. Mol. Sci. 2015, 16, 2641–2662. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.M. P53 and autophagy in cancer: Guardian of the genome meets guardian of the proteome. Eur. J. Cancer 2011, 47, 44–50. [Google Scholar] [CrossRef] [PubMed]
- White, E. Autophagy and p53. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, E.; Chiara Maiuri, M.; Morselli, E.; Criollo, A.; D’Amelio, M.; Djavaheri-Mergny, M.; Cecconi, F.; Tavernarakis, N.; Kroemer, G. A dual role of p53 in the control of autophagy. Autophagy 2008, 4, 810–814. [Google Scholar] [CrossRef] [PubMed]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. Dram, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Abrams, J. P53: The janus of autophagy? Nat. Cell Biol. 2008, 10, 637–639. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. P53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Oren, M.; Rotter, V. Mutant p53 gain-of-function in cancer. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu-Norberg, H.; Kim, M.; Xia, H.G.; Iwanicki, M.P.; Ofengeim, D.; Coloff, J.L.; Pan, L.; Ince, T.A.; Kroemer, G.; Brugge, J.S.; et al. Chaperone-mediated autophagy degrades mutant p53. Genes Dev. 2013, 27, 1718–1730. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu-Norberg, H.; Yuan, J. A degradative detour for mutant Tp53. Autophagy 2013, 9, 2158–2160. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Wileman, T. Autophagy as a defence against intracellular pathogens. Essays Biochem. 2013, 55, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- King, J.S. Mechanical stress meets autophagy: Potential implications for physiology and pathology. Trends Mol. Med. 2012, 18, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Reggiori, F.; Codogno, P. Emerging regulation and functions of autophagy. Nat. Cell Biol. 2013, 15, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, S.R.; Simonsen, A. Membrane dynamics in autophagosome biogenesis. J. Cell Sci. 2015, 128, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Mijaljica, D.; Prescott, M.; Devenish, R.J. Microautophagy in mammalian cells: Revisiting a 40-year-old conundrum. Autophagy 2011, 7, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Oku, M.; Maeda, Y.; Kagohashi, Y.; Kondo, T.; Yamada, M.; Fujimoto, T.; Sakai, Y. Evidence for escrt- and clathrin-dependent microautophagy. J. Cell Biol. 2017, 216, 3263–3274. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Orenstein, S.J.; Cuervo, A.M. Chaperone-mediated autophagy: Molecular mechanisms and physiological relevance. Semin. Cell Dev. Biol. 2010, 21, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Kraft, C.; Peter, M.; Hofmann, K. Selective autophagy: Ubiquitin-mediated recognition and beyond. Nat. Cell Biol. 2010, 12, 836–841. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. P62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Mizushima, N. Autophagy and human diseases. Cell Res. 2014, 24, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Fleming, A.; Rubinsztein, D.C. Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 2015, 16, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.L.; Cuervo, A.M. Liver autophagy: Much more than just taking out the trash. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Madrigal-Matute, J.; Cuervo, A.M. Regulation of liver metabolism by autophagy. Gastroenterology 2016, 150, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Mariño, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [PubMed]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Kimmelman, A.C. The dynamic nature of autophagy in cancer. Genes Dev. 2011, 25, 1999–2010. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the Beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [PubMed]
- Manic, G.; Obrist, F.; Kroemer, G.; Vitale, I.; Galluzzi, L. Chloroquine and hydroxychloroquine for cancer therapy. Mol. Cell. Oncol. 2014, 1, e29911. [Google Scholar] [CrossRef] [PubMed]
- Chude, C.I.; Amaravadi, R.K. Targeting autophagy in cancer: Update on clinical trials and novel inhibitors. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef]
- Lin, L.; Baehrecke, E.H. Autophagy, cell death, and cancer. Mol. Cell. Oncol. 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Chi, K.H.; Wang, Y.S.; Huang, Y.C.; Chiang, H.C.; Chi, M.S.; Chi, C.H.; Wang, H.E.; Kao, S.J. Simultaneous activation and inhibition of autophagy sensitizes cancer cells to chemotherapy. Oncotarget 2016, 7, 58075–58088. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kaçmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the dna damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed]
- Moynahan, M.E.; Jasin, M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 2010, 11, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Stracker, T.H.; Petrini, J.H. The mre11 complex: Starting from the ends. Nat. Rev. Mol. Cell Biol. 2011, 12, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F.; Kozlov, S. ATM activation and dna damage response. Cell Cycle 2007, 6, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F. ATM and the MRE11 complex combine to recognize and signal DNA double-strand breaks. Oncogene 2007, 26, 7749–7758. [Google Scholar] [CrossRef] [PubMed]
- Sung, P.; Robberson, D.L. DNA strand exchange mediated by a RAD51-ssDNA nucleoprotein filament with polarity opposite to that of RECA. Cell 1995, 82, 453–461. [Google Scholar] [CrossRef]
- Benson, F.E.; Baumann, P.; West, S.C. Synergistic actions of RAD51 and RAD52 in recombination and DNA repair. Nature 1998, 391, 401–404. [Google Scholar] [PubMed]
- Jensen, R.B.; Carreira, A.; Kowalczykowski, S.C. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 2010, 467, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Heyer, W.D.; Ehmsen, K.T.; Liu, J. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 2010, 44, 113–139. [Google Scholar] [CrossRef] [PubMed]
- San Filippo, J.; Sung, P.; Klein, H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008, 77, 229–257. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous dna end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Kass, E.M.; Jasin, M. Collaboration and competition between DNA double-strand break repair pathways. FEBS Lett. 2010, 584, 3703–3708. [Google Scholar] [CrossRef] [PubMed]
- David, S.S.; O’Shea, V.L.; Kundu, S. Base-excision repair of oxidative DNA damage. Nature 2007, 447, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C. A history of the DNA repair and mutagenesis field: The discovery of base excision repair. DNA Repair 2016, 37, A35–A39. [Google Scholar] [CrossRef] [PubMed]
- Shafirovich, V.; Geacintov, N.E. Removal of oxidatively generated DNA damage by overlapping repair pathways. Free Radic. Biol. Med. 2017, 107, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Wilson, D.M. Overview of base excision repair biochemistry. Curr. Mol. Pharmacol. 2012, 5, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Dianov, G.; Bischoff, C.; Piotrowski, J.; Bohr, V.A. Repair pathways for processing of 8-oxoguanine in DNA by mammalian cell extracts. J. Biol. Chem. 1998, 273, 33811–33816. [Google Scholar] [CrossRef] [PubMed]
- Klungland, A.; Lindahl, T. Second pathway for completion of human DNA base excision-repair: Reconstitution with purified proteins and requirement for DNase IV (FEN1). EMBO J. 1997, 16, 3341–3348. [Google Scholar] [CrossRef] [PubMed]
- Gillet, L.C.; Schärer, O.D. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem. Rev. 2006, 106, 253–276. [Google Scholar] [CrossRef] [PubMed]
- Menck, C.F.; Munford, V. DNA repair diseases: What do they tell us about cancer and aging? Genet. Mol. Biol. 2014, 37, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Iyama, T.; Wilson, D.M. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair 2013, 12, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Fagbemi, A.F.; Orelli, B.; Schärer, O.D. Regulation of endonuclease activity in human nucleotide excision repair. DNA Repair 2011, 10, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Mocquet, V.; Lainé, J.P.; Riedl, T.; Yajin, Z.; Lee, M.Y.; Egly, J.M. Sequential recruitment of the repair factors during NER: The role of XPG in initiating the resynthesis step. EMBO J. 2008, 27, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Ogi, T.; Limsirichaikul, S.; Overmeer, R.M.; Volker, M.; Takenaka, K.; Cloney, R.; Nakazawa, Y.; Niimi, A.; Miki, Y.; Jaspers, N.G.; et al. Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol. Cell 2010, 37, 714–727. [Google Scholar] [CrossRef] [PubMed]
- Moser, J.; Kool, H.; Giakzidis, I.; Caldecott, K.; Mullenders, L.H.; Fousteri, M.I. Sealing of chromosomal DNA nicks during nucleotide excision repair requires XRCC1 and DNA ligase III α in a cell-cycle-specific manner. Mol. Cell 2007, 27, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Jiricny, J. Postreplicative mismatch repair. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Modrich, P. Mechanisms in eukaryotic mismatch repair. J. Biol. Chem. 2006, 281, 30305–30309. [Google Scholar] [CrossRef] [PubMed]
- Larrea, A.A.; Lujan, S.A.; Kunkel, T.A. Snapshot: DNA mismatch repair. Cell 2010, 141. [Google Scholar] [CrossRef] [PubMed]
- Cannavo, E.; Marra, G.; Sabates-Bellver, J.; Menigatti, M.; Lipkin, S.M.; Fischer, F.; Cejka, P.; Jiricny, J. Expression of the MUTl homologue HMLH3 in human cells and its role in dna mismatch repair. Cancer Res. 2005, 65, 10759–10766. [Google Scholar] [CrossRef] [PubMed]
- Räschle, M.; Marra, G.; Nyström-Lahti, M.; Schär, P.; Jiricny, J. Identification of HMUTLβ, a heterodimer of HMLH1 and HPMS1. J. Biol. Chem. 1999, 274, 32368–32375. [Google Scholar] [CrossRef] [PubMed]
- Kadyrov, F.A.; Dzantiev, L.; Constantin, N.; Modrich, P. Endonucleolytic function of MUTLα in human mismatch repair. Cell 2006, 126, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Kadyrov, F.A.; Holmes, S.F.; Arana, M.E.; Lukianova, O.A.; O’Donnell, M.; Kunkel, T.A.; Modrich, P. Saccharomyces Cerevisiae MUTLα is a mismatch repair endonuclease. J. Biol. Chem. 2007, 282, 37181–37190. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, T.A.; Erie, D.A. Eukaryotic mismatch repair in relation to DNA replication. Annu. Rev. Genet. 2015, 49, 291–313. [Google Scholar] [CrossRef] [PubMed]
- Longley, M.J.; Pierce, A.J.; Modrich, P. DNA polymerase δ is required for human mismatch repair in vitro. J. Biol. Chem. 1997, 272, 10917–10921. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.T.; Gordenin, D.A.; Resnick, M.A. The 3′→5′ exonucleases of DNA polymerases δ and epsilon and the 5′→3′ exonuclease EXO1 have major roles in postreplication mutation avoidance in Saccharomyces Cerevisiae. Mol. Cell Biol. 1999, 19, 2000–2007. [Google Scholar] [CrossRef] [PubMed]
- Dronkert, M.L.; Kanaar, R. Repair of DNA interstrand cross-links. Mutat. Res. 2001, 486, 217–247. [Google Scholar] [CrossRef]
- Clauson, C.; Schärer, O.D.; Niedernhofer, L. Advances in understanding the complex mechanisms of DNA interstrand cross-link repair. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Gueiderikh, A.; Rosselli, F.; Neto, J.B.C. A never-ending story: The steadily growing family of the FA and FA-like genes. Genet. Mol. Biol. 2017, 40, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Legerski, R.J. Repair of DNA interstrand cross-links during s phase of the mammalian cell cycle. Environ. Mol. Mutagen. 2010, 51, 540–551. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, N.; Zhang, L.; Li, R.; Fu, W.; Ma, K.; Li, X.; Wang, L.; Wang, J.; Zhang, H.; et al. Autophagy regulates chromatin ubiquitination in DNA damage response through elimination of SQSTM1/p62. Mol. Cell 2016, 63, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Yuan, N.; Wang, Z.; Cao, Y.; Fang, Y.; Li, X.; Xu, F.; Song, L.; Wang, J.; Zhang, H.; et al. Autophagy confers DNA damage repair pathways to protect the hematopoietic system from nuclear radiation injury. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Tougeron, D.; Huang, S.; Okamoto, K.; Sinicrope, F.A. Beclin 1 and UVRAG confer protection from radiation-induced DNA damage and maintain centrosome stability in colorectal cancer cells. PLoS ONE 2014, 9, e100819. [Google Scholar]
- Xu, F.; Li, X.; Yan, L.; Yuan, N.; Fang, Y.; Cao, Y.; Xu, L.; Zhang, X.; Ge, C.; An, N.; et al. Autophagy promotes the repair of radiation-induced DNA damage in bone marrow hematopoietic cells via enhanced STAT3 signaling. Radiat. Res. 2017, 187, 382–396. [Google Scholar] [CrossRef] [PubMed]
- Tsuruma, R.; Ohbayashi, N.; Kamitani, S.; Ikeda, O.; Sato, N.; Muromoto, R.; Sekine, Y.; Oritani, K.; Matsuda, T. Physical and functional interactions between STAT3 and KAP1. Oncogene 2008, 27, 3054–3059. [Google Scholar] [CrossRef] [PubMed]
- Ziv, Y.; Bielopolski, D.; Galanty, Y.; Lukas, C.; Taya, Y.; Schultz, D.C.; Lukas, J.; Bekker-Jensen, S.; Bartek, J.; Shiloh, Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat. Cell Biol. 2006, 8, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Cann, K.L.; Dellaire, G. Heterochromatin and the DNA damage response: The need to relax. Biochem. Cell Biol. 2011, 89, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, C.; Sun, L.; Wang, D.L.; Chen, L.; Huang, Z.; Yang, Q.; Gao, J.; Yang, X.B.; Chang, J.F.; et al. RAD6 promotes homologous recombination repair by activating the autophagy-mediated degradation of heterochromatin protein HP1. Mol. Cell Biol. 2015, 35, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Baldeyron, C.; Soria, G.; Roche, D.; Cook, A.J.; Almouzni, G. HP1α recruitment to DNA damage by p150CAF-1 promotes homologous recombination repair. J. Cell Biol. 2011, 193, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wang, D.; Wu, J.; Keller, J.; Ma, T.; Yu, X. RNF168 forms a functional complex with RAD6 during the DNA damage response. J. Cell Sci. 2013, 126, 2042–2051. [Google Scholar] [CrossRef] [PubMed]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Shaid, S.; Brandts, C.H.; Serve, H.; Dikic, I. Ubiquitination and selective autophagy. Cell. Death Differ. 2013, 20, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Nedelsky, N.B.; Todd, P.K.; Taylor, J.P. Autophagy and the ubiquitin-proteasome system: Collaborators in neuroprotection. Biochim. Biophys. Acta 2008, 1782, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Uckelmann, M.; Sixma, T.K. Histone ubiquitination in the DNA damage response. DNA Repair 2017, 56, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.Y.; Xu, N.; O’Prey, J.; Lao, L.Y.; Joshi, S.; Long, J.S.; O’Prey, M.; Croft, D.R.; Beaumatin, F.; Baudot, A.D.; et al. Loss of autophagy causes a synthetic lethal deficiency in DNA repair. Proc. Natl. Acad. Sci. USA 2015, 112, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Suh, Y.; Cuervo, A.M. Regulated degradation of CHK1 by chaperone-mediated autophagy in response to DNA damage. Nat. Commun. 2015, 6, 6823. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Fang, Y.; Yan, L.; Xu, L.; Zhang, S.; Cao, Y.; Zhang, X.; Xie, J.; Jiang, G.; Ge, C.; et al. Nuclear localization of Beclin 1 promotes radiation-induced DNA damage repair independent of autophagy. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Oh, S.; Li, D.; Ni, D.; Pirooz, S.D.; Lee, J.H.; Yang, S.; Lee, J.Y.; Ghozalli, I.; Costanzo, V.; et al. A dual role for UVRAG in maintaining chromosomal stability independent of autophagy. Dev. Cell 2012, 22, 1001–1016. [Google Scholar] [CrossRef] [PubMed]
- Perelman, B.; Dafni, N.; Naiman, T.; Eli, D.; Yaakov, M.; Feng, T.L.; Sinha, S.; Weber, G.; Khodaei, S.; Sancar, A.; et al. Molecular cloning of a novel human gene encoding a 63-kDa protein and its sublocalization within the 11Q13 locus. Genomics 1997, 41, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Feng, P.; Ku, B.; Oh, B.H.; Jung, J.U. UVRAG: A new player in autophagy and tumor cell growth. Autophagy 2007, 3, 69–71. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Ni, D.; Ghozalli, I.; Pirooz, S.D.; Ma, B.; Liang, C. Uvrag: At the crossroad of autophagy and genomic stability. Autophagy 2012, 8, 1392–1393. [Google Scholar] [CrossRef] [PubMed]
- Siggens, L.; Figg, N.; Bennett, M.; Foo, R. Nutrient deprivation regulates DNA damage repair in cardiomyocytes via loss of the base-excision repair enzyme OGG1. FASEB J. 2012, 26, 2117–2124. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Lin, P.; Zhang, W.; Tan, S.; Zhou, X.; Li, R.; Pu, Q.; Koff, J.L.; Dhasarathy, A.; Ma, F.; et al. DNA repair interacts with autophagy to regulate inflammatory responses to pulmonary hyperoxia. J. Immunol. 2017, 198, 2844–2853. [Google Scholar] [PubMed]
- SenGupta, T.; Torgersen, M.L.; Kassahun, H.; Vellai, T.; Simonsen, A.; Nilsen, H. Base excision repair AP endonucleases and mismatch repair ACT together to induce checkpoint-mediated autophagy. Nat. Commun. 2013, 4, 2674. [Google Scholar] [CrossRef] [PubMed]
- Sample, A.; He, Y.Y. Autophagy in UV damage response. Photochem. Photobiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.L.; Qiang, L.; Han, W.; Ming, M.; Viollet, B.; He, Y.Y. Role of AMPK in UVB-induced DNA damage repair and growth control. Oncogene 2013, 32, 2682–2689. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Zhao, B.; Ming, M.; Wang, N.; He, T.C.; Hwang, S.; Thorburn, A.; He, Y.Y. Regulation of cell proliferation and migration by p62 through stabilization of twist1. Proc. Natl. Acad. Sci. USA 2014, 111, 9241–9246. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; He, S.; Wang, Q.; Li, F.; Kwak, M.J.; Chen, S.; O’Connell, D.; Zhang, T.; Pirooz, S.D.; Jeon, Y.; et al. Autophagic UVRAG promotes UV-induced photolesion repair by activation of the CRL4(DDB2) E3 ligase. Mol. Cell 2016, 62, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Ge, R.; Liu, L.; Dai, W.; Zhang, W.; Yang, Y.; Wang, H.; Shi, Q.; Guo, S.; Yi, X.; Wang, G.; et al. Xeroderma pigmentosum group a promotes autophagy to facilitate cisplatin resistance in melanoma cells through the activation of PARP1. J. Investig. Dermatol. 2016, 136, 1219–1228. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.E.; Kassahun, H.; SenGupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef] [PubMed]
- Scheibye-Knudsen, M.; Fang, E.F.; Croteau, D.L.; Bohr, V.A. Contribution of defective mitophagy to the neurodegeneration in DNA repair-deficient disorders. Autophagy 2014, 10, 1468–1469. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Kinsella, T.J. BNIP3 is essential for mediating 6-thioguanine- and 5-fluorouracil-induced autophagy following DNA mismatch repair processing. Cell Res. 2010, 20, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Yan, T.; Schupp, J.E.; Seo, Y.; Kinsella, T.J. DNA mismatch repair initiates 6-thioguanine--induced autophagy through p53 activation in human tumor cells. Clin. Cancer Res. 2007, 13, 1315–1321. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Kinsella, T.J. A novel role for DNA mismatch repair and the autophagic processing of chemotherapy drugs in human tumor cells. Autophagy 2007, 3, 368–370. [Google Scholar] [CrossRef] [PubMed]
- Quinsay, M.N.; Thomas, R.L.; Lee, Y.; Gustafsson, A.B. BNIP3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy 2010, 6, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Sumpter, R.; Sirasanagandla, S.; Fernández, Á.; Wei, Y.; Dong, X.; Franco, L.; Zou, Z.; Marchal, C.; Lee, M.Y.; Clapp, D.W.; et al. Fanconi anemia proteins function in mitophagy and immunity. Cell 2016, 165, 867–881. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Youle, R.J. Pink1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 2012, 125, 795–799. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomes, L.R.; Menck, C.F.M.; Leandro, G.S. Autophagy Roles in the Modulation of DNA Repair Pathways. Int. J. Mol. Sci. 2017, 18, 2351. https://doi.org/10.3390/ijms18112351
Gomes LR, Menck CFM, Leandro GS. Autophagy Roles in the Modulation of DNA Repair Pathways. International Journal of Molecular Sciences. 2017; 18(11):2351. https://doi.org/10.3390/ijms18112351
Chicago/Turabian StyleGomes, Luciana R., Carlos F. M. Menck, and Giovana S. Leandro. 2017. "Autophagy Roles in the Modulation of DNA Repair Pathways" International Journal of Molecular Sciences 18, no. 11: 2351. https://doi.org/10.3390/ijms18112351
APA StyleGomes, L. R., Menck, C. F. M., & Leandro, G. S. (2017). Autophagy Roles in the Modulation of DNA Repair Pathways. International Journal of Molecular Sciences, 18(11), 2351. https://doi.org/10.3390/ijms18112351