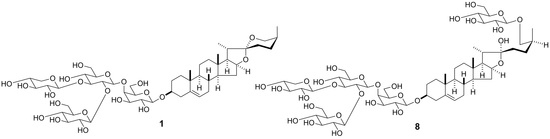
2.1. Structural Elucidation
Convallaria majalis (3.0 kg, dry weight) whole plants were extracted with hot MeOH. The MeOH extract was passed through a porous-polymer polystyrene resin (Diaion HP-20, Mitsubishi-Chemical, Tokyo, Japan) column. The MeOH-eluted and MeOH-H
2O (6:4)-eluted fractions were then subjected to silica gel and octadecylsilanized (ODS) silica gel column chromatography (CC) and reversed-phase preparative high-performance liquid chromatography (HPLC) to obtain compounds
1–
15 (
Figure 1). The structures of the known compounds
1–
3 and
7–
9 were identified as (25
S)-spirost-5-en-3β-yl
O-β-
d-glucopyranosyl-(1→2)-
O-[β-
d-xylopyranosyl-(1→3)]-
O-β-
d-glucopyranosyl-(1→4)-β-
d-galactopyranoside (
1) [
5], (25
S)-14α-hydroxyspirost-5-en-3β-yl O-β-
d-glucopyranosyl-(1→2)-
O-[β-
d-xylopyranosyl-(1→3)]-
O-β-
d-glucopyranosyl-(1→4)-β-
d-galactopyranoside (
2) [
5], (25
R)-spirost-5-en-3β-yl
O-α-L-rhamnopyranosyl-(1→4)-β-
d-glucopyranoside (
3) [
6], (25
S)-26-[(β-
d-glucopyranosyl)oxy]-22α-hydroxyfurost-5-en-3β-ol (
7) [
7], (25
S)-26-[(β-
d-glucopyranosyl)oxy]-22α-hydroxyfurost-5-en-3β-yl
O-β-
d-glucopyranosyl-(1→2)-
O-[β-
d-xylopyranosyl-(1→3)]-
O-β-
d-glucopyranosyl-(1→4)-β-
d-galactopyranoside (
8) [
8], and (25
S)-26-[(β-
d-glucopyranosyl)oxy]-14α,22α-hydroxyfurost-5-en-3β-yl
O-β-
d-glucopyranosyl-(1→2)-
O-[β-
d-xylopyranosyl-(1→3)]-
O-β-
d-glucopyranosyl-(1→4)-β-
d-galactopyranoside (
9) [
9], respectively.
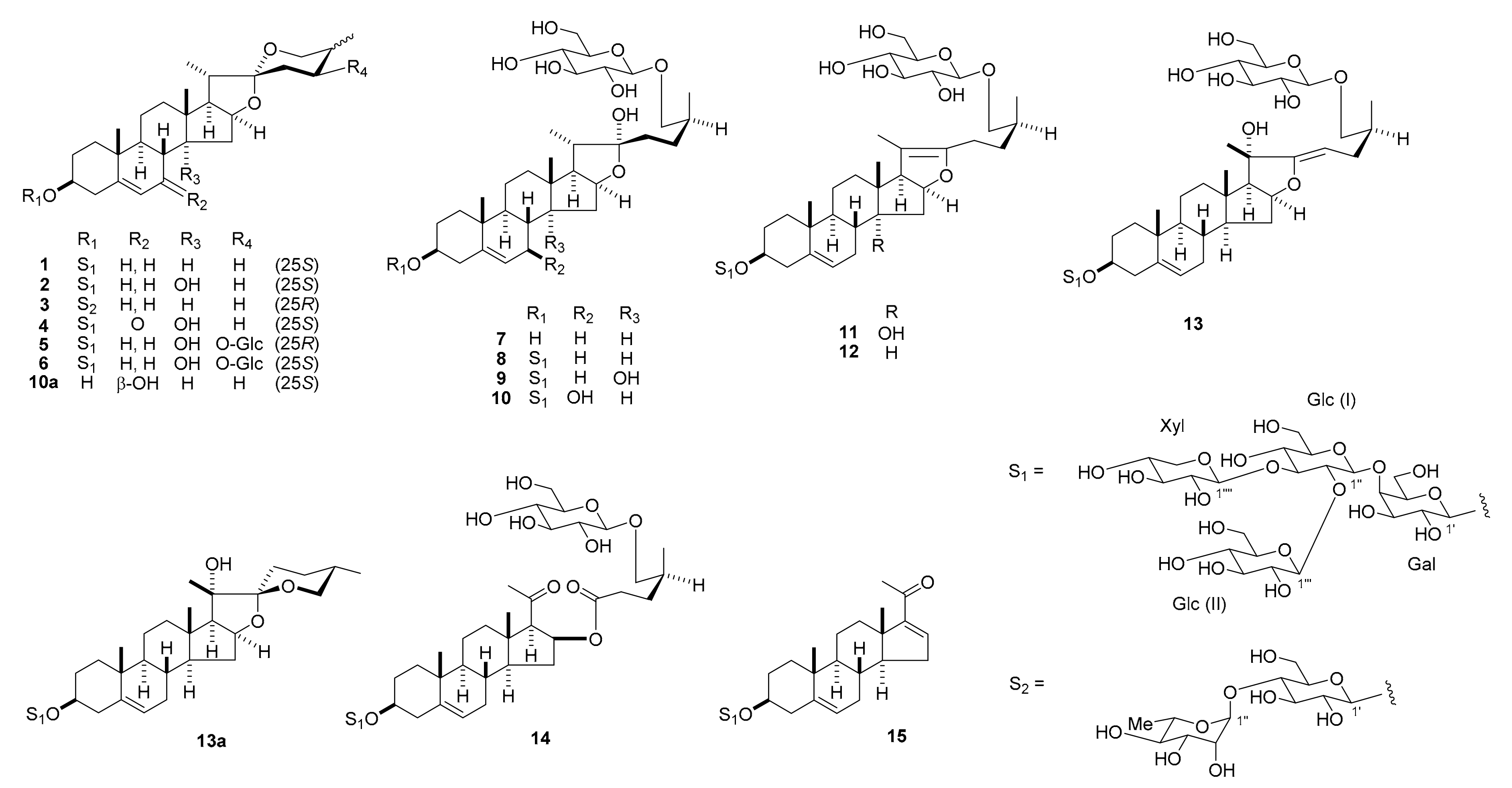
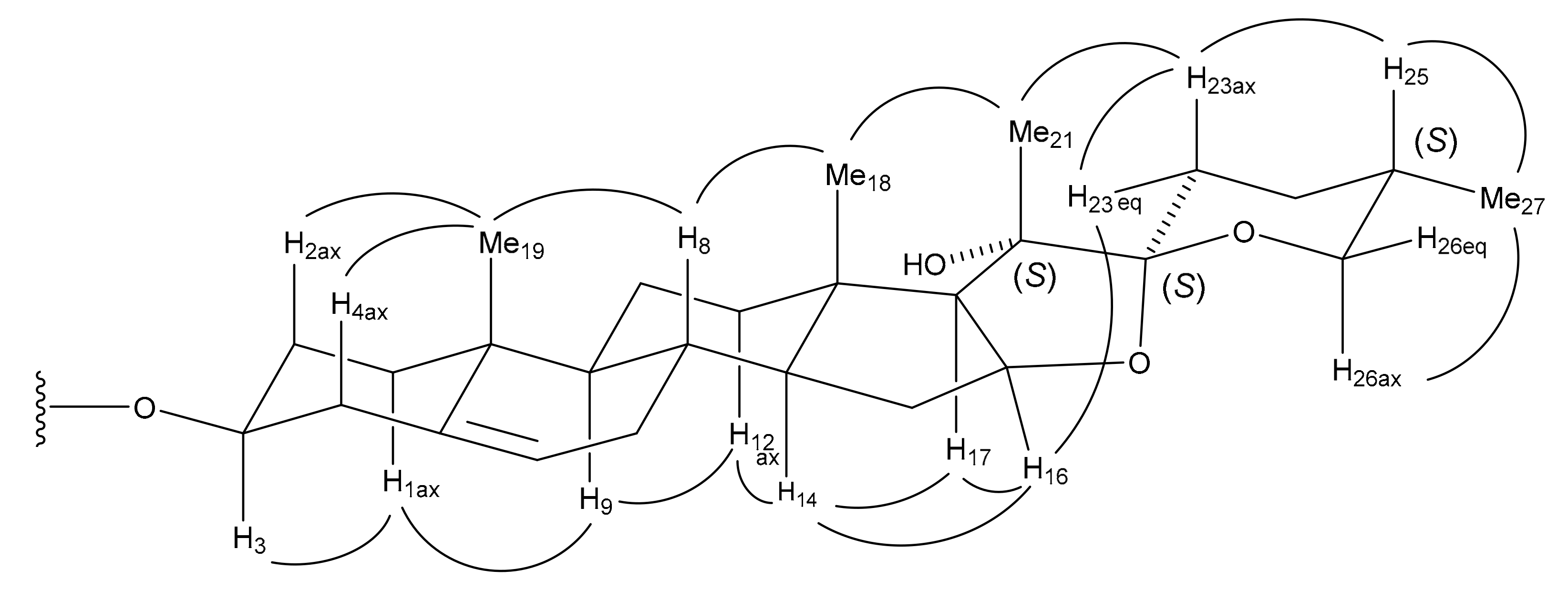
Compound
4 was obtained as an amorphous solid, and its molecular formula was identified as C
50H
78O
24, based on data from high-resolution electrospray ionization time-of-flight mass spectrometry (HR-ESI-TOF-MS;
m/
z 1085.4773 [M + Na]
+, calcd. 1085.4781) and
13C NMR (50 carbon signals) spectrum. The
1H and
13C NMR spectral features of
4 were closely related to those of
2, showing the following signals: four steroid methyl groups at δ
H 1.21 (d,
J = 7.0 Hz, Me-21), 1.07 (d,
J = 7.0 Hz, Me-27), 1.04 (s, Me-18), and 0.99 (s, Me-19), in addition to δ
C 20.4 (C-18), 17.1 (C-19), 16.3 (C-27), and 15.2 (C-21); an olefinic group at δ
H 5.75 (s, H-6) and δ
C 166.6 (C-5) and 126.9 (C-6); oxygenated protons and carbons at δ
H 3.86 (m, H-3) and δ
C 78.1 (C-3), δ
H 5.06 (dd,
J = 12.7, 9.0 Hz, H-16) and δ
C 82.2 (C-16), and δ
H 4.02 (dd,
J = 10.9, 2.5 Hz, H-26ax) and 3.31 (br d,
J = 10.9 Hz, H-26eq) and δ
C 65.0 (C-26); an oxygenated quaternary carbon at δ
C 110.0 (C-22); and four anomeric protons and carbons at δ
H 5.59 (d,
J = 7.5 Hz), 5.25 (d,
J = 7.6 Hz), 5.19 (d,
J = 7.5 Hz), and 4.83 (d,
J = 7.6 Hz) and δ
C 105.2, 105.0, 104.9, and 102.8 (
Table 1). In addition, the IR and
13C NMR spectra of
4 suggest the presence of a conjugated carbonyl group (ν
max 1650 cm
−1; δ
C 200.6) (
Table 2). In the heteronuclear multiple-bond correlation (HMBC) spectrum of
4, long-range correlations observed from H-6 at δ
H 5.75, H-9 at δ
H 2.41, and H-8 at δ
H 2.74 to C-7 at δ
C 200.6 indicated that the carbonyl group was located at C-7 of the aglycone. Nuclear Overhauser effect (NOE) correlations were found between the signals of H-8 at δ
H 2.74 and Me-18 at δ
H 1.04/Me-19 at δ
H 0.99, between the signals of H-12ax at δ
H 1.41 and H-9 at δ
H 2.41/H-17 at δ
H 2.72, between the signals of H-17 and Me-21, between the signals of H-20 at δ
H 2.02 and Me-18/H-23ax at δ
H 1.93, and between the signals of Me-27 at δ
H 1.07 and H-23ax at δ
H 1.93. These correlations in the NOE spectroscopy (NOESY) spectrum of
4 were consistent with the B/C trans, C/D trans, and D/E cis ring fusions, as well as the 14α, 20α, 22α, and 25
S configurations. The linkage of lycotetrose,
O-β-
d-glucopyranosyl-(1→2)-
O-[β-
d-xylopyranosyl-(1→3)]-
O-β-
d-glucopyranosyl-(1→4)-β-
d-galactopyranose, at C-3 of the aglycone, was ascertained by acid hydrolysis of
4 with 0.5 M HCl, obtaining
d-galactose,
d-glucose, and
d-xylose. Additionally, long-range correlations from the anomeric proton (H-1′′′) of Glc (II) at δ
H 5.59 to C-2′′ of Glc (I) at δ
C 81.3, from H-1′′′′ of Xyl at δ
H 5.25 to C-3′′ of Glc (I) at δ
C 86.7, from H-1′′ of Glc (I) at δ
H 5.19 to C-4′ of Gal at δ
C 79.8, and from H-1′ of Gal at δ
H 4.83 to C-3 of the aglycone at δ
C 78.1 in the HMBC spectrum of 4 were also observed. Thus,
4 was labeled (25
S)-3β-[(
O-β-
d-glucopyranosyl-(1→2)-
O-[β-
d-xylopyranosyl-(1→3)]-
O-β-
d-glucopyranosyl-(1→4) β-
d-galactopyranosyl)oxy]-14α-hydroxyspirost-5-en-7-one.
The 1H and 13C NMR spectral data of 5 (C56H90O29) suggest that 5 is a spirostanol glycoside resembling 2 in structure, with a lycotetrose unit at C-3 and a hydroxy group at C-14. However, the molecular formula of 5 was in excess of 2 by C6H10O6, which corresponded to a hexyloxy group. When the 1H and 13C NMR spectra of 5 were compared with those of 2, the signals for the C-24 methylene protons at δH 2.15 and 1.37 (each, m) and carbon at δC 26.2 were displaced by an oxymethine proton at δH 4.83 (ddd, J = 10.9, 5.5, 5.5 Hz) and carbon at δC 72.8. Acid hydrolysis of 5 gave d-galactose, d-glucose, and d-xylose as the sugar moieties. Analysis of the 1H-1H correlation spectroscopy (COSY) and heteronuclear multiple quantum coherence (HMQC) spectra allowed the hexosyl unit attached to C-24 to be assigned β-d-glucopyranosyl, and an HMBC correlation was observed from the anomeric proton at δH 5.04 (d, J = 7.7 Hz) to C-24 carbon at δC 72.8. In the NOESY spectrum of 5, NOE correlations between the signals of H-24 at δH 4.83 and H-25 at δH 2.24/H-26ax at δH 3.89, and J values of 3JH-24, H-23ax = 10.9 Hz, 3JH-24, H-23eq = 5.5 Hz, and 3JH-24, H-25 = 5.5 Hz provided evidence for the 24S and 25R configurations. Thus, the structure of 5 was determined to be (24S,25R)-24-[(β-d-glucopyranosyl)oxy]-14α-hydroxyspirost-5-en-3β-yl O-β-d-glucopyranosyl-(1→2)-O-[β-d-xylopyranosyl-(1→3)]-O-β-d-glucopyranosyl-(1→4)-β-d-galactopyranoside.
The molecular formula of 6 (C56H90O29) was the same as that of 5. The 1H and 13C NMR spectra of 6 showed close similarity with those of 5, suggestive of a stereoisomer with respect to the C-25 configuration. In the NOESY spectrum of 6, the NOE correlations between the signals of H-23ax at δH 2.00 and H-20 at δH 2.07/H-25 at δH 1.91, between the signals of H-26ax at δH 3.58 and H-16 at δH 5.05/H-24 at δH 4.06/Me-27 at δH 1.13, between the signals of Me-27 and H-24, and J values of 3JH-24, H-23ax = 12.4 Hz, 3JH-24, H-23eq = 5.3 Hz, and 3JH-25, H-26ax = 11.8 Hz confirmed the 24S and 25S configurations. The structure of 6 was characterized as (24S,25S)-24-[(β-d-glucopyranosyl)oxy]-14α-hydroxyspirost-5-en-3β-yl O-β-d-glucopyranosyl-(1→2)-O-[β-d-xylopyranosyl-(1→3)]-O-β-d-glucopyranosyl-(1→4)-β-d-galactopyranoside.
The hemi-acetal carbon signal at δC 110.6 and the positive color reaction in Ehrlich’s test suggest that 10 (C56H92O29) may be a 22-hydroxyfurostanol glycoside. The 1H and 13C NMR spectra of 10 were similar to those of 8; however, the molecular formula of 10 had one extra oxygen atom compared to that of 8, and a significant difference was observed in the 13C NMR signals from the B-ring portion. Enzymatic hydrolysis of 10 with β-d-glucosidase yielded the corresponding spirostanol saponin 10a. In the 1H-1H COSY spectrum of 10a, a proton signal of a hydroxy group at δH 5.73 (br d, J = 7.8 Hz), which disappeared following the addition of HCl vapor, showed a correlation peak with the C-7 oxymethine proton at δH 4.01 (m), indicating the presence of a hydroxy group at C-7. The NOE correlations between the signals of H-7 at δH 4.00 and H-9 at δH 1.05/H-12ax at δH 1.68/H-14 at δH 1.32 in the NOESY spectrum of 10a confirmed that the C-7 hydroxy group was β-oriented. Furthermore, the NOE correlations between the signals of Me-27 at δH 1.07 and H-23ax at δH 1.90 and between the signals of H-25 at δH 1.57 and H-24ax at δH 2.14 confirmed the 25S configuration. Thus, 10a was assigned as (25S)-7β-hydroxyspirost-5-en-3β-yl O-β-d-glucopyranosyl-(1→2)-O-[β-d-xylopyranosyl-(1→3)]-O-β-d-glucopyranosyl-(1→4)-β-d-galactopyranoside. The linkage of a β-d-glucopyranosyl group (Glc (III)) to the C-26 hydroxy group of the aglycone of 10 was ascertained by an HMBC correlation from H-1′′′′′ of Glc (III) at δH 4.80 (d, J = 7.7 Hz) to C-26 of the aglycone at δC 75.4. The NOE correlations between the signals of H-20 at δH 2.24 and H2-23 at δH 2.08 and 1.96 confirmed the C-22α configuration. Therefore, 10 was identified as (25S)-26-[(β-d-glucopyranosyl)oxy]-7β,22α-dihydroxyfurost-5-en-3β-yl O-β-d-glucopyranosyl-(1→2)-O-[β-d-xylopyranosyl-(1→3)]-O-β-d-glucopyranosyl-(1→4)-β-d-galactopyranoside.
The 1H and 13C NMR spectra of 11 (C56H90O28) showed similar features with those of 9; however, the molecular formula of 11 was smaller than that of 9 by H2O, and the signals assignable to H-17 and Me-21 were observed at δH 3.36 as a doublet (J = 9.7 Hz) and δH 1.67 as a singlet, respectively. Furthermore, signals for a pair of olefinic carbon were detected at δC 152.2 and 103.9, in addition to those attributable to C-5 and C-6. These data implied that 11 was the corresponding Δ20(22)-pseudo-furostanol glycoside of 9. This was confirmed by the fact that the peracetate (11a) of 11 was the same as the product obtained by treating 9 with Ac2O in pyridine at 110 °C for 3 h. Enzymatic hydrolysis of 11 with β-d-glucosidase gave the corresponding spirostanol saponin 2 and d-glucose. Accordingly, the structure of 11 was assigned (25S)-26-[(β-d-glucopyranosyl)oxy]-14α-hydroxyfurosta-5,20(22)-dien-3β-yl O-β-d-glucopyranosyl-(1→2)-O-[β-d-xylopyranosyl-(1→3)]-O-β-d-glucopyranosyl-(1→4)-β-d-galactopyranoside.
Compound 12 (C56H90O27) appeared to be the corresponding Δ20(22)-pseudo-furostanol glycoside of 8 (C56H92O28), based on the characteristic proton signals of the H-17 doublet at δH 2.44 (J = 9.9 Hz) and Me-21 singlet at δH 1.62, as well as the olefinic carbon signals at δC 152.4 and 103.5. Enzymatic hydrolysis of 12 with β-d-glucosidase gave the corresponding spirostanol saponin 1 and D-glucose. Thus, the structure of 12 was characterized as (25S)-26-[(β-d-glucopyranosyl)oxy]-furosta-5,20(22)-dien-3β-yl O-β-d-glucopyranosyl-(1→2)-O-[β-d-xylopyranosyl-(1→3)]-O-β-d-glucopyranosyl-(1→4)-β-d-galactopyranoside.
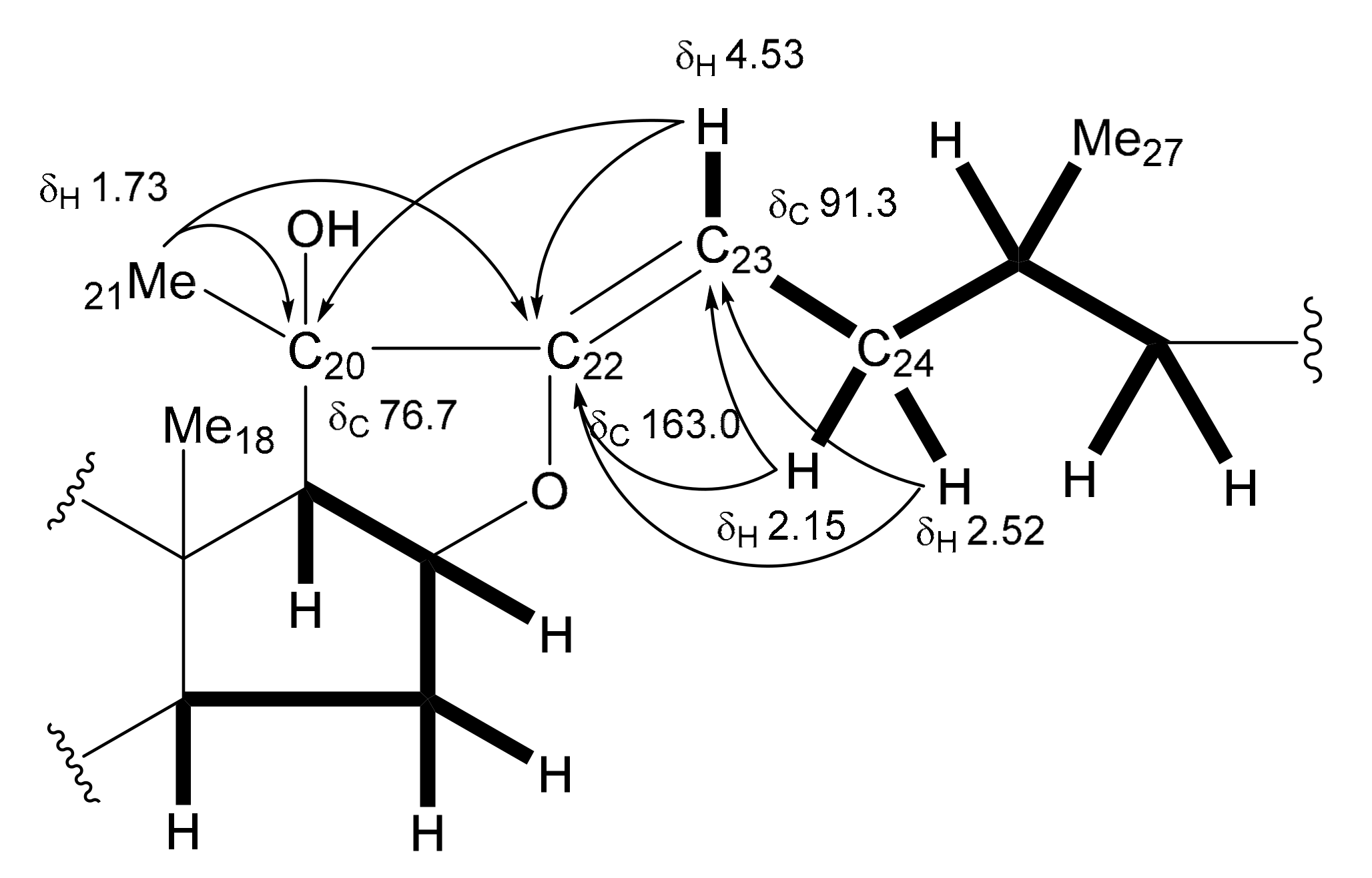
The
1H and
13C NMR spectral data for
13 (C
56H
90O
28) suggest that it is a furostanol glycoside, having a structure similar to that of
8. However, significant differences were recognized in the signals from the ring E and the side chain moiety, where a tertiary hydroxy group [δ
C 76.7 (C)] and an oxygen-bearing trisubstituted olefinic group [δ
C 163.0 (-O-
C(C)=CH-); δ
C 91.3 (-O-C(C)=
CH-)/δ
H 4.53 (br d,
J = 13.8 Hz)] were supposed to be located. The deshielded methyl singlet signal at δ
H 1.73 assignable to Me-21 and the olefinic proton at δ
H 4.53 exhibited long-range correlations with the quaternary carbon at δ
C 76.7 and the oxygen-bearing olefinic carbon at δ
C 163.0 in the HMBC spectrum of
13. The olefinic proton at δ
H 4.53 was shown to be coupled with the methylene protons at δ
H 2.52 and 2.15 (each, m) attributable to H
2-24 in the
1H-
1H COSY spectrum. These correlations allowed the tertiary hydroxy group and trisubstituted olefinic group to be placed at C-20 and between C-22 and C-23, respectively. The other correlations supporting the partial structure are depicted in
Figure 2. The geometry of the olefinic group was determined to be Z by an NOE correlation between the signals of Me-21 at δ
H 1.73 and H-23 at δ
H 4.53. Enzymatic hydrolysis of
13 with β-
d-glucosidase yielded the spirostanol saponin
13a and D-glucose. The NOE correlations between the signals of Me-21 at δ
H 1.73 and Me-18 at δ
H 1.19/H-23ax at δ
H 1.79, between the signals of H-23eq at δ
H 2.37 and H-16 at δ
H 5.06, and between the signals of H-25 at δ
H 1.69 and H-23ax/Me-27 at δ
H 0.75 in the NOESY spectrum of
13a, in addition to the large
J value of the H-26ax proton (
3JH-26ax, H-25 = 10.0 Hz), were indicative of the 20
S, 22
S, and 25
S configurations (
Figure 3). Thus, spirostanol saponin
13a was identified as (20
S,22
S,25
S)-20-hydroxyspirost-5-en-3β-yl
O-β-
d-glucopyranosyl-(1→2)-
O-[β-
d-xylopyranosyl-(1→3)]-
O-β-
d-glucopyranosyl-(1→4)-β-
d-galactopyranoside. An HMBC correlation from H-1′′′′′ of Glc (III) at δ
H 4.85 (d,
J = 7.7 Hz) to C-26 of the aglycone at δ
C 75.3 in the HMBC spectrum of
13 confirmed that a β-
d-glucopyranosyl group was attached to C-26. Therefore, the structure of
13 was elucidated to be (20
S,22
Z,25
S)-26-[(β-
d-glucopyranosyl)oxy]-20-hydroxyfurosta-5,22-dien-3β-yl
O-β-
d-glucopyranosyl-(1→2)-
O-[β-
d-xylopyranosyl-(1→3)]-
O-β-
d-glucopyranosyl-(1→4)-β-
d-galactopyranoside.
The 1H and 13C NMR data for 14 (C56H90O29) were essentially analogous to those of 12, except for the lack of signals for the C-20(22)-tetrasubstituted olefinic group at δC 103.5 and 152.4. Instead, signals for a keto carbonyl carbon at δC 205.5 and an ester carbonyl carbon at δC 173.2 were newly observed in the 13C NMR spectrum of 14. All other signals appeared at almost the same positions between the two glycosides. In the HMBC spectrum of 14, the H-17 at δH 2.48 (d, J = 7.6 Hz) and Me-21 protons at δH 2.13 (s) showed long-range correlations with the keto carbonyl carbon at δC 205.5, which was assigned to C-20. Long-range correlations from the H-16 at δH 5.66 (m) and H2-23 protons at δH 1.84 (m) and 1.47 (m) to the ester carbonyl carbon at δC 173.2 resulted in the assignment of the ester carbonyl carbon to C-22. These data suggest that 14 was formed from 12 through the oxidative cleavage of the C-20(22) double bond. This was confirmed by the fact that the peracetate (14a) of 14 was identical to the product obtained by treating 12 with Ac2O in pyridine at room temperature for 12 h and then with CrO3 in AcOH. Accordingly, the structure of 14 was determined to be 16β-[[(4S)-5-(β-d-glucopyranosyloxy)-4-methyl-1-oxo-pentyl]oxy]-3β-[(O-β-d-glucopyranosyl-(1→2)-O-[β-d-xylopyranosyl-(1→3)]-O-β-d-glucopyranosyl-(1→4)-β-d-galactopyranosyl)oxy]-pregn-5-en-20-one.
The data suggest that
15 (C
44H
68O
21) is a pregnane glycoside. Its
1H NMR spectrum showed signals for two angular methyl groups at δ
H 0.90 (s, Me-18) and 0.86 (s, Me-19), a methyl group of an acetyl moiety at δ
H 2.22 (s, Me-21), two olefinic protons at δ
H 6.57 (br s, H-16) and 5.29 (br d,
J = 5.4 Hz, H-6), and four anomeric protons at δ
H 5.58 (d,
J = 7.8 Hz, H-1′′′), 5.23 (d,
J = 7.8 Hz, H-1′′′′), 5.18 (d,
J = 7.8 Hz, H-1′′), and 4.88 (d,
J = 7.8 Hz, H-1′). The existence of an α,β-unsaturated carbonyl group was verified by the IR (1660 cm
−1), UV [239 nm (logε 3.81)], and
13C NMR [δ
C 196.1 (C=O), 155.0 (C), and 144.5 (CH)] spectra. These spectral data and comparison with those of previously reported compounds identified the aglycone of
15 as 3β-hydroxypregna-5,16-dien-20-one [
10]. Acid hydrolysis of
15 and analysis of HMBC correlations provided evidence that a lycotetrose was attached to C-3 of the aglycone. Thus, the structure of
15 was assigned 3β-[(
O-β-
d-glucopyranosyl-(1→2)-
O-[β-
d-xylopyranosyl-(1→3)]-
O-β-
d-glucopyranosyl-(1→4)-β-
d-galactopyranosyl)oxy]-pregna-5,16-dien-20-one.
2.2. Cytotoxic Activity
The isolated compounds
1–
15 were evaluated for their cytotoxic activity against HL-60 human promyelocytic leukemia cells, A549 human lung adenocarcinoma cells, HSC-4, and HSC-2 human oral squamous cell carcinoma cells. Etoposide, cisplatin, and doxorubicin were used as positive controls. Compound
1 showed cytotoxic activity against the four tumor cell lines with IC
50 values ranging from 0.96 ± 0.01 to 3.15 ± 0.43 μM. Compound
8, which is the corresponding furostanol glycoside of
1, was only cytotoxic to the adherent cell lines of A549, HSC-4, and HSC-2 cells, with IC
50 values of 2.97 ± 0.06 μM, 11.04 ± 0.25 μM, and 8.25 ± 0.20 μM, respectively (
Table 3). As we previously reported [
11,
12], the cytotoxicity of
1 compared to those of
2,
4,
5, and
6 and of
8 compared to those of
9 and
10 indicated that the introduction of polar substituents to the steroidal nuclei resulted in reduced the cytotoxicity. Compound
1 was cytotoxic to tumor cells, whereas
3, having the diglycoside did not show cytotoxic activity. These results implied that not only the structures of the aglycone moiety but also the sugar sequences in the steroidal glycosides considerably contributed to the appearance of cytotoxicity. In previous study, furostanol and pseudo-furostanol glycosides were generally inactive, and on the other hand, 3-
O-lycotetroside showed significant cytotoxic activities [
13]. In this study, correlation between the structure and cytotoxicity of steroidal glycosides have provided similar results. Isolated compounds were evaluated for their cytotoxic activity against TIG-3 normal human diploid fibroblasts. As a result, unfortunately,
1 and
8 gave IC
50 values of 1.30 ± 0.72 and 3.27 ± 0.13 µM, respectively. However, cisplatin, a clinically used anticancer agent, was also toxic to healthy cells TIG-3 with IC
50 value of 7.64 ± 0.08 μM in vitro evaluation.
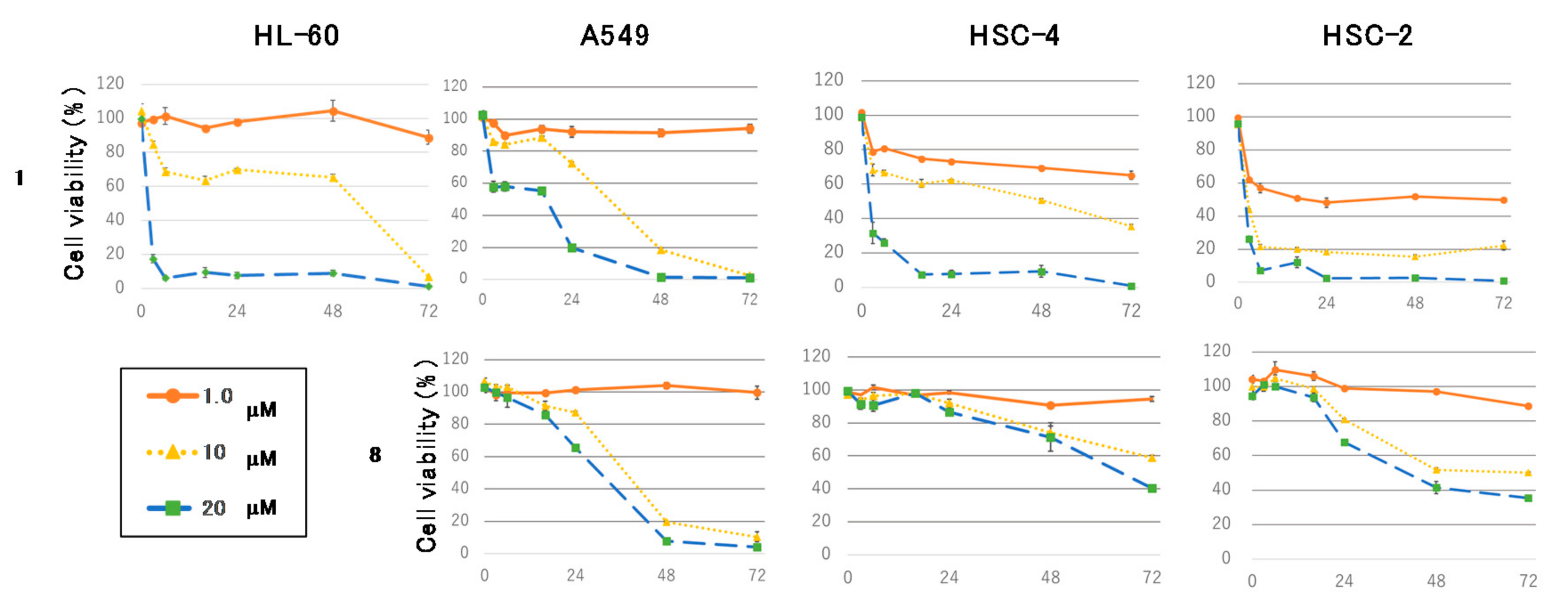
Figure 4 shows the time course of the antiproliferative effects of
1 or
8 at 1.0, 10, and 20 μM on HL-60, A549, HSC-4, and HSC-2 cells. Compound
1 decreased tumor cells viability in a dose-dependent manner within 16 h. In contrast,
8 reduced adherent cells viability in a time-dependent manner. HL-60, A549, HSC-4, and HSC-2 cells were exposed to
1 or
8 and then stained with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI). Treatment of the cells with
1 led to necrotic cell death in the four cell lines, whereas the cells exposed to
8 morphologically displayed nuclear chromatin condensation and nuclear disassembly, implying that 8 induced apoptosis in the adherent cells (
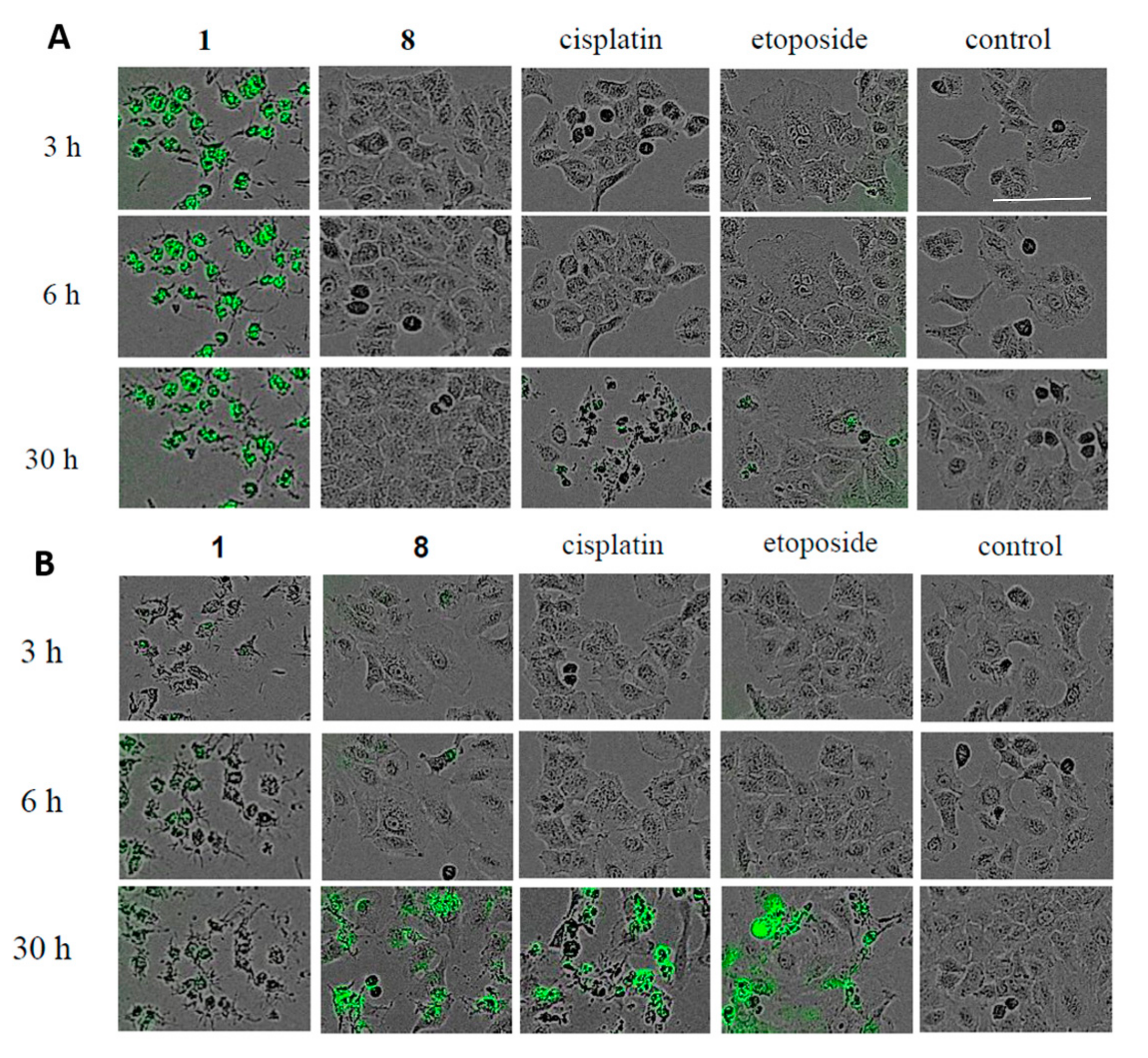
Supplementary Material 1). Next, the activation of caspase-3/7 in A549 cells treated with
1 or
8 was evaluated using the Cell Player
TM caspase-3/7 Apoptosis Assay kit. When A549 cells were treated with 20 μM of
8 for 30 h, a significant green fluorescence was detected (
Figure 5A), indicating that
8 induced apoptotic cell death in A549 cells through the activation of caspase-3/7. Alternatively, phase-contrast and fluorescent images of the A549 cells treated with 20 μM of
1 followed by staining with oxazole yellow dimer (YOYO-1), which stains the nuclear DNA in permeabilized cells [
14], showed that the cell membrane became permeable at an early stage of the necrotic cell death process (with green fluorescence) (
Figure 5B). Finally, the cell cycle distribution of A549 cells treated with
8 for 30 h was analyzed using flow cytometry. Compound 8 increased the sub-G1 cell population from 4.07% to 14.10% in A549 cells (
Table 4). These results suggest that
8 induced apoptotic death in A549 cells through caspase-3/7 activity in a time-dependent manner, whereas
1 induced necrotic cell death in A549 cells.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}