Aptamer Bioinformatics


Abstract
:
1. Introduction
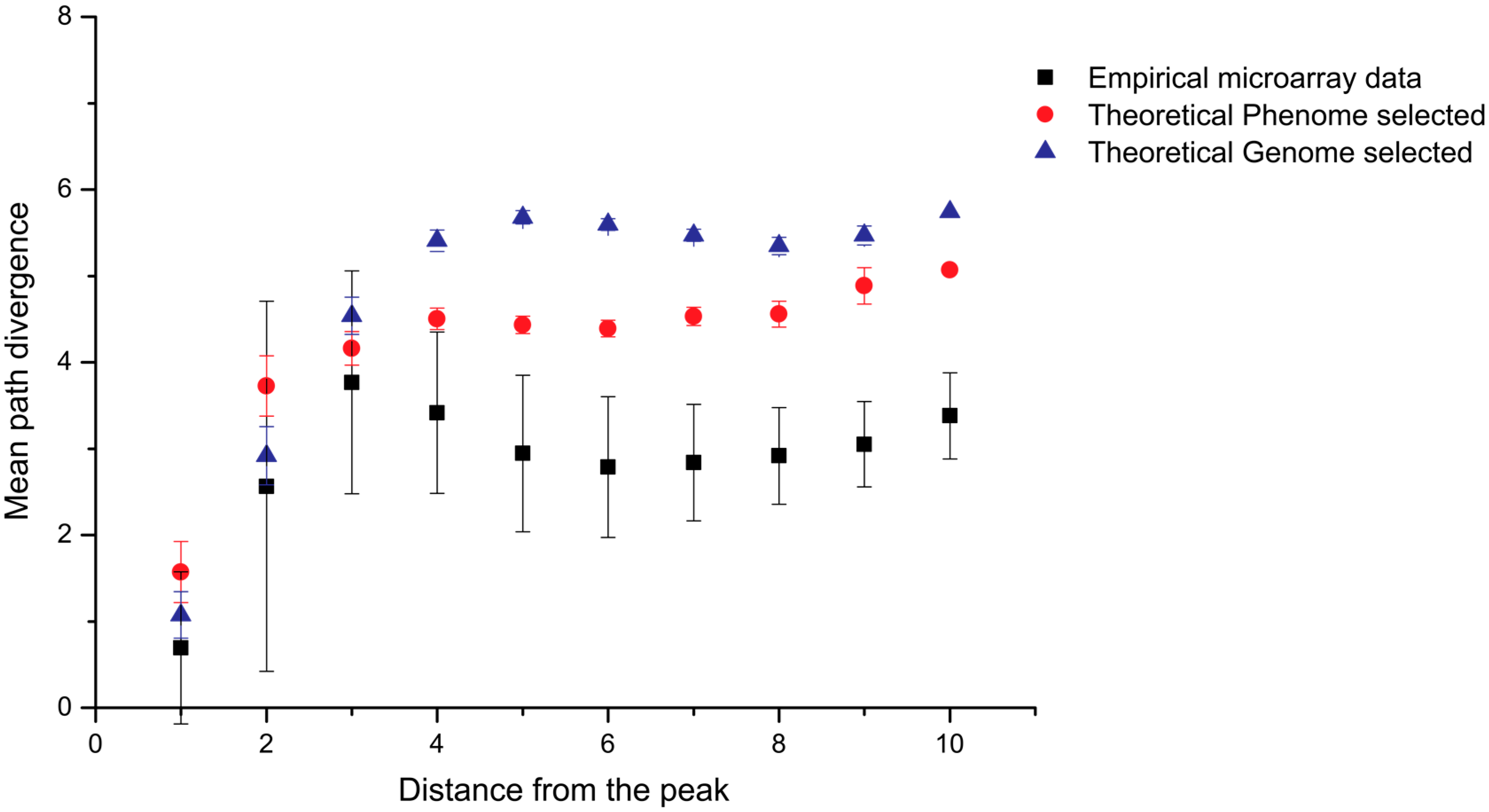
2. Simulation of Aptamer Selection
3. Aptamer Selection by Molecular Dynamics
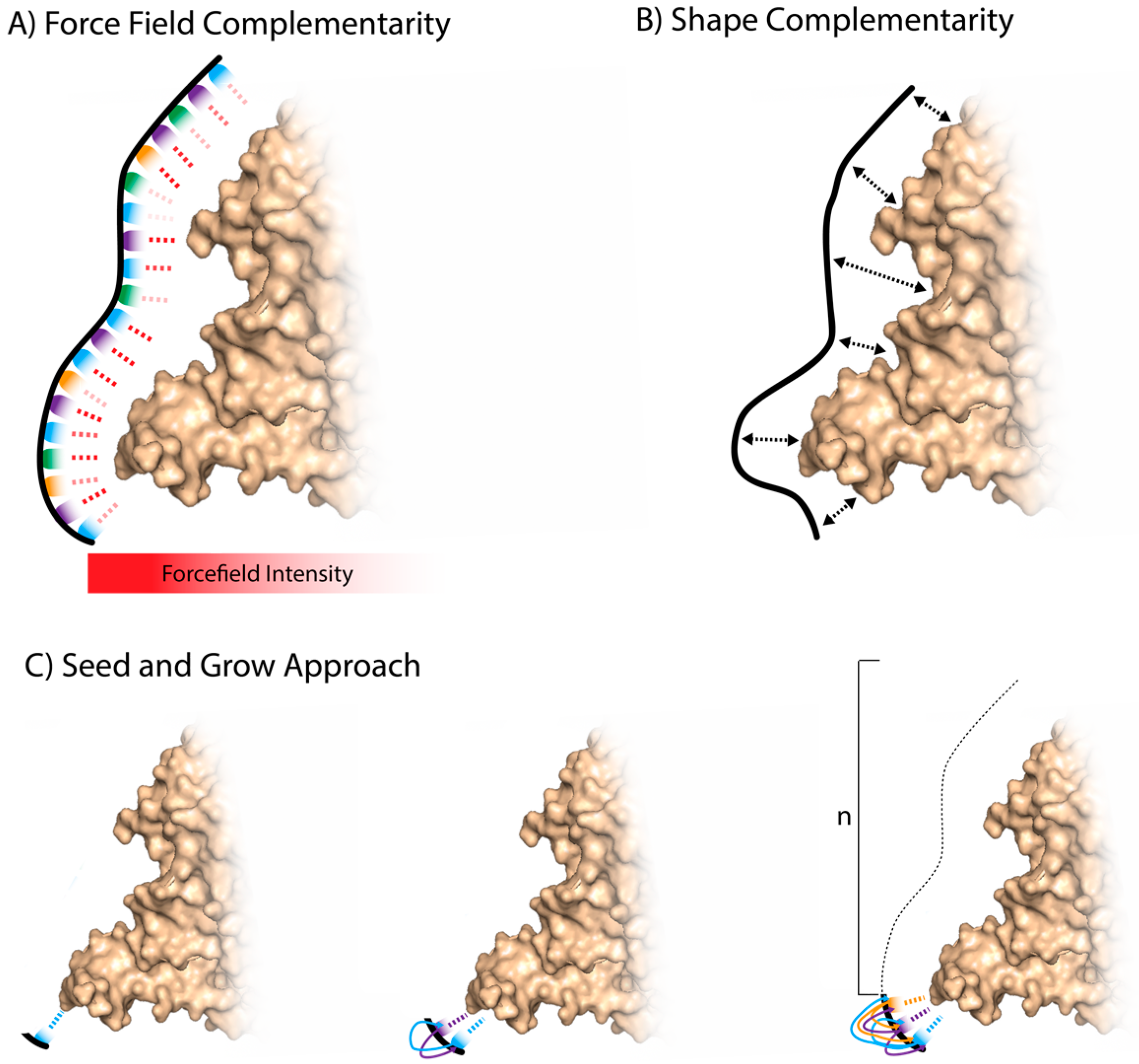
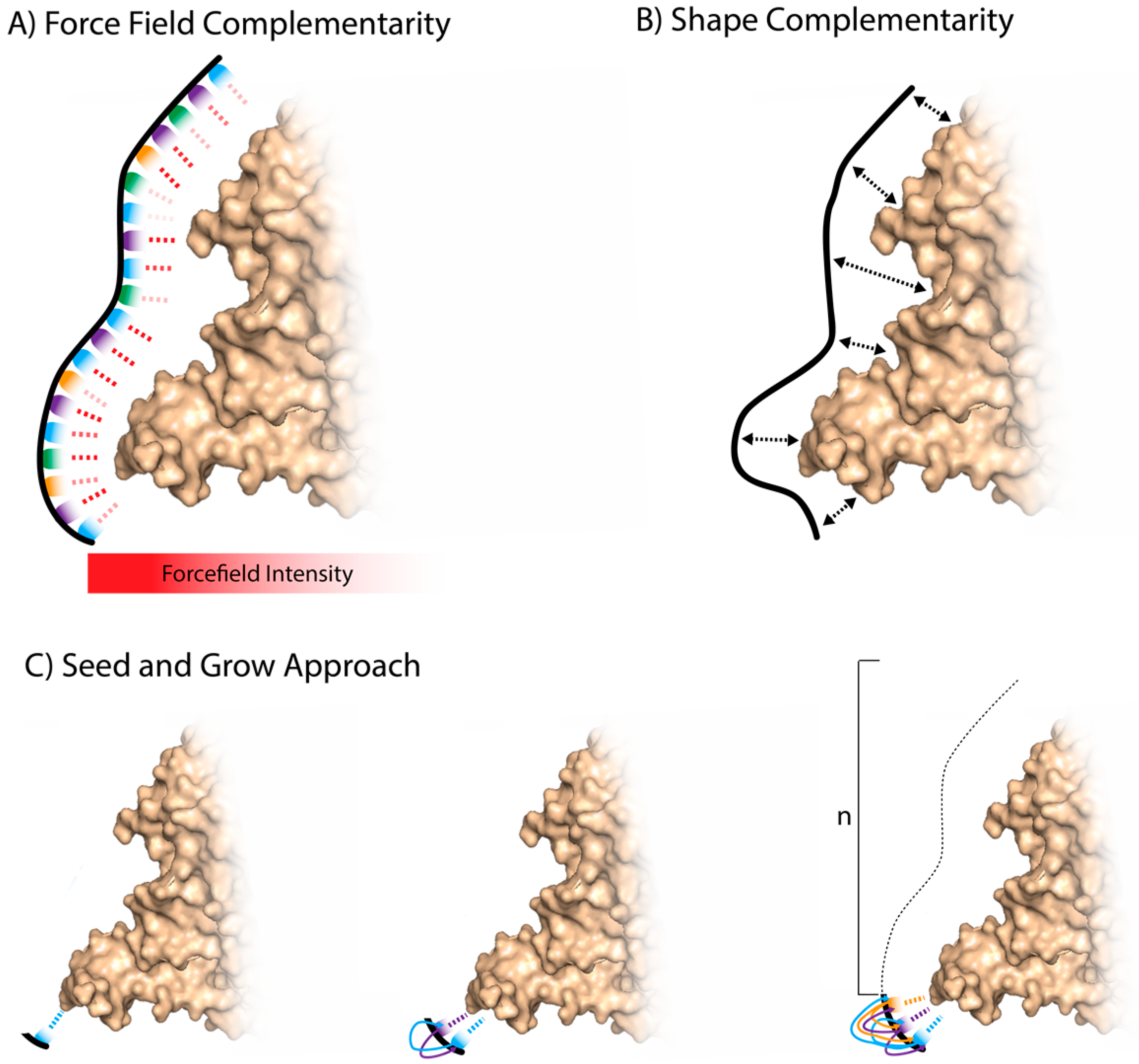
3.1. Whole Aptamer Docking
3.2. Fragment-Based Aptamer Design and Docking
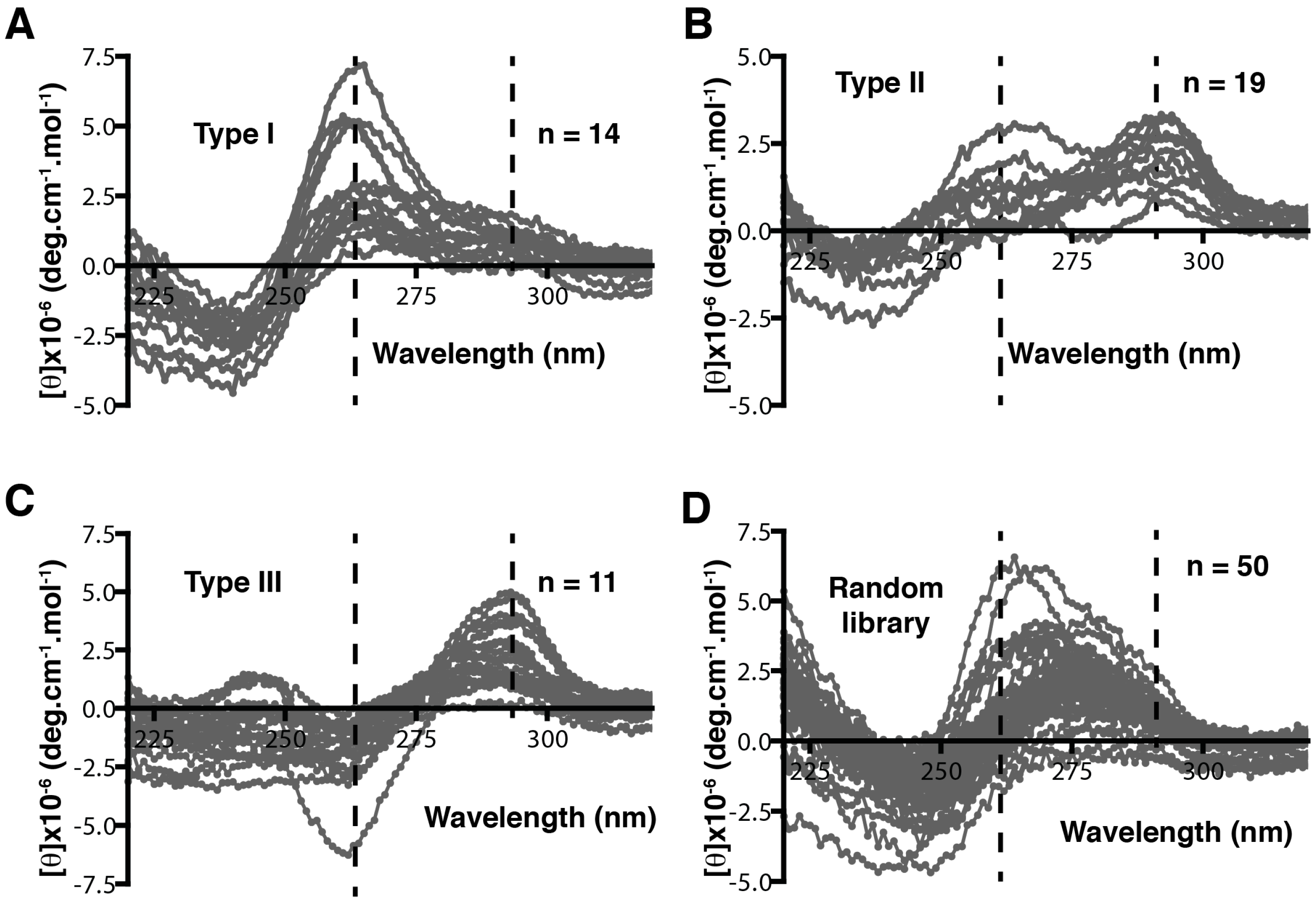
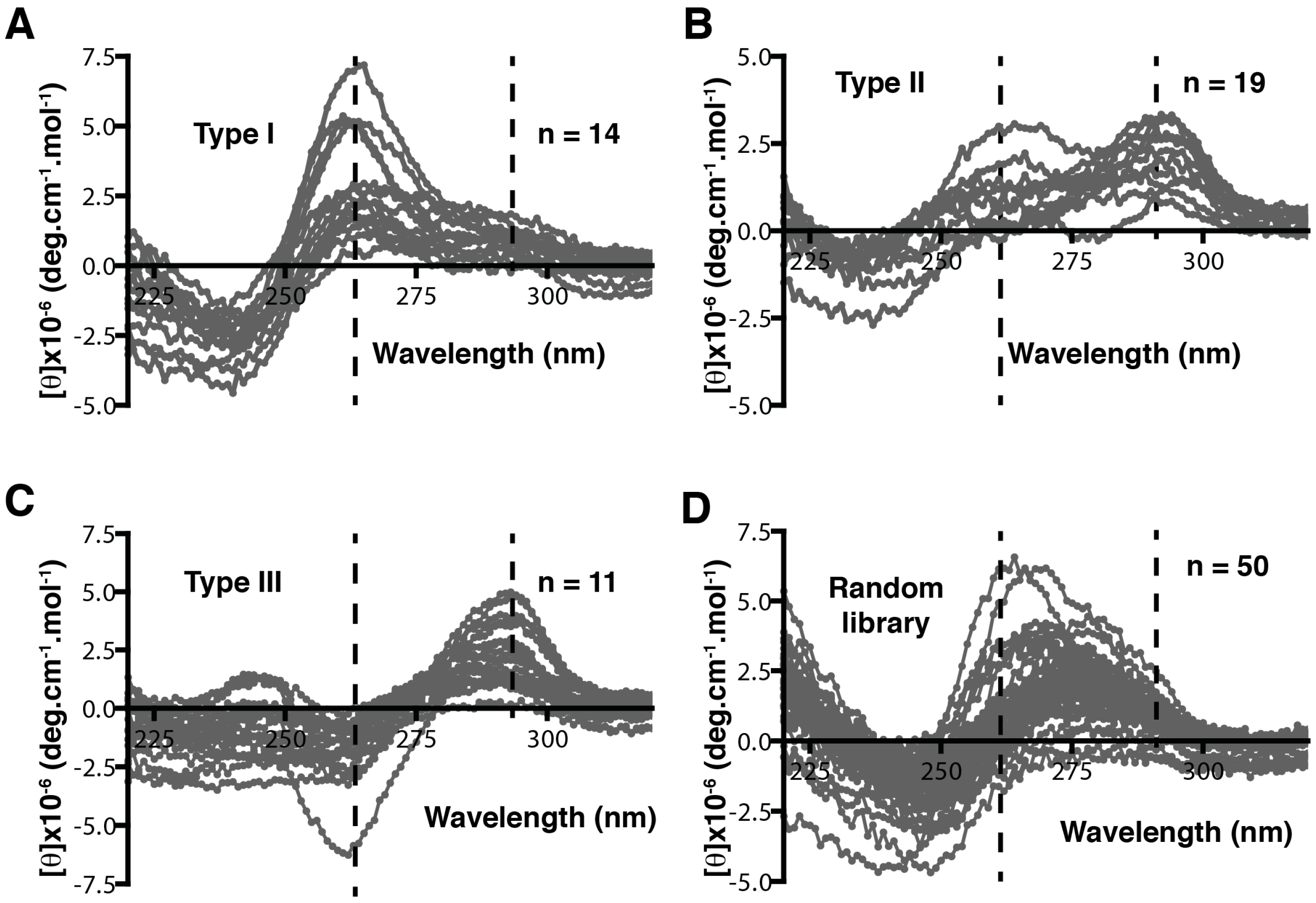
4. Patterning of Libraries
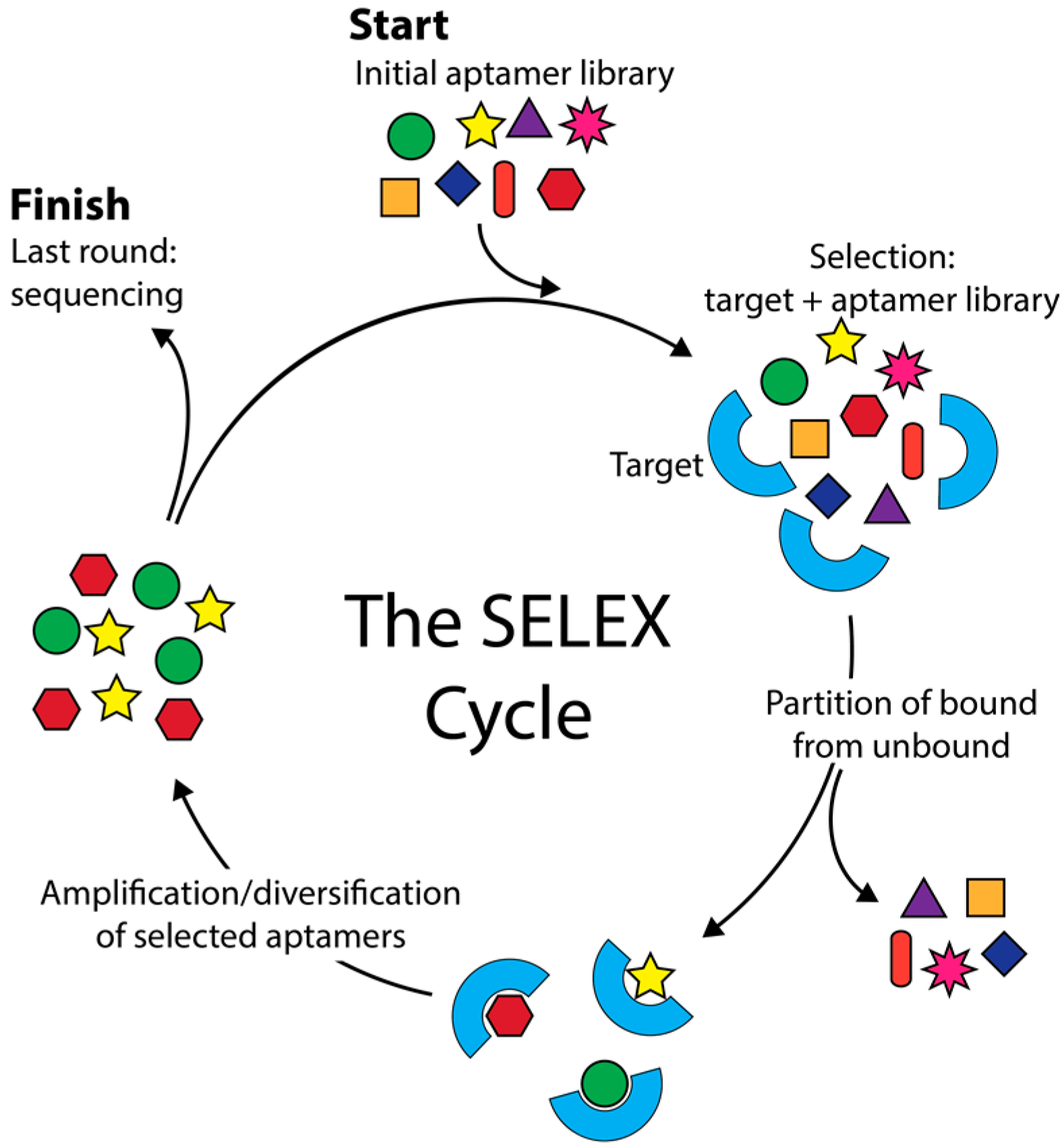
5. In Silico Aptamer Identification from High-Throughput Sequencing (HTS) Data
5.1. The Trend of Using HTS for Improving SELEX
5.2. Benchmark Toolkit for HTS SELEX Analysis
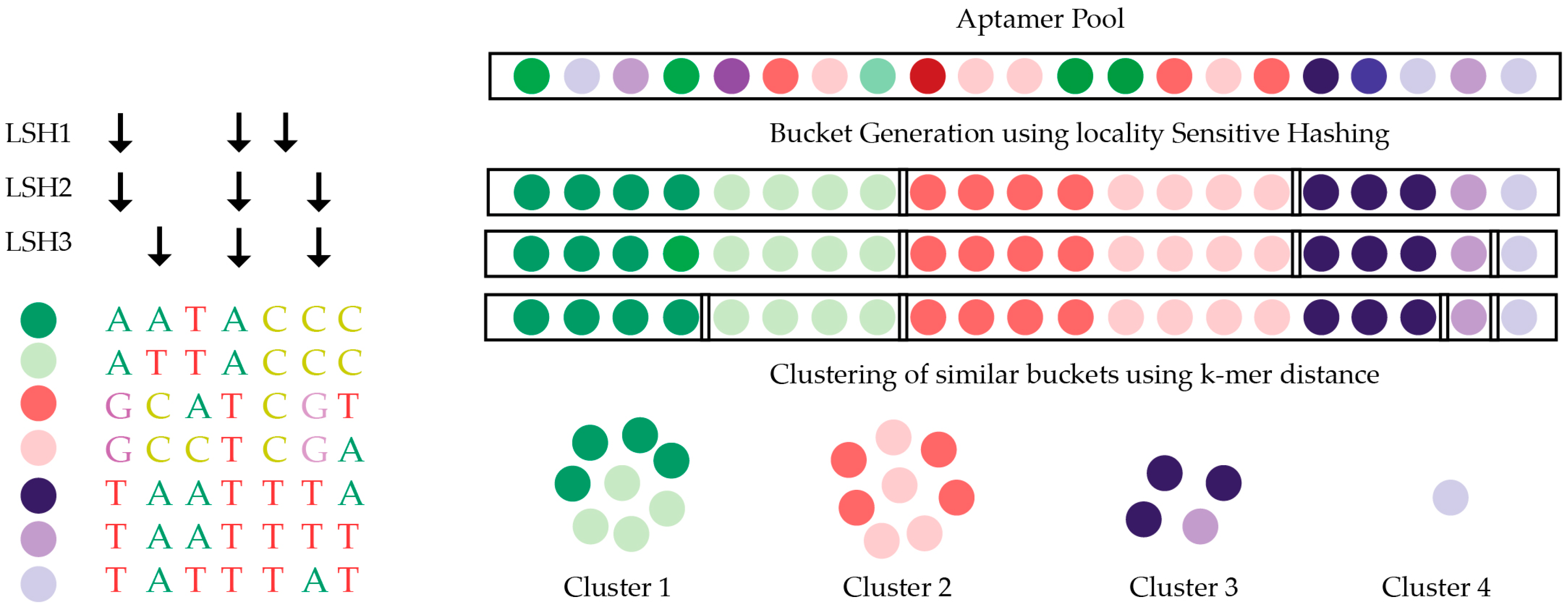
5.3. Structure Motif Clustering-Based Tools
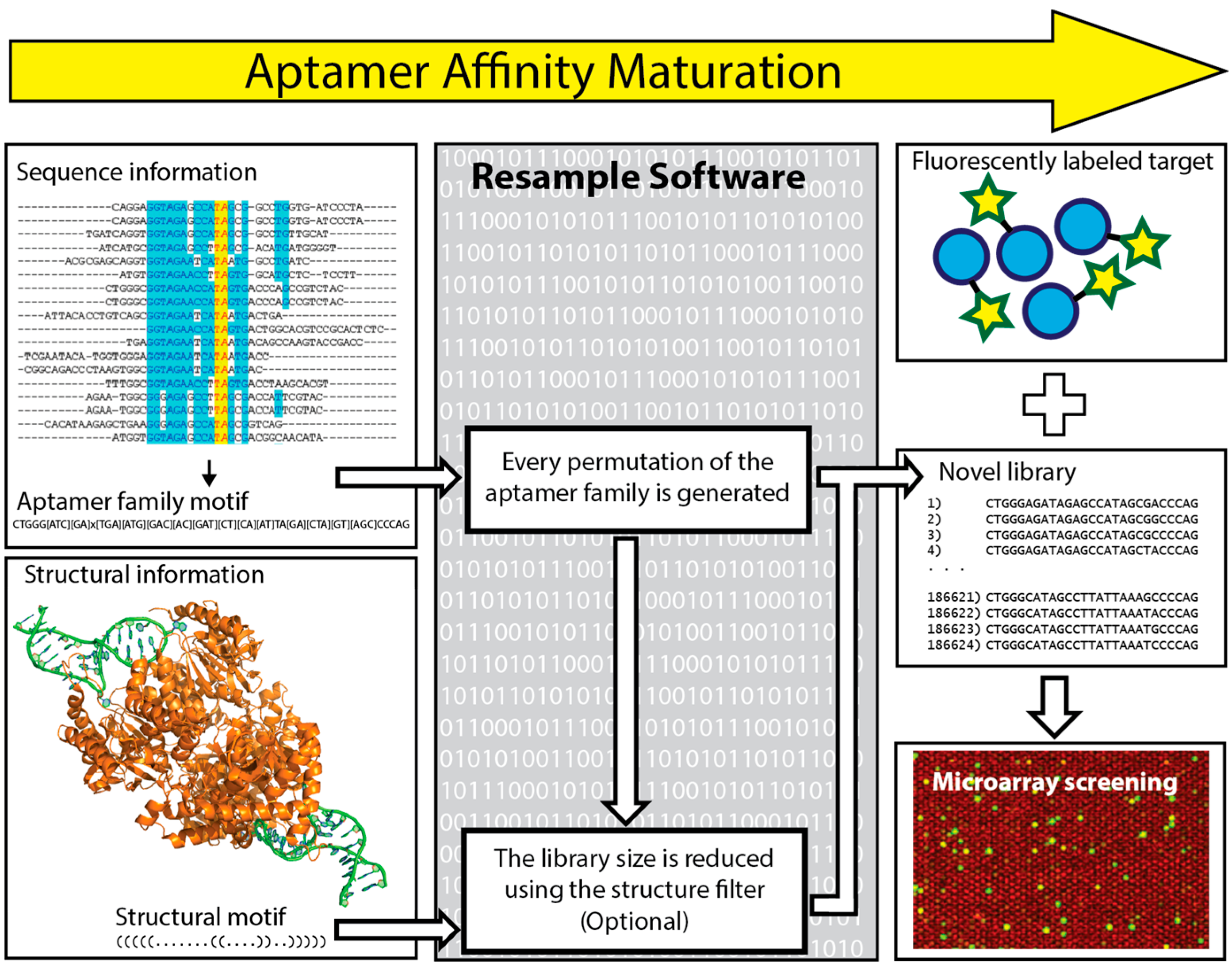
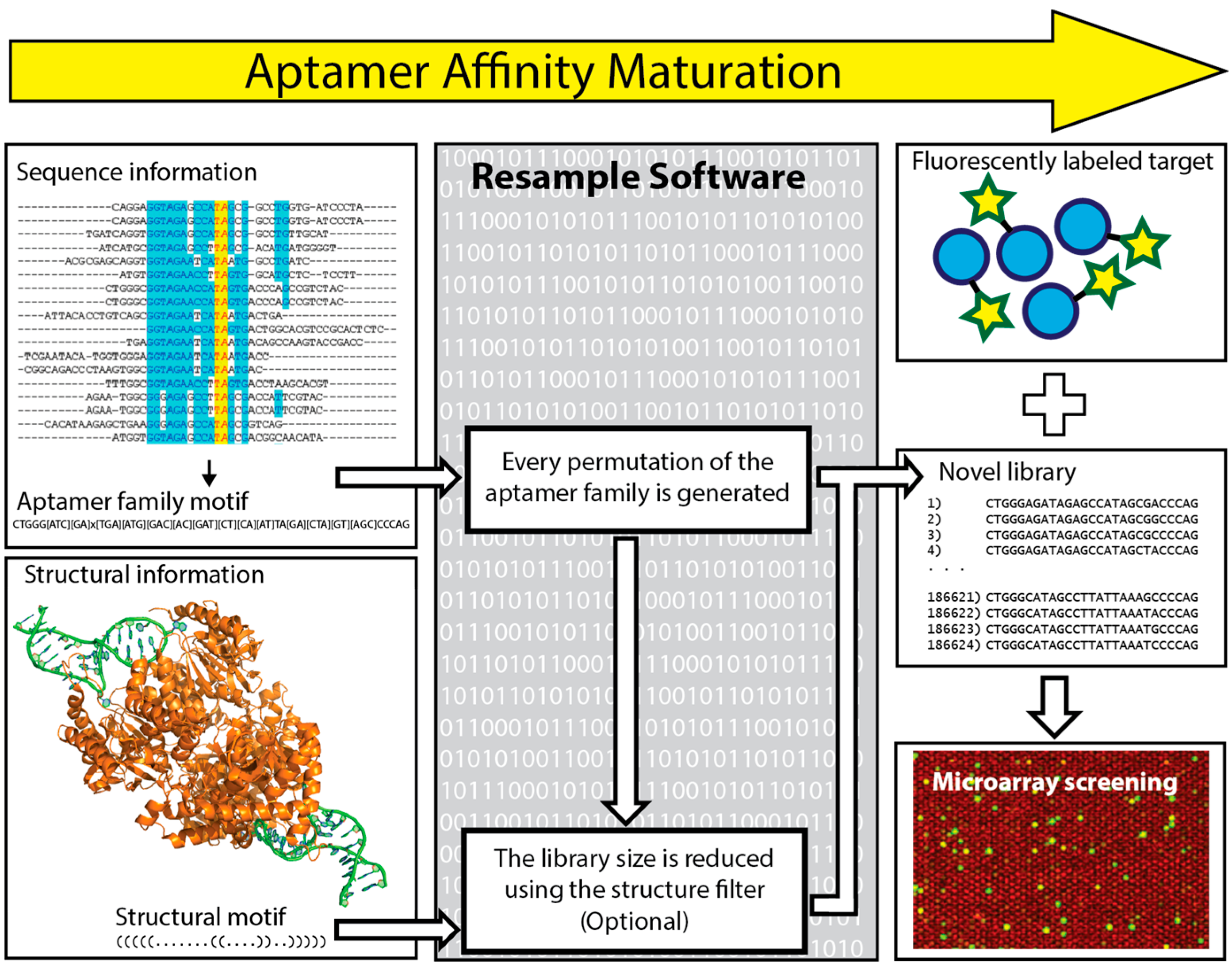
6. In Silico Aptamer Optimization
7. Conclusions and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Toledo, J.; McKeague, M.; Zhang, X.; Giamberardino, A.; McConnell, E.; Francis, T.; DeRosa, M.C.; Dumontier, M. Aptamer base: A collaborative knowledge base to describe aptamers and SELEX experiments. Database 2012. [Google Scholar] [CrossRef] [PubMed]
- McKeague, M.; McConnell, E.M.; Cruz-Toledo, J.; Bernard, E.D.; Pach, A.; Mastronardi, E.; Zhang, X.; Beking, M.; Francis, T.; Giamberardino, A. Analysis of in vitro aptamer selection parameters. J. Mol. Evol. 2015, 81, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Cheung, Y.-W.; Kwok, J.; Law, A.W.; Watt, R.M.; Kotaka, M.; Tanner, J.A. Structural basis for discriminatory recognition of Plasmodium lactate dehydrogenase by a DNA aptamer. Proc. Natl. Acad. Sci. USA 2013, 110, 15967–15972. [Google Scholar] [CrossRef] [PubMed]
- Irvine, D.; Tuerk, C.; Gold, L. SELEXION: Systematic evolution of ligands by exponential enrichment with integrated optimization by non-linear analysis. J. Mol. Biol. 1991, 222, 739–761. [Google Scholar] [CrossRef]
- Vant-Hull, B.; Payano-Baez, A.; Davis, R.H.; Gold, L. The mathematics of SELEX against complex targets. J. Mol. Biol. 1998, 278, 579–597. [Google Scholar] [CrossRef] [PubMed]
- Homann, M.; Göringer, H.U. Combinatorial selection of high affinity RNA ligands to live African trypanosomes. Nucleic Acids Res. 1999, 27, 2006–2014. [Google Scholar] [CrossRef] [PubMed]
- Coulter, L.R.; Landree, M.A.; Cooper, T.A. Identification of a new class of exonic splicing enhancers by in vivo selection. Mol. Cell. Biol. 1997, 17, 2143–2150. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-K.; Kuo, T.-L.; Chan, P.-C.; Lin, L.-Y. Subtractive SELEX against two heterogeneous target samples: Numerical simulations and analysis. Comput. Biol. Med. 2007, 37, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-K. Complex SELEX against target mixture: Stochastic computer model, simulation, and analysis. Comput. Meth. Prog. Biol. 2007, 87, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Rudzinski, J.F.; Gong, Q.; Soh, H.T.; Atzberger, P.J. Influence of target concentration and background binding on in vitro selection of affinity reagents. PLoS ONE 2012, 7, e43940. [Google Scholar] [CrossRef] [PubMed]
- Vant-Hull, B.; Gold, L.; Zichi, D.A. Theoretical principles of in vitro selection using combinatorial nucleic acid libraries. Curr. Protoc. Nucleic Acid Chem. 2000. [Google Scholar] [CrossRef]
- Zhao, Y.; Granas, D.; Stormo, G.D. Inferring binding energies from selected binding sites. PLoS Comput. Biol. 2009, 5, e1000590. [Google Scholar] [CrossRef] [PubMed]
- Spill, F.; Weinstein, Z.B.; Shemirani, A.I.; Ho, N.; Desai, D.; Zaman, M.H. Controlling uncertainty in aptamer selection. Proc. Natl. Acad. Sci. USA 2016. [Google Scholar] [CrossRef] [PubMed]
- Hoinka, J.; Berezhnoy, A.; Dao, P.; Sauna, Z.E.; Gilboa, E.; Przytycka, T.M. Large scale analysis of the mutational landscape in HT-SELEX improves aptamer discovery. Nucleic Acids Res. 2015, 43, 5699–5707. [Google Scholar] [CrossRef] [PubMed]
- Oh, I.S.; Lee, Y.-G.; McKay, R. Simulating chemical evolution. In Proceedings of the IEEE Congress on Evolutionary Computation, New Orleans, LA, USA, 5–8 July 2011; pp. 2717–2724. [Google Scholar] [CrossRef]
- Kauffman, S.; Levin, S. Towards a general theory of adaptive walks on rugged landscapes. J. Theor. Biol. 1987, 128, 11–45. [Google Scholar] [CrossRef]
- Wedge, D.C.; Rowe, W.; Kell, D.B.; Knowles, J. In silico modelling of directed evolution: Implications for experimental design and stepwise evolution. J. Theor. Biol. 2009, 257, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Deem, M.W.; Lee, H.Y. Sequence space localization in the immune system response to vaccination and disease. Phys. Rev. Lett. 2003, 91, 068101. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.; Christensen, K.; di Collobiano, S.A.; Jensen, H.J. Time-dependent extinction rate and species abundance in a tangled-nature model of biological evolution. Phys. Rev. E 2002, 66, 011904. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, S.; Macready, W. Technological evolution and adaptive organizations: Ideas from biology may find applications in economics. Complexity 1995, 1, 26–43. [Google Scholar] [CrossRef]
- Klussmann, S. Functional oligonucleotides and their applications. In The Aptamer Handbook; John Wiley & Sons: New York, NY, USA, 2006. [Google Scholar]
- Kauffman, S.A. The Origins of Order: Self Organization and Selection in Evolution; Oxford University Press: Oxford, UK, 1992; pp. 61–100. [Google Scholar]
- Altenberg, L. Evolving better representations through selective genome growth. In Proceedings of the First IEEE Conference on IEEE World Congress on Computational Intelligence, Evolutionary Computation, Orlando, FL, USA, 27–29 June 1994; pp. 182–187. [Google Scholar]
- Kinghorn, A.B.; Tanner, J.A. Selective Phenome Growth Adapted Model: A Novel Landscape to Represent Aptamer Ligand Binding. Complexity 2017, 2017, 1–12. [Google Scholar] [CrossRef]
- Wolpert, D.H.; Macready, W.G. No free lunch theorems for optimization. IEEE Trans. Evolut. Comput. 1997, 1, 67–82. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L., Jr.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar]
- MacKerell, A.D.; Nilsson, L. Molecular dynamics simulations of nucleic acid–protein complexes. Curr. Opin. Struct. Biol. 2008, 18, 194–199. [Google Scholar] [CrossRef]
- Gurtovenko, A.A.; Vattulainen, I. Pore formation coupled to ion transport through lipid membranes as induced by transmembrane ionic charge imbalance: Atomistic molecular dynamics study. J. Am. Chem. Soc. 2005, 127, 17570–17571. [Google Scholar] [CrossRef] [PubMed]
- Carothers, J.M.; Oestreich, S.C.; Szostak, J.W. Aptamers selected for higher-affinity binding are not more specific for the target ligand. J. Am. Chem. Soc. 2006, 128, 7929–7937. [Google Scholar] [CrossRef] [PubMed]
- Gevertz, J.; Gan, H.H.; Schlick, T. In vitro RNA random pools are not structurally diverse: A computational analysis. RNA 2005, 11, 853–863. [Google Scholar] [CrossRef]
- Chushak, Y.; Stone, M.O. In silico selection of RNA aptamers. Nucleic Acids Res. 2009, 37, e87. [Google Scholar] [CrossRef] [PubMed]
- Hua, W.P.; Linb, H.T.; Tsaia, J.J.; Chenc, W.Y. Investigating interactions between proteins and nucleic acids by computational approaches. In Computational Methods with Applications in Bioinformatics Analysis; World Scientific: Singapore, 2017; p. 98. [Google Scholar]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, M.; Bonvin, A.M. 3D-DART: A DNA structure modelling server. Nucleic Acids Res. 2009, 37, W235–W239. [Google Scholar] [CrossRef] [PubMed]
- Popenda, M.; Szachniuk, M.; Antczak, M.; Purzycka, K.J.; Lukasiak, P.; Bartol, N.; Blazewicz, J.; Adamiak, R.W. Automated 3D structure composition for large RNAs. Nucleic Acids Res. 2012, 40, e112. [Google Scholar] [CrossRef] [PubMed]
- Setny, P.; Bahadur, R.P.; Zacharias, M. Protein-DNA docking with a coarse-grained force field. BMC Bioinform. 2012, 13, 228. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Baker, D. Automated de novo prediction of native-like RNA tertiary structures. Proc. Natl. Acad. Sci. USA 2007, 104, 14664–14669. [Google Scholar] [CrossRef] [PubMed]
- Tsui, V.; Case, D.A. Theory and applications of the generalized Born solvation model in macromolecular simulations. Biopolymers 2000, 56, 275–291. [Google Scholar] [CrossRef]
- Zhang, S.; Kumar, K.; Jiang, X.; Wallqvist, A.; Reifman, J. DOVIS: An implementation for high-throughput virtual screening using AutoDock. BMC Bioinform. 2008, 9, 126. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Li, L.; Weng, Z. ZDOCK: An initial-stage protein-docking algorithm. Proteins 2003, 52, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.V.; Chen, W.-Y.; Tsai, J.J.; Hu, W.-P. Molecular simulation methods for selecting thrombin-binding aptamers. In Information Technology Convergence; Springer: Dordrecht, The Netherlands, 2013; Volume 253, pp. 743–749. [Google Scholar]
- White, R.R.; Shan, S.; Rusconi, C.P.; Shetty, G.; Dewhirst, M.W.; Kontos, C.D.; Sullenger, B.A. Inhibition of rat corneal angiogenesis by a nuclease-resistant RNA aptamer specific for angiopoietin-2. Proc. Natl. Acad. Sci. USA 2003, 100, 5028–5033. [Google Scholar] [CrossRef] [PubMed]
- Sarraf-Yazdi, S.; Mi, J.; Moeller, B.J.; Niu, X.; White, R.R.; Kontos, C.D.; Sullenger, B.A.; Dewhirst, M.W.; Clary, B.M. Inhibition of in vivo tumor angiogenesis and growth via systemic delivery of an angiopoietin 2-specific RNA aptamer. J. Surg. Res. 2008, 146, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.-P.; Kumar, J.V.; Huang, C.-J.; Chen, W.-Y. Computational selection of RNA aptamer against angiopoietin-2 and experimental evaluation. BioMed Res. Int. 2015, 2015, 658712. [Google Scholar] [CrossRef] [PubMed]
- Maisonpierre, P.C.; Suri, C.; Jones, P.F.; Bartunkova, S.; Wiegand, S.J.; Radziejewski, C.; Compton, D.; McClain, J.; Aldrich, T.H.; Papadopoulos, N. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 1997, 277, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Holash, J.; Maisonpierre, P.; Compton, D.; Boland, P.; Alexander, C.; Zagzag, D.; Yancopoulos, G.; Wiegand, S. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 1999, 284, 1994–1998. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Zhang, W.; Jia, S.; Guan, Z.; Yang, C.J.; Zhu, Z. Microfluidic approaches to rapid and efficient aptamer selection. Biomicrofluid 2014, 8, 041501. [Google Scholar] [CrossRef] [PubMed]
- Shangguan, D.; Bing, T.; Zhang, N. Cell-SELEX: Aptamer selection against whole cells, in Aptamers Selected by Cell-SELEX for Theranostics; Springer: Heidelberg, Germany, 2015; pp. 13–33. [Google Scholar]
- Mallikaratchy, P. Evolution of Complex Target SELEX to Identify Aptamers against Mammalian Cell-Surface Antigens. Molecules 2017, 22, 215. [Google Scholar] [CrossRef] [PubMed]
- Rabal, O.; Pastor, F.; Villanueva, H.; Soldevilla, M.M.; Hervas-Stubbs, S.; Oyarzabal, J. In Silico Aptamer Docking Studies: From a Retrospective Validation to a Prospective Case Study’TIM3 Aptamers Binding. Mol. Ther. Nucleic Acids 2016, 5, e376. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, S.; Guo, D.; Li, L.; Xiao, Y. A novel protocol for three-dimensional structure prediction of RNA-protein complexes. Sci. Rep. 2013, 3, 1887. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Tseng, C.Y.; Ashrafuzzaman, M.; Mane, J.Y.; Kapty, J.; Mercer, J.R.; Tuszynski, J.A. Entropic Fragment-Based Approach to Aptamer Design. Chem. Biol. Drug Des. 2011, 78, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ashrafuzzaman, M.; Tseng, C.-Y.; Kapty, J.; Mercer, J.R.; Tuszynski, J.A. A computationally designed DNA aptamer template with specific binding to phosphatidylserine. Nucleic Acid Ther. 2013, 23, 418–426. [Google Scholar] [CrossRef]
- Carothers, J.M.; Oestreich, S.C.; Davis, J.H.; Szostak, J.W. Informational complexity and functional activity of RNA structures. J. Am. Chem. Soc. 2004, 126, 5130–5137. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.H.; Szostak, J.W. Isolation of high-affinity GTP aptamers from partially structured RNA libraries. Proc. Natl. Acad. Sci. USA 2002, 99, 11616–11621. [Google Scholar] [CrossRef] [PubMed]
- Schultes, E.; Hraber, P.T.; LaBean, T.H. Global similarities in nucleotide base composition among disparate functional classes of single-stranded RNA imply adaptive evolutionary convergence. RNA 1997, 3, 792–806. [Google Scholar] [PubMed]
- Knight, R.; De Sterck, H.; Markel, R.; Smit, S.; Oshmyansky, A.; Yarus, M. Abundance of correctly folded RNA motifs in sequence space, calculated on computational grids. Nucleic Acids Res. 2005, 33, 5924–5935. [Google Scholar] [CrossRef] [PubMed]
- Ruff, K.M.; Snyder, T.M.; Liu, D.R. Enhanced functional potential of nucleic acid aptamer libraries patterned to increase secondary structure. J. Am. Chem. Soc. 2010, 132, 9453–9464. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.A.; Mirau, P.A.; Chushak, Y.; Chavez, J.L.; Naik, R.R.; Hagen, J.A.; Kelley-Loughnane, N. Single-Round Patterned DNA Library Microarray Aptamer Lead Identification. J. Anal. Methods Chem. 2015, 2015, 137489. [Google Scholar] [CrossRef] [PubMed]
- Hoogsteen, K. The crystal and molecular structure of a hydrogen-bonded complex between 1-methylthymine and 9-methyladenine. Acta Crystallogr. 1963, 16, 907–916. [Google Scholar] [CrossRef]
- Frank-Kamenetskii, M.D.; Mirkin, S.M. Triplex DNA structures. Annu. Rev. Biochem. 1995, 64, 65–95. [Google Scholar] [CrossRef] [PubMed]
- De Nicola, B.; Lech, C.J.; Heddi, B.; Regmi, S.; Frasson, I.; Perrone, R.; Richter, S.N.; Phan, A.T. Structure and possible function of a G-quadruplex in the long terminal repeat of the proviral HIV-1 genome. Nucleic Acids Res. 2016, 44, 6442–6451. [Google Scholar] [CrossRef] [PubMed]
- Corey, D.R. Telomeres and telomerase: From discovery to clinical trials. Chem. Biol. 2009, 16, 1219–1223. [Google Scholar] [CrossRef] [PubMed]
- Bourdoncle, A.; Estévez Torres, A.; Gosse, C.; Lacroix, L.; Vekhoff, P.; Le Saux, T.; Jullien, L.; Mergny, J.L. Quadruplex-Based Molecular Beacons as Tunable DNA Probes. J. Am. Chem. Soc. 2006, 128, 11094–11105. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhang, L.; Li, T.; Dong, S.; Wang, E. Enzyme-free unlabeled DNA logic circuits based on toehold-mediated strand displacement and split G-quadruplex enhanced fluorescence. Adv. Mater. 2013, 25, 2440–2444. [Google Scholar] [CrossRef] [PubMed]
- Webba da Silva, M. Geometric formalism for DNA quadruplex folding. Chemistry 2007, 13, 9738–9745. [Google Scholar] [CrossRef] [PubMed]
- Bates, P.J.; Kahlon, J.B.; Thomas, S.D.; Trent, J.O.; Miller, D.M. Antiproliferative activity of G-rich oligonucleotides correlates with protein binding. J. Biol. Chem. 1999, 274, 26369–26377. [Google Scholar] [CrossRef] [PubMed]
- Travascio, P.; Li, Y.; Sen, D. DNA-enhanced peroxidase activity of a DNA-aptamer-hemin complex. Chem. Biol. 1998, 5, 505–517. [Google Scholar] [CrossRef]
- Sen, D.; Poon, L.C. RNA and DNA complexes with hemin [Fe(III) heme] are efficient peroxidases and peroxygenases: How do they do it and what does it mean? Crit. Rev. Biochem. Mol. Biol. 2011, 46, 478–492. [Google Scholar] [CrossRef] [PubMed]
- Baugh, C.; Grate, D.; Wilson, C. 2.8 Å crystal structure of the malachite green aptamer11Edited by J. A. Doudna. J. Mol. Biol. 2000, 301, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Warner, K.D.; Chen, M.C.; Song, W.; Strack, R.L.; Thorn, A.; Jaffrey, S.R.; Ferre-D’Amare, A.R. Structural basis for activity of highly efficient RNA mimics of green fluorescent protein. Nat. Struct. Mol. Biol. 2014, 21, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Trachman, R.J., 3rd; Demeshkina, N.A.; Lau, M.W.L.; Panchapakesan, S.S.S.; Jeng, S.C.Y.; Unrau, P.J.; Ferre-D’Amare, A.R. Structural basis for high-affinity fluorophore binding and activation by RNA Mango. Nat. Chem. Biol. 2017, 13, 807–813. [Google Scholar] [CrossRef] [PubMed]
- McManus, S.A.; Li, Y. Assessing the amount of quadruplex structures present within G(2)-tract synthetic random-sequence DNA libraries. PLoS ONE 2013, 8, e64131. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Marian, A.J. Sequencing your genome: What does it mean? Methodist Debakey Cardiovasc. J. 2014, 10, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Blind, M.; Blank, M. Aptamer selection technology and recent advances. Mol. Ther. Nucleic Acids 2015, 4, e223. [Google Scholar] [CrossRef] [PubMed]
- Berezhnoy, A.; Stewart, C.A.; McNamara, J.O.; Thiel, W.; Giangrande, P.; Trinchieri, G.; Gilboa, E. Isolation and optimization of murine IL-10 receptor blocking oligonucleotide aptamers using high-throughput sequencing. Mol. Ther. 2012, 20, 1242–1250. [Google Scholar] [CrossRef] [PubMed]
- Thiel, W.H.; Bair, T.; Peek, A.S.; Liu, X.; Dassie, J.; Stockdale, K.R.; Behlke, M.A.; Miller, F.J., Jr.; Giangrande, P.H. Rapid identification of cell-specific, internalizing RNA aptamers with bioinformatics analyses of a cell-based aptamer selection. PLoS ONE 2012, 7, e43836. [Google Scholar] [CrossRef] [PubMed]
- Valenzano, S.; De Girolamo, A.; DeRosa, M.C.; McKeague, M.; Schena, R.; Catucci, L.; Pascale, M. Screening and Identification of DNA Aptamers to Tyramine Using in Vitro Selection and High-Throughput Sequencing. ACS Comb. Sci. 2016, 18, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Wu, X.; Ho, M.; Chomchan, P.; Rossi, J.J.; Burnett, J.C.; Zhou, J. High throughput sequencing analysis of RNA libraries reveals the influences of initial library and PCR methods on SELEX efficiency. Sci. Rep. 2016, 6, 33697. [Google Scholar] [CrossRef] [PubMed]
- Dupont, D.M.; Larsen, N.; Jensen, J.K.; Andreasen, P.A.; Kjems, J. Characterisation of aptamer-target interactions by branched selection and high-throughput sequencing of SELEX pools. Nucleic Acids Res. 2015, 43, e139. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, B.; Gesell, T.; Chen, D.; Lorenz, C.; Schroeder, R. Monitoring genomic sequences during SELEX using high-throughput sequencing: Neutral SELEX. PLoS ONE 2010, 5, e9169. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, C.; Gesell, T.; Zimmermann, B.; Schoeberl, U.; Bilusic, I.; Rajkowitsch, L.; Waldsich, C.; von Haeseler, A.; Schroeder, R. Genomic SELEX for Hfq-binding RNAs identifies genomic aptamers predominantly in antisense transcripts. Nucleic Acids Res. 2010, 38, 3794–3808. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.; Xiao, Y.; Nie, J.; Stewart, R.; Csordas, A.T.; Oh, S.S.; Thomson, J.A.; Soh, H.T. Quantitative selection of DNA aptamers through microfluidic selection and high-throughput sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 15373–15378. [Google Scholar] [CrossRef] [PubMed]
- Schütze, T.; Wilhelm, B.; Greiner, N.; Braun, H.; Peter, F.; Mörl, M.; Erdmann, V.A.; Lehrach, H.; Konthur, Z.; Menger, M. Probing the SELEX process with next-generation sequencing. PLoS ONE 2011, 6, e29604. [Google Scholar]
- Spiga, F.M.; Maietta, P.; Guiducci, C. More DNA-Aptamers for Small Drugs: A Capture-SELEX Coupled with Surface Plasmon Resonance and High-Throughput Sequencing. ACS Comb. Sci. 2015, 17, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Gotrik, M.R.; Feagin, T.A.; Csordas, A.T.; Nakamoto, M.A.; Soh, H.T. Advancements in Aptamer Discovery Technologies. Acc. Chem. Res. 2016, 49, 1903–1910. [Google Scholar] [CrossRef] [PubMed]
- Ditzler, M.A.; Lange, M.J.; Bose, D.; Bottoms, C.A.; Virkler, K.F.; Sawyer, A.W.; Whatley, A.S.; Spollen, W.; Givan, S.A.; Burke, D.H. High-throughput sequence analysis reveals structural diversity and improved potency among RNA inhibitors of HIV reverse transcriptase. Nucleic Acids Res. 2013, 41, 1873–1884. [Google Scholar] [CrossRef] [PubMed]
- Scoville, D.J.; Uhm, T.K.; Shallcross, J.A.; Whelan, R.J. Selection of DNA Aptamers for Ovarian Cancer Biomarker CA125 Using One-Pot SELEX and High-Throughput Sequencing. J. Nucleic Acids 2017, 2017, 9879135. [Google Scholar] [CrossRef] [PubMed]
- Hoon, S.; Zhou, B.; Janda, K.D.; Brenner, S.; Scolnick, J. Aptamer selection by high-throughput sequencing and informatic analysis. Biotechniques 2011, 51, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Narechania, A.; Stein, J.C.; Ware, D. A new method to compute K-mer frequencies and its application to annotate large repetitive plant genomes. BMC Genom. 2008, 9, 517. [Google Scholar] [CrossRef] [PubMed]
- Weese, D.; Emde, A.K.; Rausch, T.; Doring, A.; Reinert, K. RazerS–fast read mapping with sensitivity control. Genome Res. 2009, 19, 1646–1654. [Google Scholar] [CrossRef] [PubMed]
- Jing, M.; Bowser, M.T. Tracking the emergence of high affinity aptamers for rhVEGF165 during capillary electrophoresis-systematic evolution of ligands by exponential enrichment using high throughput sequencing. Anal. Chem. 2013, 85, 10761–10770. [Google Scholar] [CrossRef] [PubMed]
- Thiel, W.H.; Giangrande, P.H. Analyzing HT-SELEX data with the Galaxy Project tools—A web based bioinformatics platform for biomedical research. Methods 2016, 97, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Thiel, W.H. Galaxy Workflows for Web-based Bioinformatics Analysis of Aptamer High-throughput Sequencing Data. Mol. Ther. Nucleic Acids 2016, 5, e345. [Google Scholar] [CrossRef] [PubMed]
- Alam, K.K.; Chang, J.L.; Burke, D.H. FASTAptamer: A Bioinformatic Toolkit for High-throughput Sequence Analysis of Combinatorial Selections. Mol. Ther. Nucleic Acids. 2015, 4, e230. [Google Scholar] [CrossRef] [PubMed]
- Ducongé, F. Improvement of aptamers using PATTERNITY-seq (high-throughput analysis of sequence patterns and paternity relationship between them). In Proceedings of the the Aptamer Bordeaux Conference, Bordeaux, France, 24–25 June 2016. [Google Scholar]
- Nguyen Quang, N.; Perret, G.; Duconge, F. Applications of High-Throughput Sequencing for In Vitro Selection and Characterization of Aptamers. Pharmaceuticals 2016, 9, 76. [Google Scholar] [CrossRef] [PubMed]
- Hoinka, J.; Berezhnoy, A.; Sauna, Z.E.; Gilboa, E.; Przytycka, T.M. AptaCluster—A Method to Cluster HT-SELEX Aptamer Pools and Lessons from its Application. Res. Comput. Mol. Biol. 2014, 8394, 115–128. [Google Scholar] [PubMed]
- Hoinka, J.; Dao, P.; Przytycka, T.M. AptaGUI-A Graphical User Interface for the Efficient Analysis of HT-SELEX Data. Mol. Ther. Nucleic Acids. 2015, 4, e257. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Dao, P.; Hoinka, J.; Takahashi, M.; Zhou, J.; Ho, M.; Wang, Y.; Costa, F.; Rossi, J.J.; Backofen, R.; Burnett, J. AptaTRACE Elucidates RNA Sequence-Structure Motifs from Selection Trends in HT-SELEX Experiments. Cell Syst. 2016, 3, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Chan, C.Y.; Lawrence, C.E. S fold web server for statistical folding and rational design of nucleic acids. Nucleic Acids Res. 2004, 32, W135–W141. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.Y.; Lawrence, C.E.; Ding, Y. Structure clustering features on the S fold Web server. Bioinformatics 2005, 21, 3926–3928. [Google Scholar] [CrossRef] [PubMed]
- Hofacker, I.L. RNA secondary structure analysis using the Vienna RNA package. In Current Protoc Bioinformatics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009. [Google Scholar]
- Wang, J.; Gong, Q.; Maheshwari, N.; Eisenstein, M.; Arcila, M.L.; Kosik, K.S.; Soh, H.T. Particle Display: A Quantitative Screening Method for Generating High-Affinity Aptamers. Angew. Chem. Int. Ed. 2014, 53, 4796–4801. [Google Scholar] [CrossRef] [PubMed]
- Bowser, M.T. SELEX: Just another separation? Analyst 2005, 130, 128–130. [Google Scholar] [CrossRef] [PubMed]
- Katilius, E.; Flores, C.; Woodbury, N.W. Exploring the sequence space of a DNA aptamer using microarrays. Nucleic Acids Res. 2007, 35, 7626–7635. [Google Scholar] [CrossRef] [PubMed]
- Platt, M.; Rowe, W.; Knowles, J.; Day, P.J.; Kell, D.B. Analysis of aptamer sequence activity relationships. Integr. Biol. 2009, 1, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Knight, C.G.; Platt, M.; Rowe, W.; Wedge, D.C.; Khan, F.; Day, P.J.; McShea, A.; Knowles, J.; Kell, D.B. Array-based evolution of DNA aptamers allows modelling of an explicit sequence-fitness landscape. Nucleic Acids Res. 2009, 37, e6. [Google Scholar] [CrossRef] [PubMed]
- Rowe, W.; Platt, M.; Wedge, D.C.; Day, P.J.; Kell, D.B.; Knowles, J. Convergent evolution to an aptamer observed in small populations on DNA microarrays. Phys. Biol. 2010, 7, 036007. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, Y.; Yoshida, W.; Abe, K.; Ferri, S.; Schulze, H.; Bachmann, T.T.; Ikebukuro, K. Affinity improvement of a VEGF aptamer by in silico maturation for a sensitive VEGF-detection system. Anal. Chem. 2012, 85, 1132–1137. [Google Scholar] [CrossRef] [PubMed]
- Kinghorn, A.B.; Dirkzwager, R.M.; Liang, S.; Cheung, Y.-W.; Fraser, L.A.; Shiu, S.C.-C.; Tang, M.S.; Tanner, J.A. Aptamer Affinity Maturation by Resampling and Microarray Selection. Anal. Chem. 2016, 88, 6981–6985. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pattern | Library Design |
|---|---|
| 1 | (RY)3-N4-(RY)4-N3-(RY)4-N4-(RY)4-N3-(RY)3 |
| 2 | (RRYY)2-N4-(RRYY)-N3-(RRYY)-N4-(RRYY)-N3-(RRYY)-N4-(RRYY)2 |
| 3 | (RRYY)2-N4-(RRRYYY)-N4-(RRRYYY)-N4-(RRRYYY)-N4-(RRYY)2 |
| 4 | (RRYY)2-N4-(RY)3-N4-(RY)3-N4-(RY)3-N4-(RRYY)2 |
| Program | Operation System | Language | Clustering Method | Validation Experiment |
|---|---|---|---|---|
| FASTAptamer | Mac/Linux | Perl | Levenshtein distance | HIV-1 Reverse Transcriptase |
| AptaCluster/AptaGUI | Mac/Linux/PC | Java | LSH and k-mer counting | IL-10RA |
| APTANI | Linux | Python | Structure motif-based clustering | Murine IL4Ra |
| AptaTrace | Mac/Linux/PC | C++, Java | Structure motif-based clustering | C-C chemokine receptor |
| PATTERNITY-seq | No details | No details | Levenshtein distance | Annexin-A2 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kinghorn, A.B.; Fraser, L.A.; Liang, S.; Shiu, S.C.-C.; Tanner, J.A. Aptamer Bioinformatics. Int. J. Mol. Sci. 2017, 18, 2516. https://doi.org/10.3390/ijms18122516
Kinghorn AB, Fraser LA, Liang S, Shiu SC-C, Tanner JA. Aptamer Bioinformatics. International Journal of Molecular Sciences. 2017; 18(12):2516. https://doi.org/10.3390/ijms18122516
Chicago/Turabian StyleKinghorn, Andrew B., Lewis A. Fraser, Shaolin Liang, Simon Chi-Chin Shiu, and Julian A. Tanner. 2017. "Aptamer Bioinformatics" International Journal of Molecular Sciences 18, no. 12: 2516. https://doi.org/10.3390/ijms18122516
APA StyleKinghorn, A. B., Fraser, L. A., Liang, S., Shiu, S. C.-C., & Tanner, J. A. (2017). Aptamer Bioinformatics. International Journal of Molecular Sciences, 18(12), 2516. https://doi.org/10.3390/ijms18122516