
The Pleiotropic Role of L1CAM in Tumor Vasculature



Abstract
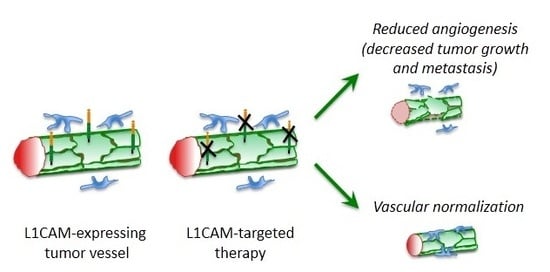
:
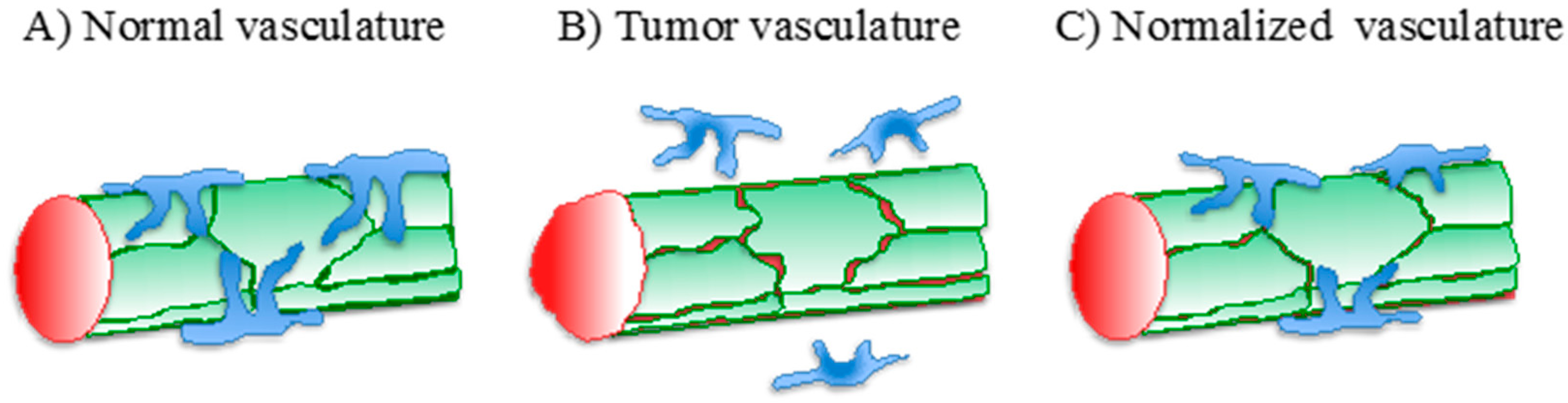
1. Tumor Angiogenesis
2. Antiangiogenic Therapy
3. Vascular Normalization
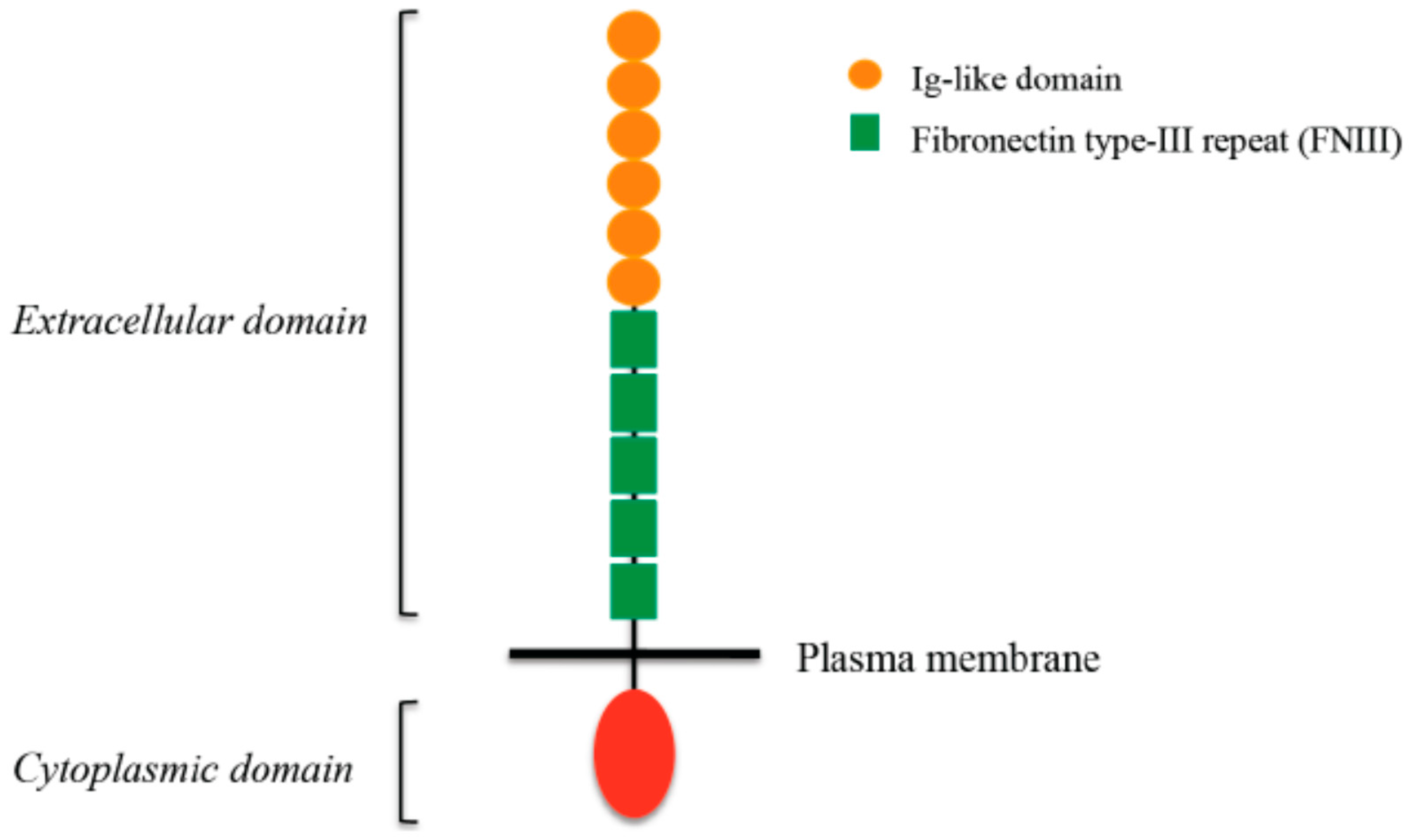
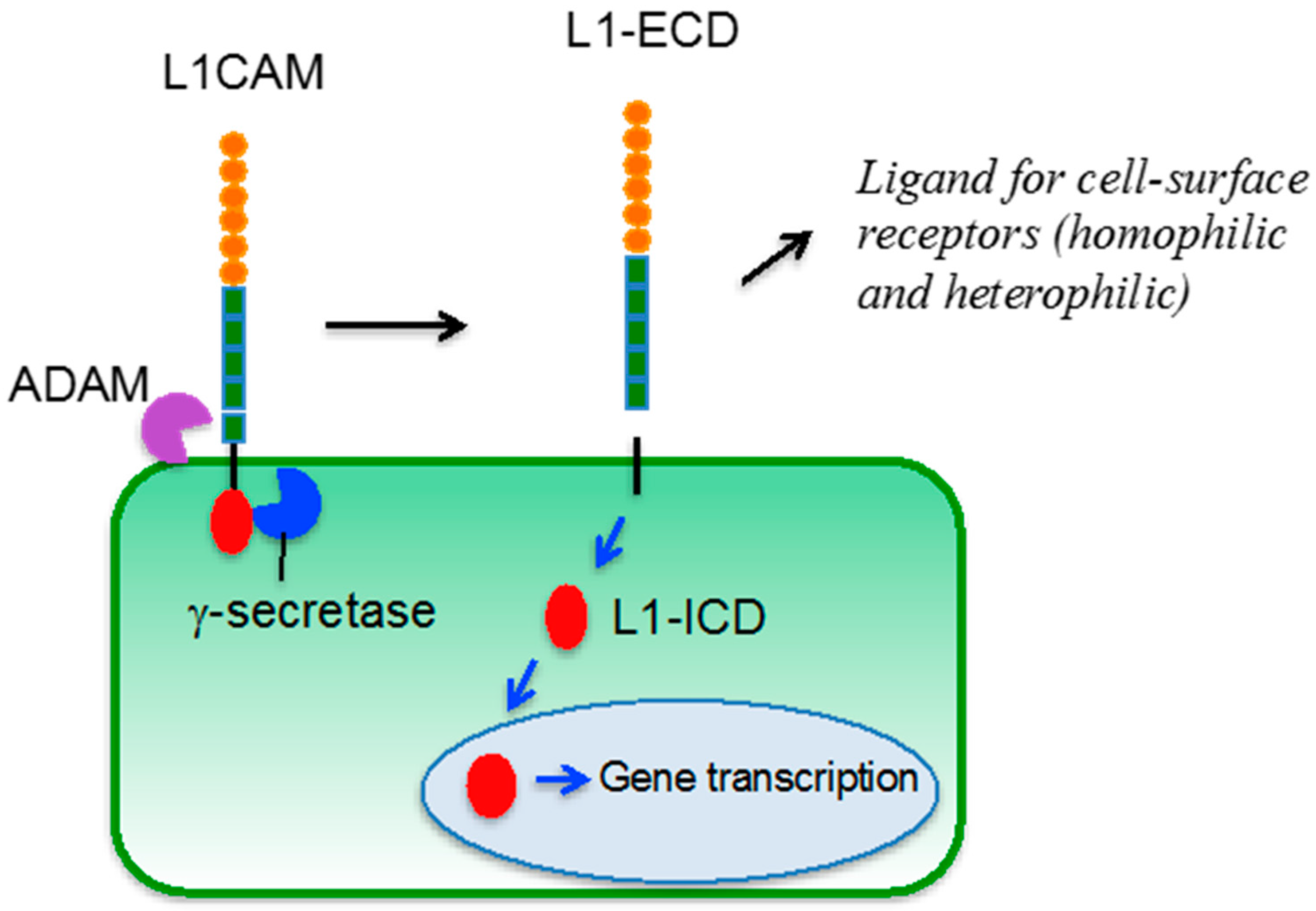
4. L1CAM
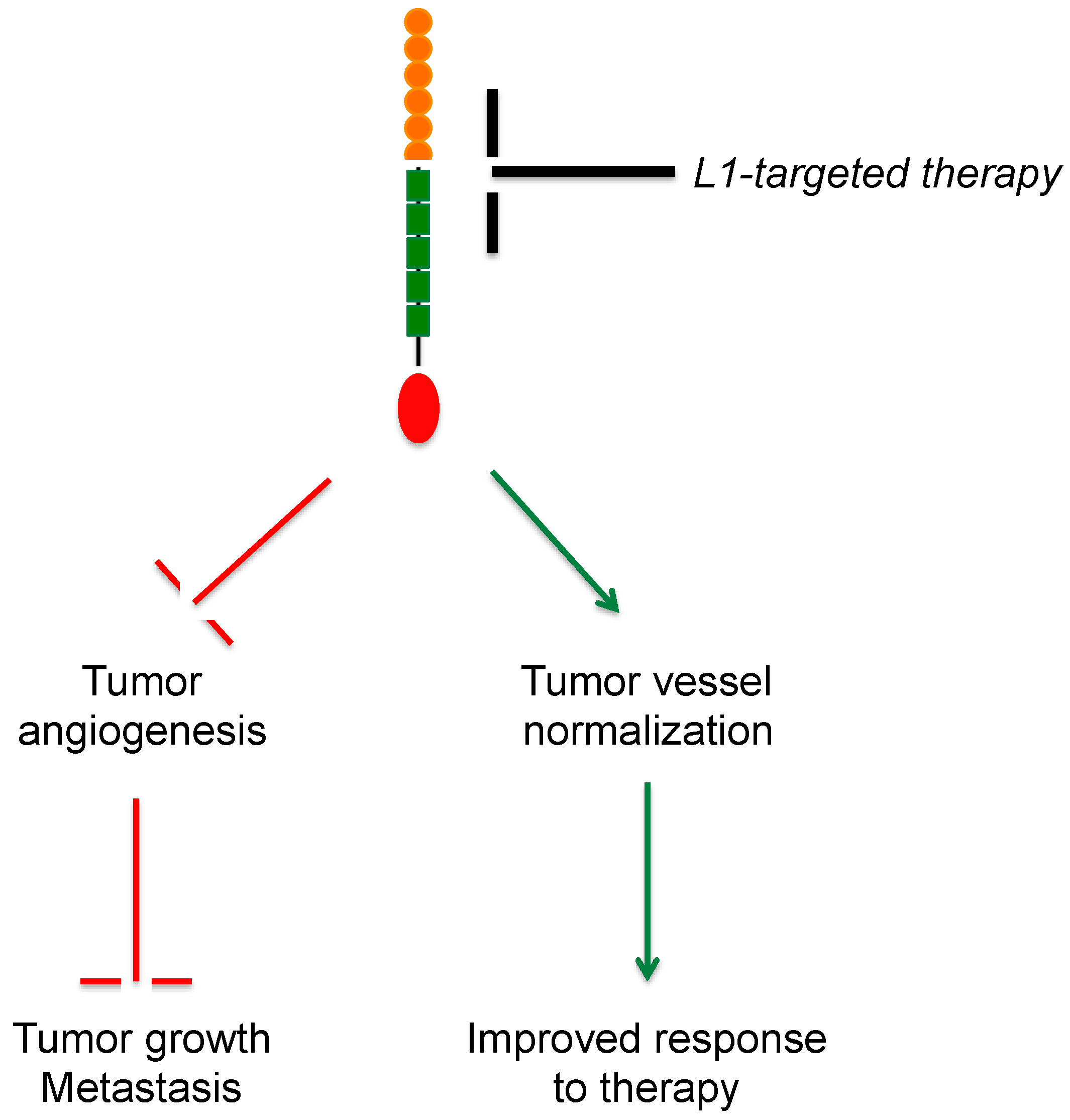
5. L1CAM in Tumor Vasculature
6. Therapeutic Perspectives
Acknowledgments
Conflicts of Interest
References
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Zhang, D.; Zhao, N.; Zhao, X. Epithelial-to-endothelial transition and cancer stem cells: Two cornerstones of vasculogenic mimicry in malignant tumors. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Donnem, T.; Hu, J.; Ferguson, M.; Adighibe, O.; Snell, C.; Harris, A.L.; Gatter, K.C.; Pezzella, F. Vessel co-option in primary human tumors and metastases: An obstacle to effective anti-angiogenic treatment? Cancer Med. 2013, 2, 427–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidner, N. Intratumor microvessel density as a prognostic factor in cancer. Am. J. Pathol. 1995, 147, 9–19. [Google Scholar] [PubMed]
- Qin, L.; Bromberg-White, J.L.; Qian, C.N. Opportunities and challenges in tumor angiogenesis research: Back and forth between bench and bed. Adv. Cancer Res. 2012, 113, 191–239. [Google Scholar] [PubMed]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [PubMed]
- Shing, Y.; Folkman, J.; Sullivan, R.; Butterfield, C.; Murray, J.; Klagsbrun, M. Heparin affinity: Purification of a tumor-derived capillary endothelial cell growth factor. Science 1984, 223, 1296–1299. [Google Scholar] [CrossRef] [PubMed]
- Claesson-Welsh, L. VEGF receptor signal transduction—A brief update. Vasc. Pharmacol. 2016, 86, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Ronca, R.; Giacomini, A.; Rusnati, M.; Presta, M. The potential of fibroblast growth factor/fibroblast growth factor receptor signaling as a therapeutic target in tumor angiogenesis. Expert Opin. Ther. Targets 2015, 19, 1361–1377. [Google Scholar] [CrossRef] [PubMed]
- Seghezzi, G.; Patel, S.; Ren, C.J.; Gualandris, A.; Pintucci, G.; Robbins, E.S.; Shapiro, R.L.; Galloway, A.C.; Rifkin, D.B.; Mignatti, P. Fibroblast growth factor-2 (FGF-2) induces vascular endothelial growth factor (VEGF) expression in the endothelial cells of forming capillaries: An autocrine mechanism contributing to angiogenesis. J. Cell Biol. 1998, 141, 1659–1673. [Google Scholar] [CrossRef] [PubMed]
- Kano, M.R.; Morishita, Y.; Iwata, C.; Iwasaka, S.; Watabe, T.; Ouchi, Y.; Miyazono, K.; Miyazawa, K. VEGF-A and FGF-2 synergistically promote neoangiogenesis through enhancement of endogenous PDGF-B-PDGFRβ signaling. J. Cell Sci. 2005, 118, 3759–3768. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Ji, H.; Feng, N.; Zhang, Y.; Yang, X.; Andersson, P.; Sun, Y.; Tritsaris, K.; Hansen, A.J.; Dissing, S.; et al. Collaborative interplay between FGF-2 and VEGF-C promotes lymphangiogenesis and metastasis. Proc. Natl. Acad. Sci. USA 2012, 109, 15894–15899. [Google Scholar] [CrossRef] [PubMed]
- Ziyad, S.; Iruela-Arispe, M.L. Molecular mechanisms of tumor angiogenesis. Genes Cancer 2011, 2, 1085–1096. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [PubMed]
- Jayson, G.C.; Kerbel, R.; Ellis, L.M.; Harris, A.L. Antiangiogenic therapy in oncology: Current status and future directions. Lancet 2016, 388, 518–529. [Google Scholar] [CrossRef]
- Khan, K.A.; Bicknell, R. Anti-angiogenic alternatives to VEGF blockade. Clin. Exp. Metastasis 2016, 33, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Antiangiogenesis strategies revisited: From starving tumors to alleviating hypoxia. Cancer Cell 2014, 26, 605–622. [Google Scholar] [CrossRef] [PubMed]
- Ye, W. The complexity of translating anti-angiogenesis therapy from basic science to the clinic. Dev. Cell 2016, 37, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Zalcman, G.; Mazieres, J.; Margery, J.; Greillier, L.; Audigier-Valette, C.; Moro-Sibilot, D.; Molinier, O.; Corre, R.; Monnet, I.; Gounant, V.; et al. Bevacizumab for newly diagnosed pleural mesothelioma in the Mesothelioma Avastin Cisplatin Pemetrexed Study (MAPS): A randomised, controlled, open-label, phase 3 trial. Lancet 2016, 387, 1405–1414. [Google Scholar] [CrossRef]
- Ebos, J.M.; Lee, C.R.; Cruz-Munoz, W.; Bjarnason, G.A.; Christensen, J.G.; Kerbel, R.S. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 2009, 15, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Paez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Vinals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009, 15, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Gacche, R.N.; Meshram, R.J. Angiogenic factors as potential drug target: Efficacy and limitations of anti-angiogenic therapy. Biochim. Biophys. Acta 2014, 1846, 161–179. [Google Scholar] [CrossRef] [PubMed]
- Du, R.; Lu, K.V.; Petritsch, C.; Liu, P.; Ganss, R.; Passegue, E.; Song, H.; Vandenberg, S.; Johnson, R.S.; Werb, Z.; et al. HIF1α induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 2008, 13, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Bridgeman, V.L.; Vermeulen, P.B.; Foo, S.; Bilecz, A.; Daley, F.; Kostaras, E.; Nathan, M.R.; Wan, E.; Frentzas, S.; Schweiger, T.; et al. Vessel co-option is common in human lung metastases and mediates resistance to anti-angiogenic therapy in preclinical lung metastasis models. J. Pathol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kuczynski, E.A.; Yin, M.; Bar-Zion, A.; Lee, C.R.; Butz, H.; Man, S.; Daley, F.; Vermeulen, P.B.; Yousef, G.M.; Foster, F.S.; et al. Co-option of liver vessels and not sprouting angiogenesis drives acquired sorafenib resistance in hepatocellular carcinoma. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed]
- Van der Schaft, D.W.; Seftor, R.E.; Seftor, E.A.; Hess, A.R.; Gruman, L.M.; Kirschmann, D.A.; Yokoyama, Y.; Griffioen, A.W.; Hendrix, M.J. Effects of angiogenesis inhibitors on vascular network formation by human endothelial and melanoma cells. J. Natl. Cancer Inst. 2004, 96, 1473–1477. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, Q.; Li, X.Y.; Yang, Q.Y.; Xu, W.W.; Liu, G.L. Short-term anti-vascular endothelial growth factor treatment elicits vasculogenic mimicry formation of tumors to accelerate metastasis. J. Exp. Clin. Cancer Res. 2012, 31, 16. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, M.J.; Seftor, E.A.; Seftor, R.E.; Chao, J.T.; Chien, D.S.; Chu, Y.W. Tumor cell vascular mimicry: Novel targeting opportunity in melanoma. Pharmacol. Ther. 2016, 159, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Hamzah, J.; Jugold, M.; Kiessling, F.; Rigby, P.; Manzur, M.; Marti, H.H.; Rabie, T.; Kaden, S.; Grone, H.J.; Hammerling, G.J.; et al. Vascular normalization in Rgs5-deficient tumours promotes immune destruction. Nature 2008, 453, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, M.; Dettori, D.; Leite de Oliveira, R.; Loges, S.; Schmidt, T.; Jonckx, B.; Tian, Y.M.; Lanahan, A.A.; Pollard, P.; Ruiz de Almodovar, C.; et al. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell 2009, 136, 839–851. [Google Scholar] [CrossRef] [PubMed]
- Cantelmo, A.R.; Conradi, L.C.; Brajic, A.; Goveia, J.; Kalucka, J.; Pircher, A.; Chaturvedi, P.; Hol, J.; Thienpont, B.; Teuwen, L.A.; et al. Inhibition of the glycolytic activator PFKFB3 in endothelium induces tumor vessel normalization, impairs metastasis, and improves chemotherapy. Cancer Cell 2016, 30, 968–985. [Google Scholar] [CrossRef] [PubMed]
- Gilles, M.E.; Maione, F.; Cossutta, M.; Carpentier, G.; Caruana, L.; di Maria, S.; Houppe, C.; Destouches, D.; Shchors, K.; Prochasson, C.; et al. Nucleolin targeting impairs the progression of pancreatic cancer and promotes the normalization of tumor vasculature. Cancer Res. 2016, 76, 7181–7193. [Google Scholar] [CrossRef] [PubMed]
- Mangala, L.S.; Wang, H.; Jiang, D.; Wu, S.Y.; Somasunderam, A.; Volk, D.E.; Lokesh, G.L.; Li, X.; Pradeep, S.; Yang, X.; et al. Improving vascular maturation using noncoding RNAs increases antitumor effect of chemotherapy. JCI Insight 2016, 1, e87754. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Liu, T.; Ma, P.; Mitteer, R.A., Jr.; Zhang, Z.; Kim, H.J.; Yeo, E.; Zhang, D.; Cai, P.; Li, C.; et al. c-Met-mediated endothelial plasticity drives aberrant vascularization and chemoresistance in glioblastoma. J. Clin. Investig. 2016, 126, 1801–1814. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.; Hamzah, J.; Payne, C.J.; Ganss, R. Tumor-targeted TNFα stabilizes tumor vessels and enhances active immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 7841–7846. [Google Scholar] [CrossRef] [PubMed]
- Johansson-Percival, A.; Li, Z.J.; Lakhiani, D.D.; He, B.; Wang, X.; Hamzah, J.; Ganss, R. Intratumoral LIGHT restores pericyte contractile properties and vessel integrity. Cell Rep. 2015, 13, 2687–2698. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.; Duquette, M.; Liu, J.; Drapkin, R.; Lawler, J.; Petrik, J. Combined therapy with thrombospondin-1 type I repeats (3TSR) and chemotherapy induces regression and significantly improves survival in a preclinical model of advanced stage epithelial ovarian cancer. FASEB J. 2015, 29, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Peterson, T.E.; Kirkpatrick, N.D.; Huang, Y.; Farrar, C.T.; Marijt, K.A.; Kloepper, J.; Datta, M.; Amoozgar, Z.; Seano, G.; Jung, K.; et al. Dual inhibition of Ang-2 and VEGF receptors normalizes tumor vasculature and prolongs survival in glioblastoma by altering macrophages. Proc. Natl. Acad. Sci. USA 2016, 113, 4470–4475. [Google Scholar] [CrossRef] [PubMed]
- Avallone, A.; Pecori, B.; Bianco, F.; Aloj, L.; Tatangelo, F.; Romano, C.; Granata, V.; Marone, P.; Leone, A.; Botti, G.; et al. Critical role of bevacizumab scheduling in combination with pre-surgical chemo-radiotherapy in MRI-defined high-risk locally advanced rectal cancer: Results of the BRANCH trial. Oncotarget 2015, 6, 30394–30407. [Google Scholar] [PubMed]
- Tolaney, S.M.; Boucher, Y.; Duda, D.G.; Martin, J.D.; Seano, G.; Ancukiewicz, M.; Barry, W.T.; Goel, S.; Lahdenrata, J.; Isakoff, S.J.; et al. Role of vascular density and normalization in response to neoadjuvant bevacizumab and chemotherapy in breast cancer patients. Proc. Natl. Acad. Sci. USA 2015, 112, 14325–14330. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Yoshihara, F.; Tojima, T.; Ooashi, N.; Yoon, W.; Mikoshiba, K.; Bennett, V.; Kamiguchi, H. L1-dependent neuritogenesis involves ankyrinB that mediates L1CAM coupling with retrograde actin flow. J. Cell Biol. 2003, 163, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Gil, O.D.; Sakurai, T.; Bradley, A.E.; Fink, M.Y.; Cassella, M.R.; Kuo, J.A.; Felsenfeld, D.P. Ankyrin binding mediates L1CAM interactions with static components of the cytoskeleton and inhibits retrograde movement of L1CAM on the cell surface. J. Cell Biol. 2003, 162, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Dickson, T.C.; Mintz, C.D.; Benson, D.L.; Salton, S.R. Functional binding interaction identified between the axonal CAM L1 and members of the ERM family. J. Cell Biol. 2002, 157, 1105–1112. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, A.W.; Kamei, Y.; Kamiguchi, H.; Wong, E.V.; Rapoport, I.; Kirchhausen, T.; Beach, C.M.; Landreth, G.; Lemmon, S.K.; Lemmon, V. L1 endocytosis is controlled by a phosphorylation-dephosphorylation cycle stimulated by outside-in signaling by L1. J. Cell Biol. 2002, 157, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Mechtersheimer, S.; Gutwein, P.; Agmon-Levin, N.; Stoeck, A.; Oleszewski, M.; Riedle, S.; Postina, R.; Fahrenholz, F.; Fogel, M.; Lemmon, V.; et al. Ectodomain shedding of L1 adhesion molecule promotes cell migration by autocrine binding to integrins. J. Cell Biol. 2001, 155, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Maretzky, T.; Schulte, M.; Ludwig, A.; Rose-John, S.; Blobel, C.; Hartmann, D.; Altevogt, P.; Saftig, P.; Reiss, K. L1 is sequentially processed by two differently activated metalloproteases and presenilin/γ-secretase and regulates neural cell adhesion, cell migration, and neurite outgrowth. Mol. Cell. Biol. 2005, 25, 9040–9053. [Google Scholar] [CrossRef] [PubMed]
- Riedle, S.; Kiefel, H.; Gast, D.; Bondong, S.; Wolterink, S.; Gutwein, P.; Altevogt, P. Nuclear translocation and signalling of L1CAM in human carcinoma cells requires ADAM10 and presenilin/γ-secretase activity. Biochem. J. 2009, 420, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Maness, P.F.; Schachner, M. Neural recognition molecules of the immunoglobulin superfamily: Signaling transducers of axon guidance and neuronal migration. Nat. Neurosci. 2007, 10, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Kenwrick, S.; Watkins, A.; Angelis, E.D. Neural cell recognition molecule L1: Relating biological complexity to human disease mutations. Hum. Mol. Genet. 2000, 9, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Thor, G.; Probstmeier, R.; Schachner, M. Characterization of the cell adhesion molecules L1, N-CAM and J1 in the mouse intestine. EMBO J. 1987, 6, 2581–2586. [Google Scholar] [PubMed]
- Nolte, C.; Moos, M.; Schachner, M. Immunolocalization of the neural cell adhesion molecule L1 in epithelia of rodents. Cell Tissue Res. 1999, 298, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Debiec, H.; Christensen, E.I.; Ronco, P.M. The cell adhesion molecule L1 is developmentally regulated in the renal epithelium and is involved in kidney branching morphogenesis. J. Cell Biol. 1998, 143, 2067–2079. [Google Scholar] [CrossRef] [PubMed]
- Zecchini, S.; Bianchi, M.; Colombo, N.; Fasani, R.; Goisis, G.; Casadio, C.; Viale, G.; Liu, J.; Herlyn, M.; Godwin, A.K.; et al. The differential role of L1 in ovarian carcinoma and normal ovarian surface epithelium. Cancer Res. 2008, 68, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Pancook, J.D.; Reisfeld, R.A.; Varki, N.; Vitiello, A.; Fox, R.I.; Montgomery, A.M. Expression and regulation of the neural cell adhesion molecule L1 on human cells of myelomonocytic and lymphoid origin. J. Immunol. 1997, 158, 4413–4421. [Google Scholar] [PubMed]
- Maddaluno, L.; Verbrugge, S.E.; Martinoli, C.; Matteoli, G.; Chiavelli, A.; Zeng, Y.; Williams, E.D.; Rescigno, M.; Cavallaro, U. The adhesion molecule L1 regulates transendothelial migration and trafficking of dendritic cells. J. Exp. Med. 2009, 206, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Kallunki, P.; Edelman, G.M.; Jones, F.S. Tissue-specific expression of the L1 cell adhesion molecule is modulated by the neural restrictive silencer element. J. Cell Biol. 1997, 138, 1343–1354. [Google Scholar] [CrossRef] [PubMed]
- Colombo, F.; Meldolesi, J. L1CAM and NCAM: From adhesion proteins to pharmacological targets. Trends Pharmacol. Sci. 2015, 36, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Mikulak, J.; Negrini, S.; Klajn, A.; D’Alessandro, R.; Mavilio, D.; Meldolesi, J. Dual REST-dependence of L1CAM: From gene expression to alternative splicing governed by Nova2 in neural cells. J. Neurochem. 2012, 120, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Altevogt, P.; Doberstein, K.; Fogel, M. L1CAM in human cancer. Int. J. Cancer 2016, 138, 1565–1576. [Google Scholar] [CrossRef] [PubMed]
- Hua, T.; Liu, S.; Xin, X.; Jin, Z.; Liu, Q.; Chi, S.; Wang, X.; Wang, H. Prognostic significance of L1 cell adhesion molecule in cancer patients: A systematic review and meta-analysis. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Van der Putten, L.J.; Visser, N.C.; van de Vijver, K.; Santacana, M.; Bronsert, P.; Bulten, J.; Hirschfeld, M.; Colas, E.; Gil-Moreno, A.; Garcia, A.; et al. L1CAM expression in endometrial carcinomas: An ENITEC collaboration study. Br. J. Cancer 2016, 115, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Pasanen, A.; Tuomi, T.; Isola, J.; Staff, S.; Butzow, R.; Loukovaara, M. L1 cell adhesion molecule as a predictor of disease-specific survival and patterns of relapse in endometrial cancer. Int. J. Gynecol. Cancer 2016, 26, 1465–1471. [Google Scholar] [CrossRef] [PubMed]
- Kiefel, H.; Bondong, S.; Hazin, J.; Ridinger, J.; Schirmer, U.; Riedle, S.; Altevogt, P. L1CAM: A major driver for tumor cell invasion and motility. Cell Adhes. Migr. 2012, 6, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Valiente, M.; Obenauf, A.C.; Jin, X.; Chen, Q.; Zhang, X.H.; Lee, D.J.; Chaft, J.E.; Kris, M.G.; Huse, J.T.; Brogi, E.; et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 2014, 156, 1002–1016. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wong, C.C.; Wei, H.; Gilkes, D.M.; Korangath, P.; Chaturvedi, P.; Schito, L.; Chen, J.; Krishnamachary, B.; Winnard, P.T., Jr.; et al. HIF-1-dependent expression of angiopoietin-like 4 and L1CAM mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene 2012, 31, 1757–1770. [Google Scholar] [CrossRef] [PubMed]
- Burgett, M.E.; Lathia, J.D.; Roth, P.; Nowacki, A.S.; Galileo, D.S.; Pugacheva, E.; Huang, P.; Vasanji, A.; Li, M.; Byzova, T.; et al. Direct contact with perivascular tumor cells enhances integrin αvβ3 signaling and migration of endothelial cells. Oncotarget 2016, 7, 43852–43867. [Google Scholar] [PubMed]
- Felding-Habermann, B.; Silletti, S.; Mei, F.; Siu, C.H.; Yip, P.M.; Brooks, P.C.; Cheresh, D.A.; O’Toole, T.E.; Ginsberg, M.H.; Montgomery, A.M. A single immunoglobulin-like domain of the human neural cell adhesion molecule L1 supports adhesion by multiple vascular and platelet integrins. J. Cell Biol. 1997, 139, 1567–1581. [Google Scholar] [CrossRef] [PubMed]
- Thies, A.; Schachner, M.; Moll, I.; Berger, J.; Schulze, H.J.; Brunner, G.; Schumacher, U. Overexpression of the cell adhesion molecule L1 is associated with metastasis in cutaneous malignant melanoma. Eur. J. Cancer 2002, 38, 1708–1716. [Google Scholar] [CrossRef]
- Kaifi, J.T.; Strelow, A.; Schurr, P.G.; Reichelt, U.; Yekebas, E.F.; Wachowiak, R.; Quaas, A.; Strate, T.; Schaefer, H.; Sauter, G.; et al. L1 (CD171) is highly expressed in gastrointestinal stromal tumors. Mod. Pathol. 2006, 19, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Rawnaq, T.; Quaas, A.; Zander, H.; Gros, S.J.; Reichelt, U.; Blessmann, M.; Wilzcak, W.; Schachner, M.; Sauter, G.; Izbicki, J.R.; et al. L1 is highly expressed in tumors of the nervous system: A study of over 8000 human tissues. J. Surg. Res. 2012, 173, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Magrini, E.; Villa, A.; Angiolini, F.; Doni, A.; Mazzarol, G.; Rudini, N.; Maddaluno, L.; Komuta, M.; Topal, B.; Prenen, H.; et al. Endothelial deficiency of L1 reduces tumor angiogenesis and promotes vessel normalization. J. Clin. Investig. 2014, 124, 4335–4350. [Google Scholar] [CrossRef] [PubMed]
- Issa, Y.; Nummer, D.; Seibel, T.; Muerkoster, S.S.; Koch, M.; Schmitz-Winnenthal, F.H.; Galindo, L.; Weitz, J.; Beckhove, P.; Altevogt, P. Enhanced L1CAM expression on pancreatic tumor endothelium mediates selective tumor cell transmigration. J. Mol. Med. 2009, 87, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Voura, E.B.; Ramjeesingh, R.A.; Montgomery, A.M.; Siu, C.H. Involvement of integrin αvβ3 and cell adhesion molecule L1 in transendothelial migration of melanoma cells. Mol. Biol. Cell 2001, 12, 2699–2710. [Google Scholar] [CrossRef] [PubMed]
- Castellani, V.; de Angelis, E.; Kenwrick, S.; Rougon, G. Cis and trans interactions of L1 with neuropilin-1 control axonal responses to semaphorin 3A. EMBO J. 2002, 21, 6348–6357. [Google Scholar] [CrossRef] [PubMed]
- Dippel, V.; Milde-Langosch, K.; Wicklein, D.; Schumacher, U.; Altevogt, P.; Oliveira-Ferrer, L.; Janicke, F.; Schroder, C. Influence of L1CAM expression of breast cancer cells on adhesion to endothelial cells. J. Cancer Res. Clin. Oncol. 2013, 139, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Friedli, A.; Fischer, E.; Novak-Hofer, I.; Cohrs, S.; Ballmer-Hofer, K.; Schubiger, P.A.; Schibli, R.; Grunberg, J. The soluble form of the cancer-associated L1 cell adhesion molecule is a pro-angiogenic factor. Int. J. Biochem. Cell Biol. 2009, 41, 1572–1580. [Google Scholar] [CrossRef] [PubMed]
- Doberstein, K.; Pfeilschifter, J.; Gutwein, P. The transcription factor PAX2 regulates ADAM10 expression in renal cell carcinoma. Carcinogenesis 2011, 32, 1713–1723. [Google Scholar] [CrossRef] [PubMed]
- Hall, H.; Hubbell, J.A. Matrix-bound sixth Ig-like domain of cell adhesion molecule L1 acts as an angiogenic factor by ligating αvβ3 integrin and activating VEGF-R2. Microvasc. Res. 2004, 68, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Reidy, M.; Zihlmann, P.; Hubbell, J.A.; Hall, H. Activation of cell-survival transcription factor NFκB in L1Ig6-stimulated endothelial cells. J. Biomed. Mater. Res. A 2006, 77, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Magrini, E.; Cavallaro, U.; Bianchi, F. Microarray profiling of L1-overexpressing endothelial cells reveals STAT3 activation via IL-6/IL-6Rα axis. Genom. Data 2015, 4, 137–139. [Google Scholar] [CrossRef] [PubMed]
- Potenta, S.; Zeisberg, E.; Kalluri, R. The role of endothelial-to-mesenchymal transition in cancer progression. Br. J. Cancer 2008, 99, 1375–1379. [Google Scholar] [CrossRef] [PubMed]
- Silletti, S.; Yebra, M.; Perez, B.; Cirulli, V.; McMahon, M.; Montgomery, A.M. Extracellular signal-regulated kinase (ERK)-dependent gene expression contributes to L1 cell adhesion molecule-dependent motility and invasion. J. Biol. Chem. 2004, 279, 28880–28888. [Google Scholar] [CrossRef] [PubMed]
- Gast, D.; Riedle, S.; Issa, Y.; Pfeifer, M.; Beckhove, P.; Sanderson, M.P.; Arlt, M.; Moldenhauer, G.; Fogel, M.; Kruger, A.; et al. The cytoplasmic part of L1CAM controls growth and gene expression in human tumors that is reversed by therapeutic antibodies. Oncogene 2008, 27, 1281–1289. [Google Scholar] [CrossRef] [PubMed]
- Demircioglu, F.; Hodivala-Dilke, K. αvβ3 Integrin and tumour blood vessels-learning from the past to shape the future. Curr. Opin. Cell Biol. 2016, 42, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Hall, H.; Djonov, V.; Ehrbar, M.; Hoechli, M.; Hubbell, J.A. Heterophilic interactions between cell adhesion molecule L1 and αvβ3 integrin induce HUVEC process extension in vitro and angiogenesis in vivo. Angiogenesis 2004, 7, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Hoefnagel, C.A.; Rutgers, M.; Buitenhuis, C.K.; Smets, L.A.; de Kraker, J.; Meli, M.; Carrel, F.; Amstutz, H.; Schubiger, P.A.; Novak-Hofer, I. A comparison of targeting of neuroblastoma with mIBG and anti L1CAM antibody mAb chCE7: Therapeutic efficacy in a neuroblastoma xenograft model and imaging of neuroblastoma patients. Eur. J. Nucl. Med. 2001, 28, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Novak-Hofer, I. The L1 cell adhesion molecule as a target for radioimmunotherapy. Cancer Biother. Radiopharm. 2007, 22, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Grunberg, J.; Novak-Hofer, I.; Honer, M.; Zimmermann, K.; Knogler, K.; Blauenstein, P.; Ametamey, S.; Maecke, H.R.; Schubiger, P.A. In vivo evaluation of 177Lu- and 67/64Cu-labeled recombinant fragments of antibody chCE7 for radioimmunotherapy and PET imaging of L1CAM-positive tumors. Clin. Cancer Res. 2005, 11, 5112–5120. [Google Scholar] [CrossRef] [PubMed]
- Grunberg, J.; Lindenblatt, D.; Dorrer, H.; Cohrs, S.; Zhernosekov, K.; Koster, U.; Turler, A.; Fischer, E.; Schibli, R. Anti-L1CAM radioimmunotherapy is more effective with the radiolanthanide terbium-161 compared to lutetium-177 in an ovarian cancer model. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 1907–1915. [Google Scholar] [CrossRef] [PubMed]
- Lindenblatt, D.; Fischer, E.; Cohrs, S.; Schibli, R.; Grunberg, J. Paclitaxel improved anti-L1CAM lutetium-177 radioimmunotherapy in an ovarian cancer xenograft model. EJNMMI Res. 2014, 4, 54. [Google Scholar] [CrossRef] [PubMed]
- Novak-Hofer, I.; Amstutz, H.P.; Macke, H.R.; Schwarzbach, R.; Zimmermann, K.; Morgenthaler, J.J.; Schubiger, P.A. Cellular processing of copper-67-labeled monoclonal antibody chCE7 by human neuroblastoma cells. Cancer Res. 1995, 55, 46–50. [Google Scholar] [PubMed]
- Weidle, U.H.; Eggle, D.; Klostermann, S. L1CAM as a target for treatment of cancer with monoclonal antibodies. Anticancer Res. 2009, 29, 4919–4931. [Google Scholar] [PubMed]
- Arlt, M.J.; Novak-Hofer, I.; Gast, D.; Gschwend, V.; Moldenhauer, G.; Grunberg, J.; Honer, M.; Schubiger, P.A.; Altevogt, P.; Kruger, A. Efficient inhibition of intra-peritoneal tumor growth and dissemination of human ovarian carcinoma cells in nude mice by anti-L1-cell adhesion molecule monoclonal antibody treatment. Cancer Res. 2006, 66, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Meier, F.; Busch, S.; Gast, D.; Goppert, A.; Altevogt, P.; Maczey, E.; Riedle, S.; Garbe, C.; Schittek, B. The adhesion molecule L1 (CD171) promotes melanoma progression. Int. J. Cancer 2006, 119, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Knogler, K.; Grunberg, J.; Zimmermann, K.; Cohrs, S.; Honer, M.; Ametamey, S.; Altevogt, P.; Fogel, M.; Schubiger, P.A.; Novak-Hofer, I. Copper-67 radioimmunotherapy and growth inhibition by anti-L1-cell adhesion molecule monoclonal antibodies in a therapy model of ovarian cancer metastasis. Clin. Cancer Res. 2007, 13, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Min, J.K.; Kim, J.M.; Li, S.; Lee, J.W.; Yoon, H.; Ryu, C.J.; Jeon, S.H.; Lee, J.H.; Kim, J.Y.; Yoon, H.K.; et al. L1 cell adhesion molecule is a novel therapeutic target in intrahepatic cholangiocarcinoma. Clin. Cancer Res. 2010, 16, 3571–3580. [Google Scholar] [CrossRef] [PubMed]
- Wolterink, S.; Moldenhauer, G.; Fogel, M.; Kiefel, H.; Pfeifer, M.; Luttgau, S.; Gouveia, R.; Costa, J.; Endell, J.; Moebius, U.; et al. Therapeutic antibodies to human L1CAM: Functional characterization and application in a mouse model for ovarian carcinoma. Cancer Res. 2010, 70, 2504–2515. [Google Scholar] [CrossRef] [PubMed]
- Sung, S.Y.; Wu, I.H.; Chuang, P.H.; Petros, J.A.; Wu, H.C.; Zeng, H.J.; Huang, W.C.; Chung, L.W.; Hsieh, C.L. Targeting L1 cell adhesion molecule expression using liposome-encapsulated siRNA suppresses prostate cancer bone metastasis and growth. Oncotarget 2014, 5, 9911–9929. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Preclinical Model | Outcome | References |
|---|---|---|---|
| chCE7 (radioactively labelled) | Neuroblastoma; Ovarian cancer | High tumor uptake; Improved response to chemotherapy; Reduced tumor growth | [89,90,91] |
| A10-A3 | Cholangiocarcinoma | Reduced tumor growth | [97] |
| L1-11A | Ovarian cancer; Melanoma | Inhibition of cancer cell invasion; Reduced tumor growth and dissemination | [94,95] |
| L1-9.3 | Ovarian cancer | Reduced tumor growth; Macrophage infiltration | [98] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angiolini, F.; Cavallaro, U. The Pleiotropic Role of L1CAM in Tumor Vasculature. Int. J. Mol. Sci. 2017, 18, 254. https://doi.org/10.3390/ijms18020254
Angiolini F, Cavallaro U. The Pleiotropic Role of L1CAM in Tumor Vasculature. International Journal of Molecular Sciences. 2017; 18(2):254. https://doi.org/10.3390/ijms18020254
Chicago/Turabian StyleAngiolini, Francesca, and Ugo Cavallaro. 2017. "The Pleiotropic Role of L1CAM in Tumor Vasculature" International Journal of Molecular Sciences 18, no. 2: 254. https://doi.org/10.3390/ijms18020254
APA StyleAngiolini, F., & Cavallaro, U. (2017). The Pleiotropic Role of L1CAM in Tumor Vasculature. International Journal of Molecular Sciences, 18(2), 254. https://doi.org/10.3390/ijms18020254