
An Integrated View of Aristolochic Acid Nephropathy: Update of the Literature

Abstract
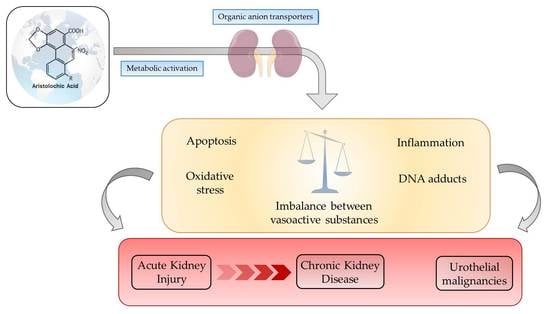
:
1. Introduction
2. The Facts—A Belgian Story of New Renal Disease
3. Epidemiology of AAN and BEN: A True Link?
3.1. Aristolochic Acid Nephropathy
3.2. Balkan Endemic Nephropathy
4. Clinical Features of AAN
4.1. Nephrotoxicity
4.2. Urothelial Malignancies
4.3. Diagnostic Criteria of AAN
5. Properties of AA and Mechanisms of Nephrotoxicity and Carcinogenicity
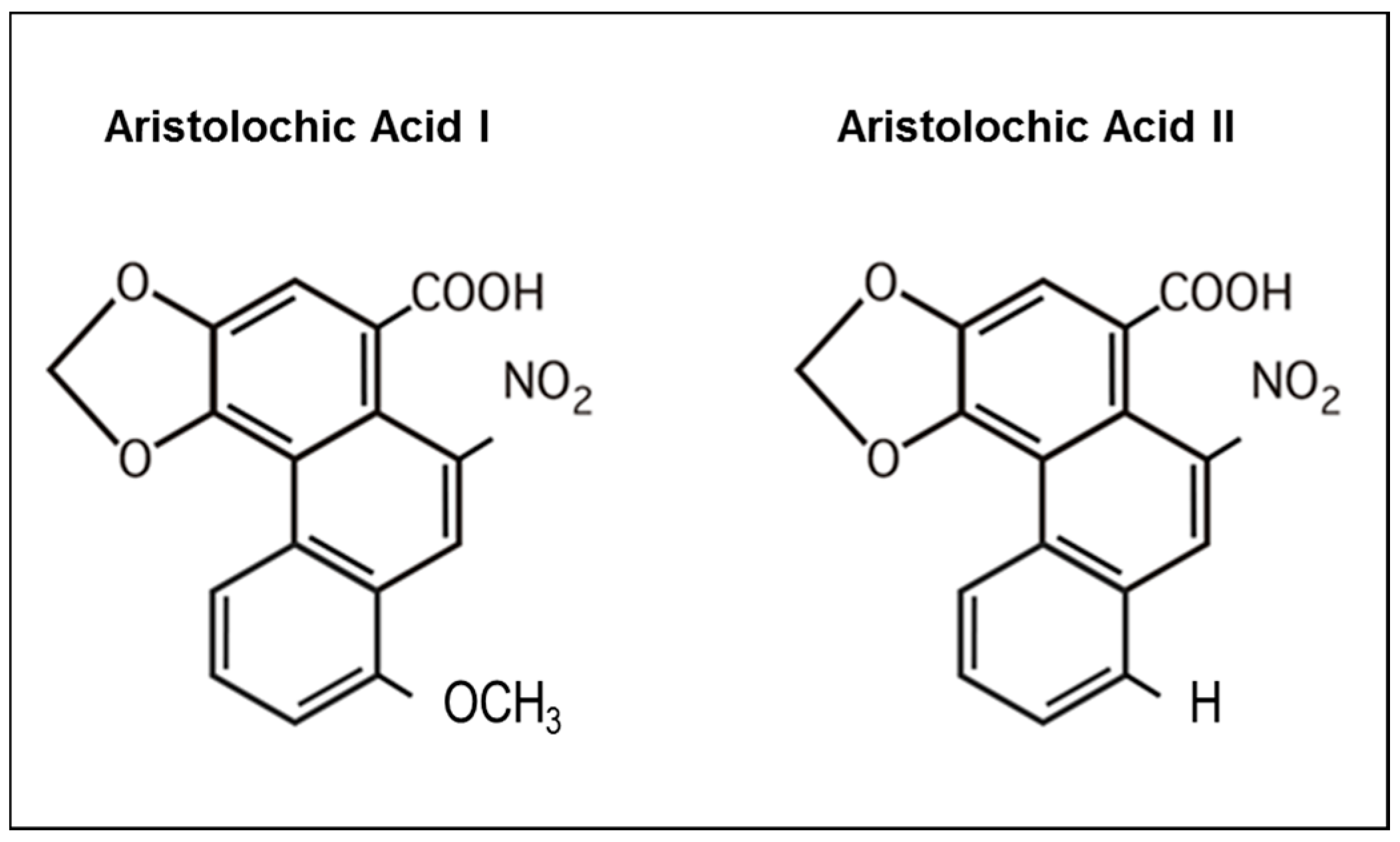
5.1. AA Structure-Activity Relationship
5.2. Exposition to AA
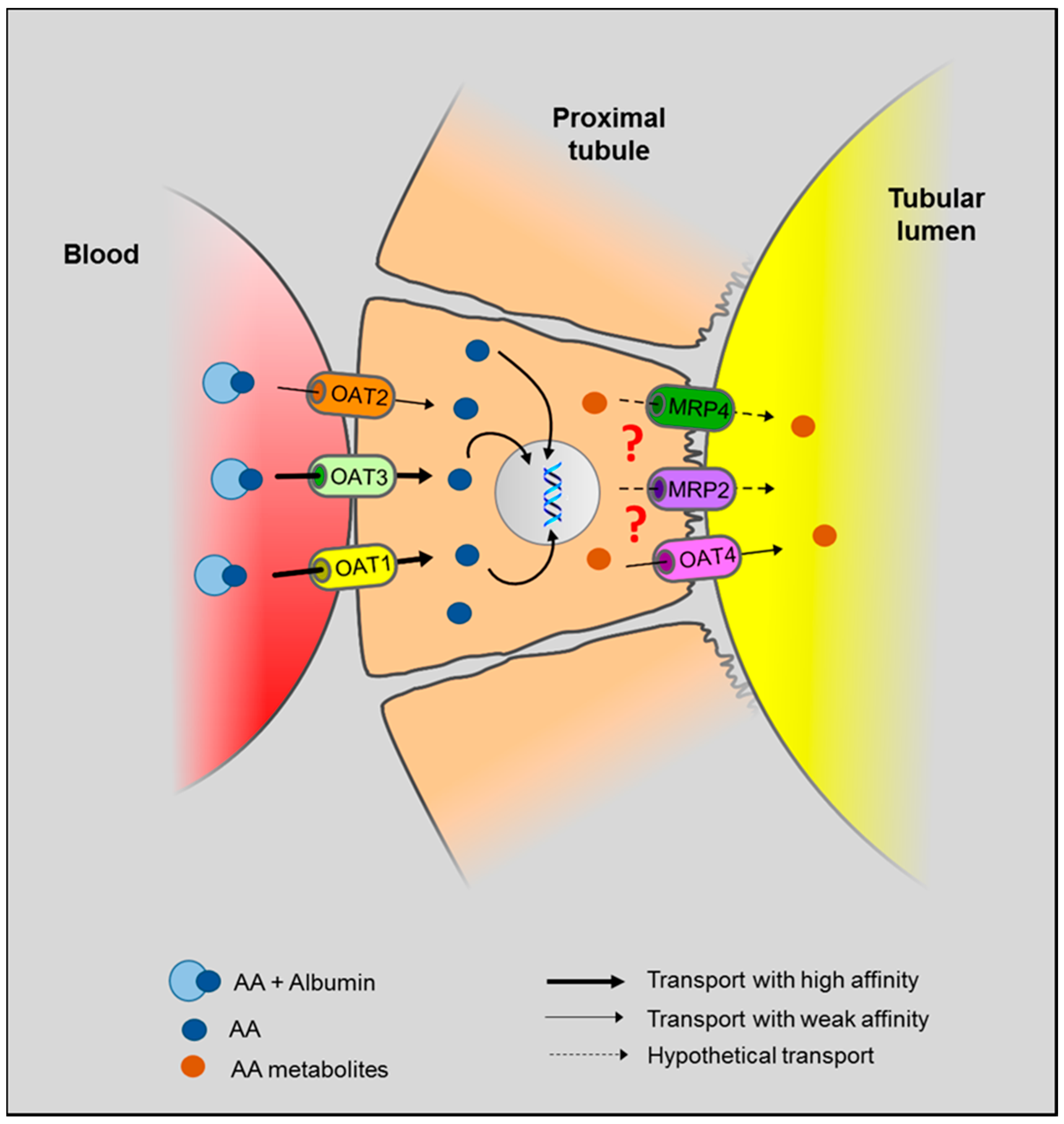
5.3. Role of OAT in AA-Induced Toxicity
5.4. Metabolic Activation of AA—How That Works?
5.5. Pathogenesis of AAN—What Do We Know from Experimental Studies?
5.5.1. A Biphasic Evolution
5.5.2. Cytotoxicity
DNA Adducts
Oxidative Stress
Apoptosis
5.5.3. Inflammation
5.5.4. Vascular and Tubular Compartments in AAN: The Egg or the Chicken?
5.5.5. Fibrosis
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vanherweghem, J.L.; Depierreux, M.; Tielemans, C.; Abramowicz, D.; Dratwa, M.; Jadoul, M.; Richard, C.; Vandervelde, D.; Verbeelen, D.; Vanhaelen-Fastre, R. Rapidly progressive interstitial renal fibrosis in young women: Association with slimming regimen including Chinese herbs. Lancet 1993, 341, 387–391. [Google Scholar] [CrossRef]
- Depierreux, M.; Van Damme, B.; Vanden Houte, K.; Vanherweghem, J.L. Pathologic aspects of a newly described nephropathy related to the prolonged use of Chinese herbs. Am. J. Kidney Dis. 1994, 24, 172–180. [Google Scholar] [CrossRef]
- Vanhaelen, M.; Vanhaelen-Fastre, R.; But, P.; Vanherweghem, J.-L. Identification of aristolochic acid in Chinese herbs. Lancet 1994, 343, 174. [Google Scholar] [CrossRef]
- Cosyns, J.; Jadoul, M.; Squifflet, J.-P.; Van Cangh, P.; van Ypersele de Strihou, C. Urothelial malignancy in nephropathy due to Chinese herbs. Lancet 1994, 344, 188. [Google Scholar] [CrossRef]
- Nortier, J.L.; Martinez Muniz, M.-C.; Schmeiser, H.H.; Arlt, V.M.; Bieler, C.A.; Petein, M.; Depierreux, M.F.; de Pauw, L.; Abramowicz, D.; Vereerstraeten, P.; et al. Urothelial carcinoma associated with the use of a Chinese herb. N. Engl. J. Med. 2000, 342, 1686–1692. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Some Traditional Herbal Medicines, Some Mycotoxins, Naphthalene and Styrene. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC Press: Lyon, France, 2002; Volume 82. [Google Scholar]
- Debelle, F.D.; Vanherweghem, J.-L.; Nortier, J.L. Aristolochic acid nephropathy: A worldwide problem. Kidney Int. 2008, 74, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-H.; Dickman, K.G.; Moriya, M.; Zavadil, J.; Sidorenko, V.S.; Edwards, K.L.; Gnatenko, D.V.; Wu, L.; Turesky, R.J.; Wu, X.-R.; et al. Aristolochic acid-associated urothelial cancer in Taiwan. Proc. Natl. Acad. Sci. USA 2012, 109, 8241–8246. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, S.-C.; Lin, I.-H.; Tseng, W.-L.; Lee, C.-H.; Wang, J.-D. Prescription profile of potentially aristolochic acid containing Chinese herbal products: An analysis of National Health Insurance data in Taiwan between 1997 and 2003. Chin. Med. 2008, 3, 13. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.N.; Lai, J.N.; Chen, P.C.; Tseng, W.L.; Chen, Y.Y.; Hwang, J.S.; Wang, J.D. Increased risks of chronic kidney disease associated with prescribed Chinese herbal products suspected to contain aristolochic acid. Nephrology 2009, 14, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Ng, Y.Y.; Yu, S.; Chen, T.W.; Wu, S.C.; Yang, A.H.; Yang, W.C. Interstitial renal fibrosis in a young woman: Association with a Chinese preparation given for irregular menses. Nephrol. Dial. Transplant. 1998, 13, 2115–2117. [Google Scholar] [CrossRef] [PubMed]
- Cosyns, J.; Jadoul, M.; Squifflet, J.; Plaen, J.; Ferluga, D.; van Ypersele de Strihou, C. Chinese herbs nephropathy: A clue to Balkan endemic nephropathy? Kidney Int. 1994, 45, 1680–1688. [Google Scholar] [CrossRef] [PubMed]
- Arlt, V.M.; Ferluga, D.; Stiborova, M.; Pfohl-Leszkowicz, A.; Vukelic, M.; Ceovic, S.; Schmeiser, H.H.; Cosyns, J.-P. Is aristolochic acid a risk factor for Balkan endemic nephropathy-associated urothelial cancer? Int. J. Cancer 2002, 101, 500–502. [Google Scholar] [CrossRef] [PubMed]
- Arlt, V.M.; Alunni-Perret, V.; Quatrehomme, G.; Ohayon, P.; Albano, L.; Gaïd, H.; Michiels, J.-F.; Meyrier, A.; Cassuto, E.; Wiessler, M.; et al. Aristolochic acid (AA)-DNA adduct as marker of AA exposure and risk factor for AA nephropathy-associated cancer. Int. J. Cancer 2004, 111, 977–980. [Google Scholar] [CrossRef] [PubMed]
- Nortier, J.L.; Pozdzik, A.; Roumeguere, T.; Vanherweghem, J.-L. Néphropathie aux acides aristolochiques (“Néphropathie aux herbes chinoises”). Néphrol. Thér. 2015, 11, 574–588. [Google Scholar] [CrossRef] [PubMed]
- Mengs, U. Acute toxicity of aristolochic acid in rodents. Arch. Toxicol. 1987, 59, 328–331. [Google Scholar] [CrossRef] [PubMed]
- Schmeiser, H.H.; Bieler, C.A.; Wiessler, M.; van Ypersele de, S.C.; Cosyns, J.P. Detection of DNA adducts formed by aristolochic acid in renal tissue from patients with Chinese herbs nephropathy. Cancer Res. 1996, 56, 2025–2028. [Google Scholar] [PubMed]
- Martinez, M.-C.M.; Nortier, J.; Vereerstraeten, P.; Vanherweghem, J.-L. Progression rate of Chinese herb nephropathy: Impact of Aristolochia fangchi ingested dose. Nephrol. Dial. Transplant. 2002, 17, 408–412. [Google Scholar] [CrossRef] [PubMed]
- Cosyns, J.P.; Goebbels, R.M.; Liberton, V.; Schmeiser, H.H.; Bieler, C.A.; Bernard, A.M. Chinese herbs nephropathy-associated slimming regimen induces tumours in the forestomach but no interstitial nephropathy in rats. Arch. Toxicol. 1998, 72, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Gillerot, G.; Jadoul, M.; Arlt, V.M.; van Ypersele de Strihou, C.; Schmeiser, H.H.; But, P.P.; Bieler, C.A.; Cosyns, J.P. Aristolochic acid nephropathy in a Chinese patient: Time to abandon the term “Chinese herbs nephropathy”? Am. J. Kidney Dis. 2001, 38, E26. [Google Scholar] [CrossRef] [PubMed]
- Vanherweghem, J.-L. Misuse of Herbal Remedies: The case of an outbreak of terminal renal failure in Belgium (Chinese Herbs Nephropathy). J. Altern. Complement. Med. 1998, 4, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Pena, J.; Borras, M.; Ramos, J.; Montoliu, J. Rapidly progressive interstitial renal fibrosis due to a chronic intake of a herb (Aristolochia pistolochia) infusion. Nephrol. Dial. Transplant. 1996, 11, 1359–1360. [Google Scholar] [CrossRef] [PubMed]
- Stengel, B.; Jones, E. End-Stage Renal Insufficiency Associated With Chinese Herbal Consumption in France. Nephrologie 1998, 19, 15–20. [Google Scholar] [PubMed]
- Lord, G.M.; Tagore, R.; Cook, T.; Gower, P.; Pusey, C.D. Nephropathy caused by Chinese herbs in the UK. Lancet 1999, 354, 481–482. [Google Scholar] [CrossRef]
- Krumme, B.; Endmeir, R.; Vanhaelen, M.; Walb, D. Reversible Fanconi syndrome after ingestion of a Chinese herbal “remedy” containing aristolochic acid. Nephrol. Dial. Transplant. 2001, 16, 400–402. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.M.; Chen, T.P.; Bennett, W.M. Chinese herb nephropathy. Proc. (Bayl. Univ. Med. Cent.) 2000, 13, 334–337. [Google Scholar]
- Chau, W.; Ross, R.; Li, J.Y.Z.; Yong, T.Y.; Klebe, S.; Barbara, J.A. Nephropathy associated with use of a Chinese herbal product containing aristolochic acid. Med. J. Aust. 2011, 194, 367–368. [Google Scholar] [PubMed]
- Chen, W.; Chen, Y.; Li, A. The clinical and pathological manifestations of aristolochic acid nephropathy—The report of 58 cases. Zhonghua Yi Xue Za Zhi 2001, 81, 1101–1105. [Google Scholar] [PubMed]
- Lo, S.H.K.; Mo, K.L.; Wong, K.S.; Poon, S.P.; Chan, C.K.; Lai, C.K.; Chan, A. Aristolohic acid nephropathy complicating a patient with focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2004, 19, 1913–1915. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, X.; Wang, H. Possible mechanisms explaining the tendency towards interstitial fibrosis in aristolochic acid-induced acute tubular necrosis. Nephrol. Dial. Transplant. 2007, 22, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Su, T.; Li, X.M.; Wang, X.; Cai, S.Q.; Meng, L.Q.; Zou, W.Z.; Wang, H.Y. Aristolochic acid nephropathy: Variation in presentation and prognosis. Nephrol. Dial. Transplant. 2012, 27, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Lin, C.H.; Chang, S.H.; Hsu, H.C. Rapidly progressive fibrosing interstitial nephritis associated with Chinese herbal drugs. Am. J. Kidney Dis. 2000, 35, 313–318. [Google Scholar] [CrossRef]
- Lee, C.T.; Wu, M.S.; Lu, K.; Hsu, K.T. Renal tubular acidosis, hypokalemic paralysis, rhabdomyolysis, and acute renal failure—A rare presentation of Chinese herbal nephropathy. Ren. Fail. 1999, 21, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.T.; Fu, L.S.; Chung, L.H.; Hung, S.C.; Huang, Y.T.; Chi, C.S. Fanconi’s syndrome, interstitial fibrosis and renal failure by aristolochic acid in Chinese herbs. Pediatr. Nephrol. 2006, 21, 577–579. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Nishida, R.; Sawai, K.; Nagae, T.; Shinkai, S.; Ishikawa, M.; Maeda, K.; Murata, M.; Seta, K.; Okuda, J.; et al. Traditional remedy-induced Chinese herbs nephropathy showing rapid deterioration of renal function. Nihon Jinzo Gakkai Shi 1997, 39, 794–797. [Google Scholar] [PubMed]
- Tanaka, A.; Nishida, R.; Maeda, K.; Sugawara, A.; Kuwahara, T. Aristolochic acid—Induced Fanconi’s syndrome and nephropathy presenting as hypokalemic paralysis. Clin. Nephrol. 2000, 39, 521116. [Google Scholar]
- Kazama, I.; Matsubara, M.; Michimata, M.; Suzuki, M.; Hatano, R.; Sato, H.; Ito, S. Adult onset Fanconi syndrome: Extensive tubulo-interstitial lesions and glomerulopathy in the early stage of Chinese herbs nephropathy. Clin. Exp. Nephrol. 2004, 8, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Michl, J.; Jennings, H.M.; Kite, G.C.; Ingrouille, M.J.; Simmonds, M.S.J.; Heinrich, M. Is aristolochic acid nephropathy a widespread problem in developing countries: A case study of Aristolochia indica L. in Bangladesh using an ethnobotanical-phytochemical approach. J. Ethnopharmacol. 2013, 149, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, T.; Lee, B.; Choi, H.; Yang, M.; Ihm, C.; Kim, M. Fanconi’s syndrome and subsequent progressive renal failure caused by a Chinese herb containing aristolochic acid. Nephrology 2004, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Poon, W.T.; Lai, C.K.; Chan, A.Y.W. Aristolochic acid nephropathy: The Hong Kong perspective. Hong Kong J. Nephrol. 2007, 9, 7–14. [Google Scholar] [CrossRef]
- Nortier, J.L.; Vanherweghem, J.-L. For patients taking herbal therapy—Lessons from aristolochic acid nephropathy. Nephrol. Dial. Transplant. 2007, 22, 1512–1517. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.; Chan, J.; Wanke, S.; Neinhuis, C.; Simmonds, M.S.J. Local uses of Aristolochia species and content of nephrotoxic aristolochic acid 1 and 2—A global assessment based on bibliographic sources. J. Ethnopharmacol. 2009, 125, 108–144. [Google Scholar] [CrossRef] [PubMed]
- National Toxicology Program. Aristolochic Acids. Rep. Carcinog. 2011, 12, 45–49. [Google Scholar]
- Stiborová, M.; Arlt, V.M.; Schmeiser, H.H. Balkan endemic nephropathy: An update on its aetiology. Arch. Toxicol. 2016, 90, 2595–2615. [Google Scholar] [CrossRef] [PubMed]
- Pavlović, N.M. Balkan endemic nephropathy—Current status and future perspectives. Clin. Kidney J. 2013, 6, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Batuman, V. Fifty years of Balkan endemic nephropathy: Daunting questions, elusive answers. Kidney Int. 2006, 69, 644–646. [Google Scholar] [CrossRef] [PubMed]
- Stefanović, V.; Polenaković, M. Fifty years of research in balkan endemic nephropathy: Where are we now? Nephron Clin. Pract. 2009, 112, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, P.; Tsolova, S.; Georgieva, R.; Bozhilova, D.; Simeonov, V.; Bonev, A.; Karmaus, W. Clinical markers in adult offspring of families with and without Balkan Endemic Nephropathy. Kidney Int. 2006, 69, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Pavlovic, N.M.; Maksimovic, V.; Maksimovic, J.D.; Orem, W.H.; Tatu, C.A.; Lerch, H.E.; Bunnell, J.E.; Kostic, E.N.; Szilagyi, D.N.; Paunescu, V. Possible health impacts of naturally occurring uptake of aristolochic acids by maize and cucumber roots: Links to the etiology of endemic (Balkan) nephropathy. Environ. Geochem. Health 2013, 35, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Stefanovic, V.; Radovanovic, Z. Balkan endemic nephropathy and associated urothelial cancer. Nat. Clin. Pract. Urol. 2008, 5, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Miletić-Medved, M.; Domijan, A.M.; Peraica, M. Recent data on endemic nephropathy and related urothelial tumors in Croatia. Wien. Klin. Wochenschr. 2005, 117, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Grollman, A.P.; Jelaković, B. Role of environmental toxins in endemic (Balkan) nephropathy. October 2006, Zagreb, Croatia. J. Am. Soc. Nephrol. 2007, 18, 2817–2823. [Google Scholar] [CrossRef] [PubMed]
- Grollman, A.P. Aristolochic Acid Nephropathy: Harbinger of a Global Iatrogenic Disease. Environ. Mol. Mutagen. 2013, 51, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Jelaković, B.; Nikolić, J.; Radovanović, Z.; Nortier, J.; Cosyns, J.P.; Grollman, A.P.; Bašić-Jukić, N.; Belicza, M.; Bukvić, D.; Čavaljuga, S.; et al. Consensus statement on screening, diagnosis, classification and treatment of endemic (Balkan) nephropathy. Nephrol. Dial. Transplant. 2014, 29, 2020–2027. [Google Scholar] [CrossRef] [PubMed]
- Arlt, V.M.; Stiborova, M.; Schmeiser, H.H. Aristolochic acid as a probable human cancer hazard in herbal remedies: A review. Mutagenesis 2002, 17, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Hranjec, T.; Kovac, A.; Kos, J.; Mao, W.; Chen, J.J.; Grollman, A.P.; Jelaković, B. Endemic nephropathy: The case for chronic poisoning by Aristolochia. Croat. Med. J. 2005, 46, 116–125. [Google Scholar] [PubMed]
- De Broe, M.E. Chinese herbs nephropathy and Balkan endemic nephropathy: Toward a single entity, aristolochic acid nephropathy. Kidney Int. 2012, 81, 513–515. [Google Scholar] [CrossRef] [PubMed]
- Arlt, V.M.; Stiborova, M.; vom Brocke, J.; Simoes, M.L.; Lord, G.M.; Nortier, J.L.; Hollstein, M.; Phillips, D.H.; Schmeiser, H.H. Aristolochic acid mutagenesis: Molecular clues to the aetiology of Balkan endemic nephropathy-associated urothelial cancer. Carcinogenesis 2007, 28, 2253–2261. [Google Scholar] [CrossRef] [PubMed]
- Yun, B.H.; Rosenquist, T.A.; Sidorenko, V.; Iden, C.R.; Chen, C.H.; Pu, Y.S.; Bonala, R.; Johnson, F.; Dickman, K.G.; Grollman, A.P.; et al. Biomonitoring of aristolactam-DNA adducts in human tissues using ultra-performance liquid chromatography/ion-trap mass spectrometry. Chem. Res. Toxicol. 2012, 25, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.; Pavlović, N.M.; Li, W.; Chan, C.-K.; Liu, J.; Deng, K.; Wang, Y.; Milosavljević, B.; Kostić, E.N. Quantitation of Aristolochic Acids in Corn, Wheat Grain, and Soil Samples Collected in Serbia: Identifying a Novel Exposure Pathway in the Etiology of Balkan Endemic Nephropathy. J. Agric. Food Chem. 2016, 64, 5928–5934. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Hu, Q.; Chan, W. Uptake and Accumulation of Nephrotoxic and Carcinogenic Aristolochic Acids in Food Crops Grown in Aristolochia clematitis-Contaminated Soil and Water. J. Agric. Food Chem. 2016, 64, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Grollman, A.P.; Shibutani, S.; Moriya, M.; Miller, F.; Wu, L.; Moll, U.; Suzuki, N.; Fernandes, A.; Rosenquist, T.; Medverec, Z.; et al. Aristolochic acid and the etiology of endemic (Balkan) nephropathy. Proc. Natl. Acad. Sci. USA 2007, 104, 12129–12134. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Nishida, R.; Maeda, K.; Sugawara, A.; Kuwahara, T. Chinese herb nephropathy in Japan presents adult-onset Fanconi syndrome: Could different components of aristolochic acids cause a different type of Chinese herb nephropathy? Clin. Nephrol. 2000, 53, 301–306. [Google Scholar] [PubMed]
- Reginster, F.; Jadoul, M.; van Ypersele de Strihou, C. Chinese herbs nephropathy presentation, natural history and fate after transplantation. Nephrol. Dial. Transplant. 1997, 12, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Kabanda, A.; Jadoul, M.; Lauwerys, R.; Bernard, A.; van Ypersele de Strihou, C. Low molecular weight proteinuria in Chinese herbs nephropathy. Kidney Int. 1995, 48, 1571–1576. [Google Scholar] [CrossRef] [PubMed]
- Nortier, J.L.; Deschodt-Lanckman, M.M.; Simon, S.; Thielemans, N.O.; de Prez, E.G.; Depierreux, M.F.; Tielemans, C.L.; Richard, C.; Lauwerys, R.R.; Bernard, A.M.; et al. Proximal tubular injury in Chinese herbs nephropathy: Monitoring by neutral endopeptidase enzymuria. Kidney Int. 1997, 51, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Lord, G.M.; Cook, T.; Arlt, V.M.; Schmeiser, H.H.; Williams, G.; Pusey, C.D. Urothelial malignant disease and Chinese herbal nephropathy. Lancet 2001, 358, 1515–1516. [Google Scholar] [CrossRef]
- Mengs, U.; Lang, W.; Poch, J.A. The carcinogenic action of aristolochic acid in rats. Arch. Toxicol. 1982, 51, 107–119. [Google Scholar] [CrossRef]
- Mengs, U. Tumor induction in mice following exposure to aristolochic acid. Arch. Toxicol. 1988, 61, 504–505. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Liu, Z.H.; Qiu, Q.; Li, H.; Li, L.S. Tumour induction in rats following exposure to short-term high dose aristolochic acid I. Mutagenesis 2005, 20, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Cosyns, J.P.; Dehoux, J.P.; Guiot, Y.; Goebbels, R.M.; Robert, A.; Bernard, A.M.; van Ypersele de Strihou, C. Chronic aristolochic acid toxicity in rabbits: A model of Chinese herbs nephropathy? Kidney Int. 2001, 59, 2164–2173. [Google Scholar] [CrossRef] [PubMed]
- Lemy, A.; Wissing, K.M.; Rorive, S.; Zlotta, A.; Roumeguere, T.; Muniz Martinez, M.C.; Decaestecker, C.; Salmon, I.; Abramowicz, D.; Vanherweghem, J.L.; et al. Late Onset of Bladder Urothelial Carcinoma after Kidney Transplantation for End-Stage Aristolochic Acid Nephropathy: A Case Series With 15-Year Follow-up. Am. J. Kidney Dis. 2008, 51, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Gokmen, M.; Cosyns, J.; Arlt, V.M.; Stiborova, M.; Phillips, D.; Schmeiser, H.H.; Simmonds, M.S.J.; Cook, T.; Vanherweghem, J.-L.; Nortier, J.; et al. The Epidemiology, Diagnosis, and Management of Aristolochic Acid Nephropathy. Ann. Intern. Med. 2013, 158, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Scarpellini, A.; Funck, M.; Verderio, E.A.M.; Johnson, T.S. Development of a Chronic Kidney Disease Model in C57BL/6 Mice with Relevance to Human Pathology. Nephron Extra 2013, 33, 12–29. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Takahashi, D.; Chen, S.-M.; Tsuchiya, R.; Mukoyama, T.; Yamagata, S.; Ogawa, M.; Yoshida, M.; Kondo, S.; Satoh, N.; et al. Acute nephrotoxicity of aristolochic acids in mice. J. Pharm. Pharmacol. 2004, 56, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Nortier, J.L.; Schmeiser, H.H.; Muniz Martinez, M.C.; Arlt, V.M.; Vervaet, C.; Garbar, C.H.; Daelemans, P.; Vabherweghem, J.L. Invasive urothelial carcinoma after exposure to Chinese herbal medicine containing aristolochic acid may occur without severe renal failure. Nephrol. Dial. Transplant. 2003, 18, 426–428. [Google Scholar] [CrossRef] [PubMed]
- Dickman, K.G.; Sweet, D.H.; Bonala, R.; Ray, T.; Wu, A. Physiological and molecular characterization of aristolochic acid transport by the kidney. J. Pharmacol. Exp. Ther. 2011, 338, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, P.; Wei, F.; Lin, R.; Khan, I.A.; Pasco, D.S. Structure activity relationships of aristolochic acid analogues: Toxicity in cultured renal epithelial cells. Kidney Int. 2005, 67, 1797–1805. [Google Scholar] [CrossRef] [PubMed]
- Shibutani, S.; Dong, H.; Suzuki, N.; Ueda, S.; Miller, F.; Grollman, A.P. Selective toxicity of aristolochic acids I and II. Drug Metab. Dispos. 2007, 35, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- Krumbiegel, G.; Hallensleben, J.; Mennicke, W.; Rittmann, N.; Roth, H. Studies on the metabolism of aristolochic acids I and II. Xenobiotica 1987, 17, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.; Luo, H.B.; Zheng, Y.; Cheng, Y.K.; Cai, Z. Investigation of the metabolism and reductive activation of carcinogenic aristolochic acids in rats. Drug Metab. Dispos. 2007, 35, 866–874. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.M.K.; Chan, W. Noninvasive measurement of aristolochic acid-DNA adducts in urine samples from aristolochic acid-treated rats by liquid chromatography coupled tandem mass spectrometry: Evidence for DNA repair by nucleotide-excision repair mechanisms. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2014, 766–767, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.M.K.; Chan, W. Quantification of aristolochic acid-RNA adducts in the urine of aristolochic acid-treated rats by liquid chromatography-tandem mass spectrometry. Chem. Res. Toxicol. 2015, 28, 567–569. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Gong, L.; Qi, X.; Xing, G.; Luan, Y.; Wu, Y.; Xiao, Y.; Yao, J.; Li, Y.; Xue, X.; et al. Inhibition of renal NQO1 activity by dicoumarol suppresses nitroreduction of aristolochic acid I and attenuates its nephrotoxicity. Toxicol. Sci. 2011, 122, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Wu, H.; Yue, H.; Lin, S.; Lai, Y.; Cai, Z. A novel and specific method for the determination of aristolochic acid-derived DNA adducts in exfoliated urothelial cells by using ultra performance liquid chromatography-triple quadrupole mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Babu, E.; Takeda, M.; Nishida, R.; Noshiro-Kofuji, R.; Yoshida, M.; Ueda, S.; Fukutomi, T.; Anzai, N.; Endou, H. Interactions of human organic anion transporters with aristolochic acids. J. Pharmacol. Sci. 2010, 196, 192–196. [Google Scholar] [CrossRef]
- Hagos, Y.; Wolff, N.A. Assessment of the role of renal organic anion transporters in drug-induced nephrotoxicity. Toxins (Basel) 2010, 2, 2055–2082. [Google Scholar] [CrossRef] [PubMed]
- Sekine, T.; Miyazaki, H.; Endou, H. Molecular physiology of renal organic anion transporters. Am. J. Physiol. Ren. Physiol. 2006, 290, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Nigam, S.K.; Bush, K.T.; Martovetsky, G.; Ahn, S.-Y.; Liu, H.C.; Richard, E.; Bhatnagar, V.; Wu, W. The Organic Anion Transporter (OAT) Family: A Systems Biology Perspective. Physiol. Rev. 2015, 95, 83–123. [Google Scholar] [CrossRef] [PubMed]
- Otani, N.; Ouchi, M.; Hayashi, K.; Jutabha, P.; Anzai, N. Roles of organic anion transporters (OATs) in renal proximal tubules and their localization. Anat. Sci. Int. 2016. [Google Scholar] [CrossRef] [PubMed]
- Bakhiya, N.; Arlt, V.M.; Bahn, A.; Burckhardt, G.; Phillips, D.H.; Glatt, H. Molecular evidence for an involvement of organic anion transporters (OATs) in aristolochic acid nephropathy. Toxicology 2009, 264, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.-Y.; Nigam, S.K. Toward a systems level understanding of organic anion and other multispecific drug transporters: A remote sensing and signaling hypothesis. Mol. Pharmacol. 2009, 76, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Anzai, N.; Endou, H. Renal drug transporters and nephrotoxicity. Life Sci. 2008, 447–452. [Google Scholar]
- Yin, J.; Wang, J. Renal drug transporters and their significance in drug–drug interactions. Acta Pharm. Sin. B 2016, 6, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Gong, L.-K.; Maeda, K.; Luan, Y.; Qi, X.-M.; Sugiyama, Y.; Ren, J. Critical role of organic anion transporters 1 and 3 in kidney accumulation and toxicity of aristolochic acid I. Mol. Pharm. 2011, 8, 2183–2192. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Zhang, R.; Wu, J.; Liu, M.; Peng, W.; Yu, X.; Yang, X. Organic anion transporter 1 (OAT1) involved in renal cell transport of aristolochic acid I. Hum. Exp. Toxicol. 2012, 31, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Baudoux, T.E.R.; Pozdzik, A.A.; Arlt, V.M.; de Prez, E.G.; Antoine, M.-H.; Quellard, N.; Goujon, J.-M.; Nortier, J.L. Probenecid prevents acute tubular necrosis in a mouse model of aristolochic acid nephropathy. Kidney Int. 2012, 82, 1105–1113. [Google Scholar] [CrossRef] [PubMed]
- Rosenquist, T.A.; Grollman, A.P. Mutational signature of aristolochic acid: Clue to the recognition of a global disease. DNA Repair (Amst.) 2016, 44, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Frei, E.; Arlt, V.M.; Schmeiser, H.H. Metabolic activation of carcinogenic aristolochic acid, a risk factor for Balkan endemic nephropathy. Mutat. Res. Rev. Mutat. Res. 2008, 658, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Stiborova, M.; Frei, E.; Arlt, V.M.; Schmeiser, H.H. The role of biotransformation enzymes in the development of renal injury and urothelial cancer caused by aristolochic acid: Urgent questions and difficult answers. Biomed. Pap. 2009, 153, 5–12. [Google Scholar] [CrossRef]
- Stiborová, M.; Frei, E.; Schmeiser, H.H.; Arlt, V.M.; Martínek, V. Mechanisms of enzyme-catalyzed reduction of two carcinogenic nitro-aromatics, 3-nitrobenzanthrone and aristolochic acid I: Experimental and theoretical approaches. Int. J. Mol. Sci. 2014, 15, 10271–10295. [Google Scholar] [CrossRef] [PubMed]
- Arlt, V.M.; Levová, K.; Bárta, F.; Shi, Z.; Evans, J.D.; Frei, E.; Schmeiser, H.H.; Nebert, D.W.; Phillips, D.H.; Stiborová, M. Role of P450 1A1 and P450 1A2 in bioactivation versus detoxication of the renal carcinogen aristolochic acid I: Studies in Cyp1a1(−/−), Cyp1a2(−/−), and Cyp1a1/1a2(−/−) mice. Chem. Res. Toxicol. 2011, 24, 1710–1719. [Google Scholar] [CrossRef] [PubMed]
- Bieler, C.A.; Stiborova, M.; Wiessler, M.; Cosyns, J.P.; van Ypersele de Strihou, C.; Schmeiser, H.H. 32P-post-labelling analysis of DNA adducts formed by aristolochic acid in tissues from patients with Chinese herbs nephropathy. Carcinogenesis 1997, 18, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Martínek, V.; Frei, E.; Arlt, V.M.; Schmeiser, H.H. Enzymes Metabolizing Aristolochic Acid and their Contribution to the Development of Aristolochic Acid Nephropathy and Urothelial Cancer. Curr. Drug Metab. 2013, 14, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Milichovký, J.; Bárta, F.; Schmeiser, H.; Arlt, V.; Frei, E.; Stiborová, M.; Martínek, V. Active Site Mutations as a Suitable Tool Contributing to Explain a Mechanism of Aristolochic Acid I Nitroreduction by Cytochromes P450 1A1, 1A2 and 1B1. Int. J. Mol. Sci. 2016, 17, 213. [Google Scholar] [CrossRef] [PubMed]
- Schmeiser, H.H.; Nortier, J.L.; Singh, R.; da Costa, G.G.; Sennesael, J.; Cassuto-Viguier, E.; Ambrosetti, D.; Rorive, S.; Pozdzik, A.; Phillips, D.H.; et al. Exceptionally long-term persistence of DNA adducts formed by carcinogenic aristolochic acid I in renal tissue from patients with aristolochic acid nephropathy. Int. J. Cancer 2014, 135, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Yun, B.H.; Sidorenko, V.S.; Rosenquist, T.A.; Dickman, K.G.; Grollman, A.P.; Turesky, R.J. New Approaches for Biomonitoring Exposure to the Human Carcinogen Aristolochic Acid. Toxicol. Res. (Camb.) 2015, 4, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.M.K.; Chan, W. Comparison of DNA and RNA Adduct Formation: Significantly Higher Levels of RNA than DNA Modifications in the Internal Organs of Aristolochic Acid-Dosed Rats. Chem. Res. Toxicol. 2015, 28, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Lebeau, C.; Debelle, F.D.; Arlt, V.M.; Pozdzik, A.; De Prez, E.G.; Phillips, D.H.; Deschodt-Lanckman, M.M.; Vanherweghem, J.L.; Nortier, J.L. Early proximal tubule injury in experimental aristolochic acid nephropathy: Functional and histological studies. Nephrol. Dial. Transplant. 2005, 20, 2321–2332. [Google Scholar] [CrossRef] [PubMed]
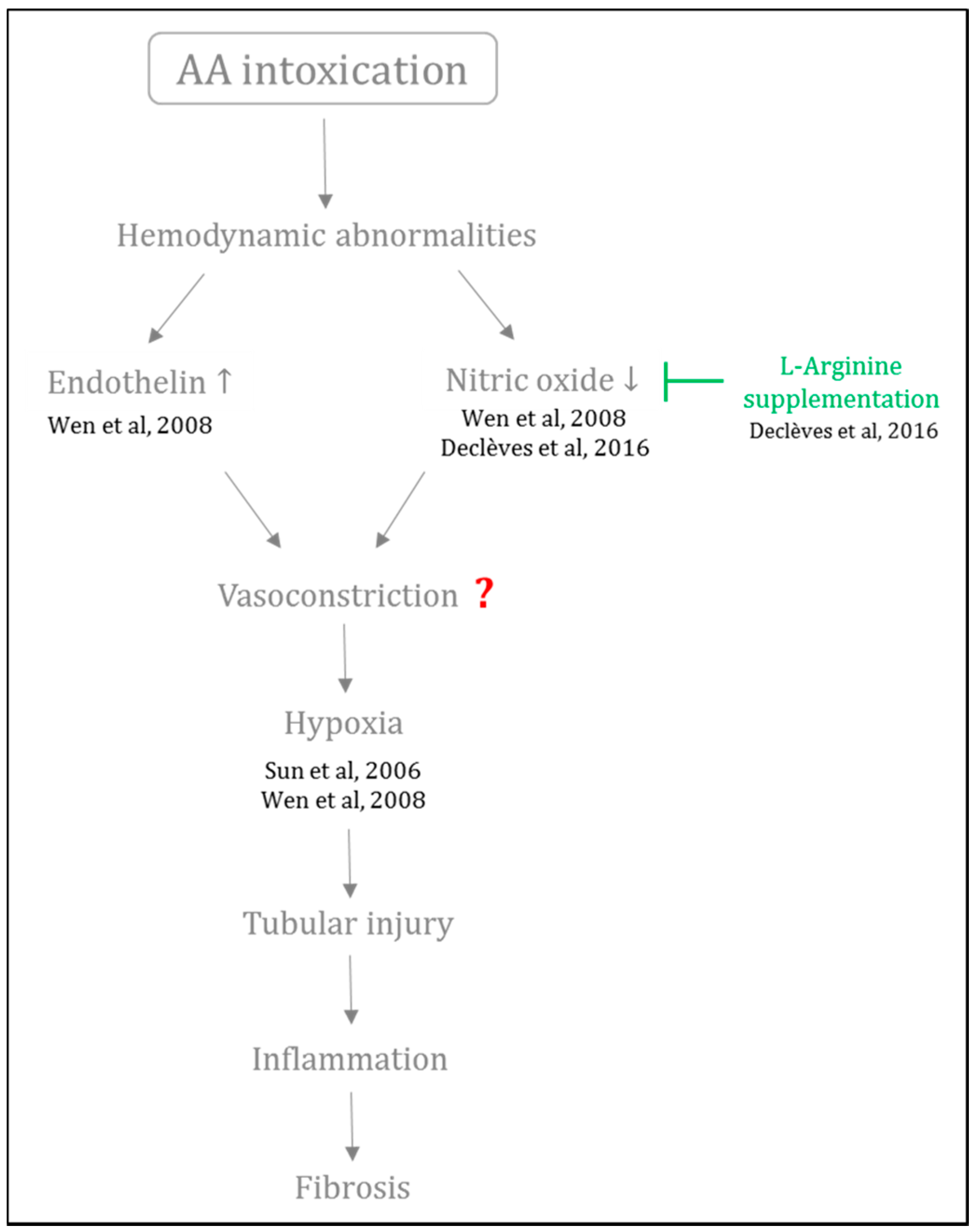
- Declèves, A.-E.; Jadot, I.; Colombaro, V.; Martin, B.; Voisin, V.; Nortier, J.; Caron, N. Protective effect of nitric oxide in aristolochic acid-induced toxic acute kidney injury: An old friend with new assets. Exp. Physiol. 2016, 101, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Pozdzik, A.A.; Salmon, I.J.; Husson, C.P.; Decaestecker, C.; Rogier, E.; Bourgeade, M.-F.; Deschodt-Lanckman, M.M.; Vanherweghem, J.-L.; Nortier, J.L. Patterns of interstitial inflammation during the evolution of renal injury in experimental aristolochic acid nephropathy. Nephrol. Dial. Transplant. 2008, 23, 2480–2491. [Google Scholar] [CrossRef] [PubMed]
- Pozdzik, A.A.; Salmon, I.J.; Debelle, F.D.; Decaestecker, C.; van den Branden, C.; Verbeelen, D.; Deschodt-Lanckman, M.M.; Vanherweghem, J.-L.; Nortier, J.L. Aristolochic acid induces proximal tubule apoptosis and epithelial to mesenchymal transformation. Kidney Int. 2008, 73, 595–607. [Google Scholar] [CrossRef] [PubMed]
- Diamond, J.R.; Pallone, T.L. Acute interstitial nephritis following use of tung shueh pills. Am. J. Kidney Dis. 1994, 24, 219–221. [Google Scholar] [CrossRef]
- Jelaković, B.; Karanović, S.; Vuković-Lela, I.; Miller, F.; Edwards, K.L.; Nikolić, J.; Tomić, K.; Slade, N.; Brdar, B.; Turesky, R.J.; et al. Aristolactam-DNA adducts are a biomarker of environmental exposure to aristolochic acid. Kidney Int. 2012, 81, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Schmeiser, H.H.; Janssen, J.W.G.; Lyons, J.; Scherf, H.R.; Pfau, W.; Buchmann, A.; Bartram, C.R.; Wiessler, M. Aristolochic Acid Activates ras Genes in Rat Tumors at Deoxyadenosine Residues. Cancer Res. 1990, 50, 5464–5469. [Google Scholar] [PubMed]
- Lord, G.M.; Hollstein, M.; Arlt, V.M.; Roufosse, C.; Pusey, C.D.; Cook, T.; Schmeiser, H.H. DNA adducts and p53 mutations in a patient with aristolochic acid-associated nephropathy. Am. J. Kidney Dis. 2004, 43, e18.1–e18.7. [Google Scholar] [CrossRef]
- Hollstein, M.; Moriya, M.; Grollman, A.P.; Olivier, M. Analysis of TP53 mutation spectra reveals the fingerprint of the potent environmental carcinogen, aristolochic acid. Mutat. Res. Rev. Mutat. Res. 2013, 753, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Slade, N.; Moll, U.M.; Brdar, B.; Zorić, A.; Jelaković, B. p53 mutations as fingerprints for aristolochic acid—An environmental carcinogen in endemic (Balkan) nephropathy. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2009, 663, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hoang, M.L.; Chen, C.-H.; Sidorenko, V.S.; He, J.; Dickman, K.G.; Yun, B.H.; Moriya, M.; Niknafs, N.; Douville, C.; Karchin, R.; et al. Mutational Signature of Aristolochic Acid Exposure as Revealed by Whole-Exome Sequencing. Sci. Transl. Med. 2013, 5, 197ra102. [Google Scholar] [CrossRef] [PubMed]
- Moriya, M.; Slade, N.; Brdar, B.; Medverec, Z.; Tomic, K.; Jelaković, B.; Wu, L.; Truong, S.; Fernandes, A.; Grollman, A.P. TP53 Mutational signature for aristolochic acid: An environmental carcinogen. Int. J. Cancer 2011, 129, 1532–1536. [Google Scholar] [CrossRef] [PubMed]
- Aydin, S.; Dekairelle, A.-F.; Ambroise, J.; Durant, J.-F.; Heusterspreute, M.; Cosyns, Y.G.J.-P.; Gala, J.-L. Unambiguous Detection of Multiple TP53 Gene Mutations in AAN-Associated Urothelial Cancer in Belgium Using Laser Capture Microdissection. PLoS ONE 2014, 9, e106301. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.-Y.; Wu, T.-S.; Chen, T.-W.; Liu, B.-H. Aristolochic acid I induced oxidative DNA damage associated with glutathione depletion and ERK1/2 activation in human cells. Toxicol. In Vitro 2011, 25, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Romanov, V.; Whyard, T.C.; Waltzer, W.C.; Grollman, A.P.; Rosenquist, T. Aristolochic acid-induced apoptosis and G2 cell cycle arrest depends on ROS generation and MAP kinases activation. Arch. Toxicol. 2015, 89, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.K.; Wei, C.W.; Pan, Y.R.; Cherng, S.H.; Chang, W.J.; Wang, H.F.; Yu, Y.L. Vitamin C attenuates the toxic effect of aristolochic acid on renal tubular cells via decreasing oxidative stress mediated cell death pathways. Mol. Med. Rep. 2015, 12, 6086–6092. [Google Scholar] [CrossRef] [PubMed]
- Bunel, V.; Antoine, M.-H.; Stévigny, C.; Nortier, J.; Duez, P. New in vitro insights on a cell death pathway induced by magnolol and honokiol in aristolochic acid tubulotoxicity. Food Chem. Toxicol. 2016, 87, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Tsai, S.H.; Chen, S.M.; Chang, Y.M.; Huang, T.C.; Huang, Y.P.; Chang, C.T.; Lee, J.A. Aristolochic acid-induced accumulation of methylglyoxal and N-(carboxymethyl)lysine: An important and novel pathway in the pathogenic mechanism for aristolochic acid nephropathy. Biochem. Biophys. Res. Commun. 2012, 423, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.C.; Chen, S.-M.; Li, Y.-C.; Lee, J.-A. Increased renal semicarbazide-sensitive amine oxidase activity and methylglyoxal levels in aristolochic acid-induced nephrotoxicity. Life Sci. 2014, 114, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Watanabe, Y.; Inoue, T.; Kobayashi, T.; Kanno, Y.; Shiota, G.; Nakamura, T.; Sugaya, T.; Fukamizu, A.; Suzuki, H. Transgene-derived hepatocyte growth factor attenuates reactive renal fibrosis in aristolochic acid nephrotoxicity. Nephrol. Dial. Transplant. 2003, 18, 2515–2523. [Google Scholar] [CrossRef] [PubMed]
- Hsin, Y.H.; Cheng, C.H.; Tzen, J.T.C.; Wu, M.J.; Shu, K.H.; Chen, H.C. Effect of aristolochic acid on intracellular calcium concentration and its links with apoptosis in renal tubular cells. Apoptosis 2006, 11, 2167–2177. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Cai, Y.; Gong, L.; Liu, L.; Chen, F.; Xiao, Y.; Wu, X.; Li, Y.; Xue, X.; Ren, J. Role of mitochondrial permeability transition in human renal tubular epithelial cell death induced by aristolochic acid. Toxicol. Appl. Pharmacol. 2007, 222, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.C.; Maruyama, S.; Mizuno, M.; Morita, Y.; Hanaki, S.; Yuzawa, Y.; Matsuo, S. The nephrotoxicity of Aristolochia manshuriensis in rats is attributable to its aristolochic acids. Clin. Exp. Nephrol. 2003, 7, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Fu, P.; Huang, X.R.; Liu, F.; Lai, K.N.; Lan, H.Y. Activation of p53 promotes renal injury in acute aristolochic acid nephropathy. J. Am. Soc. Nephrol. 2010, 21, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Ma, L.; Zhou, L.; Fu, P. Renal Protective Effects of 17β-Estradiol on Mice with Acute Aristolochic Acid Nephropathy. Molecules 2016, 21, 1391. [Google Scholar] [CrossRef] [PubMed]
- Debelle, F.D.; Nortier, J.L.; Husson, C.P.; de Prez, E.G.; Vienne, A.R.; Rombaut, K.; Salmon, I.J.; Deschodt-Lanckman, M.M.; Vanherweghem, J.-L. The renin-angiotensin system blockade does not prevent renal interstitial fibrosis induced by aristolochic acids. Kidney Int. 2004, 66, 1815–1825. [Google Scholar] [CrossRef] [PubMed]
- Pozdzik, A.A.; Berton, A.; Schmeiser, H.H.; Missoum, W.; Decaestecker, C.; Salmon, I.J.; Vanherweghem, J.L.; Nortier, J.L. Aristolochic acid nephropathy revisited: A place for innate and adaptive immunity? Histopathology 2010, 56, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.-J.; Qu, L.; Li, X.-M. Ischemic injury underlies the pathogenesis of aristolochic acid-induced acute kidney injury. Transl. Res. 2008, 152, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Vanherweghem, J.L. Renal failure and urinary tract carcinoma secondary to the intake of certain Chinese herbal medicines. Med. Sci. 2002, 18, 1095–1101. [Google Scholar]
- Sun, D.; Feng, J.; Dai, C.; Sun, L.; Jin, T.; Ma, J.; Wang, L. Role of peritubular capillary loss and hypoxia in progressive tubulointerstitial fibrosis in a rat model of aristolochic acid nephropathy. Am. J. Nephrol. 2006, 26, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Zuk, A.; Bonventre, J. Acute Kidney Injury. Annu. Rev. Med. 2016, 67, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Gewin, L. NO clue to pathogenesis of aristolochic acid nephropathy. Exp. Physiol. 2016, 101, 33. [Google Scholar] [CrossRef] [PubMed]
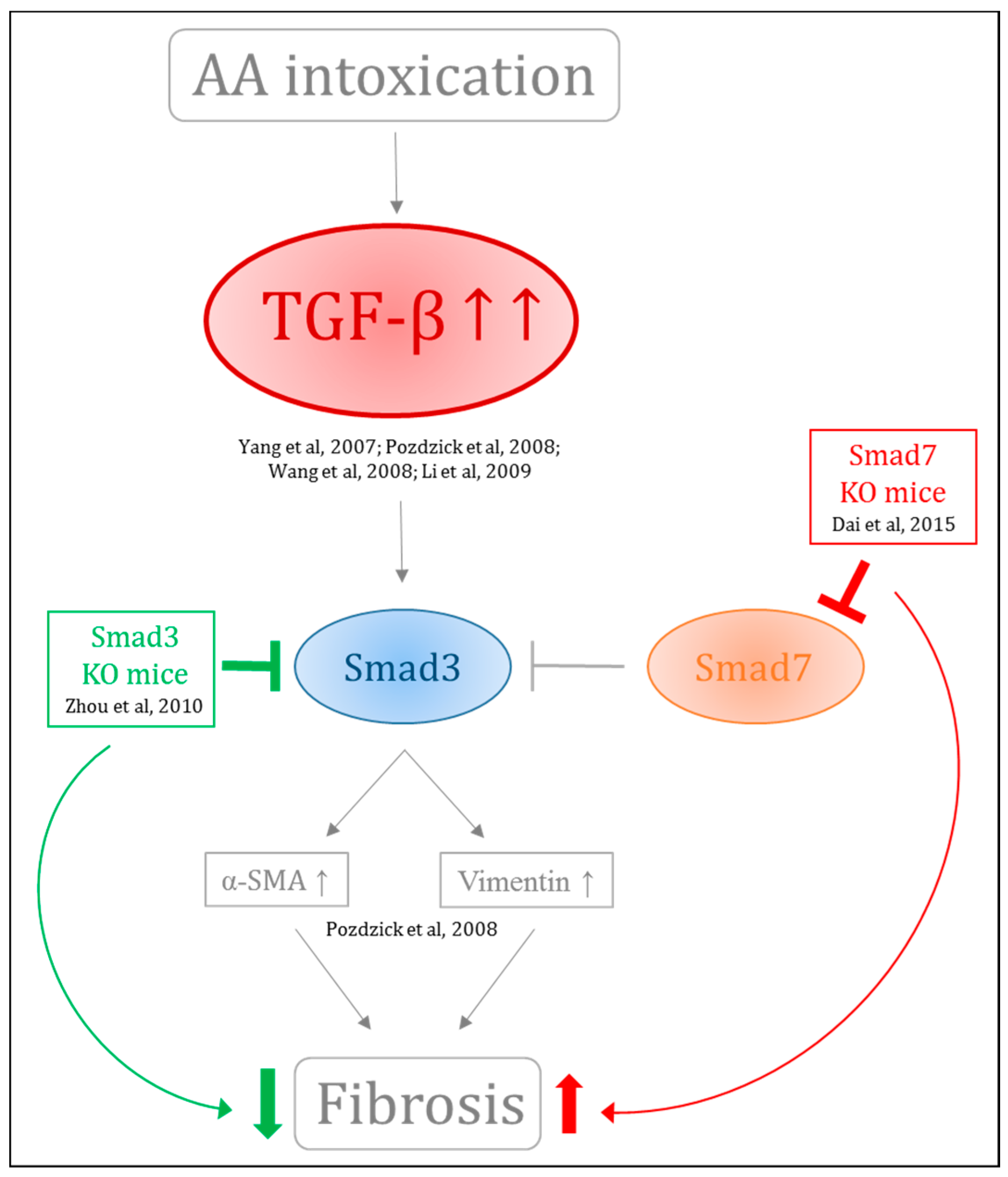
- Li, J.; Zhang, Z.; Wang, D.; Wang, Y.; Li, Y.; Wu, G. TGF-β 1/Smads signaling stimulates renal interstitial fibrosis in experimental AAN. J. Recept. Signal Transduct. Res. 2009, 29, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Z.; Shen, H.; Lu, Y.; Li, H.; Ren, X.; Wu, G. TGF-β1/Smad7 Signaling Stimulates Renal Tubulointerstitial Fibrosis Induced by AAI. J. Recept. Signal Transduct. 2008, 28, 413–428. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Fu, P.; Huang, X.R.; Liu, F.; Chung, A.C.K.; Lai, K.N.; Lan, H.Y. Mechanism of chronic aristolochic acid nephropathy: Role of Smad3. Am. J. Physiol. Ren. Physiol. 2010, 298, F1006–F1017. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.-Y.; Zhou, L.; Huang, X.-R.; Fu, P.; Lan, H.-Y. Smad7 protects against chronic aristolochic acid nephropathy in mice. Oncotarget 2015, 6, 11930–11944. [Google Scholar] [CrossRef] [PubMed]
- Antoine, M.-H.; Debelle, F.; Piccirilli, J.; El Kaddouri, F.; Declèves, A.-E.; De Prez, E.; Husson, C.; Mies, F.; Bourgeade, M.-F.; Nortier, J.L. Human bone morphogenetic protein-7 does not counteract aristolochic acid-induced renal toxicity. J. Appl. Toxicol. 2015, 35, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- Samarakoon, R.; Rehfuss, A.; Khakoo, N.S.; Falke, L.L.; Dobberfuhl, A.D.; Helo, S.; Overstreet, J.M.; Goldschmeding, R.; Higgins, P.J. Loss of expression of protein phosphatase magnesium-dependent 1A during kidney injury promotes fibrotic maladaptive repair. FASEB J. 2016, 30, 3308–3320. [Google Scholar] [CrossRef] [PubMed]
- Pozdzik, A.A.; Giordano, L.; Li, G.; Antoine, M.-H.; Quellard, N.; Godet, J.; de Prez, E.; Husson, C.; Declèves, A.-E.; Arlt, V.M.; et al. Blocking TGF-β Signaling Pathway Preserves Mitochondrial Proteostasis and Reduces Early Activation of PDGFRβ+ Pericytes in Aristolochic Acid Induced Acute Kidney Injury in Wistar Male Rats. PLoS ONE 2016, 11, e0157288. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.Y.; Huang, X.R.; Zhou, L.; Zhang, L.; Fu, P.; Manthey, C. Targeting c-fms kinase attenuates chronic aristolochic acid nephropathy in mice. Oncotarget 2016, 7, 10841–10856. [Google Scholar] [PubMed]
- Chawla, L.S.; Amdur, R.L.; Amodeo, S.; Kimmel, P.L.; Palant, C.E. The severity of acute kidney injury predicts progression to chronic kidney disease. Kidney Int. 2011, 79, 1361–1369. [Google Scholar] [CrossRef] [PubMed]
- Chawla, L.S.; Eggers, P.W.; Star, R.A.; Kimmel, P.L. Acute kidney injury and chronic kidney disease as interconnected syndromes. N. Engl. J. Med. 2014, 371, 58–66. [Google Scholar] [CrossRef] [PubMed]

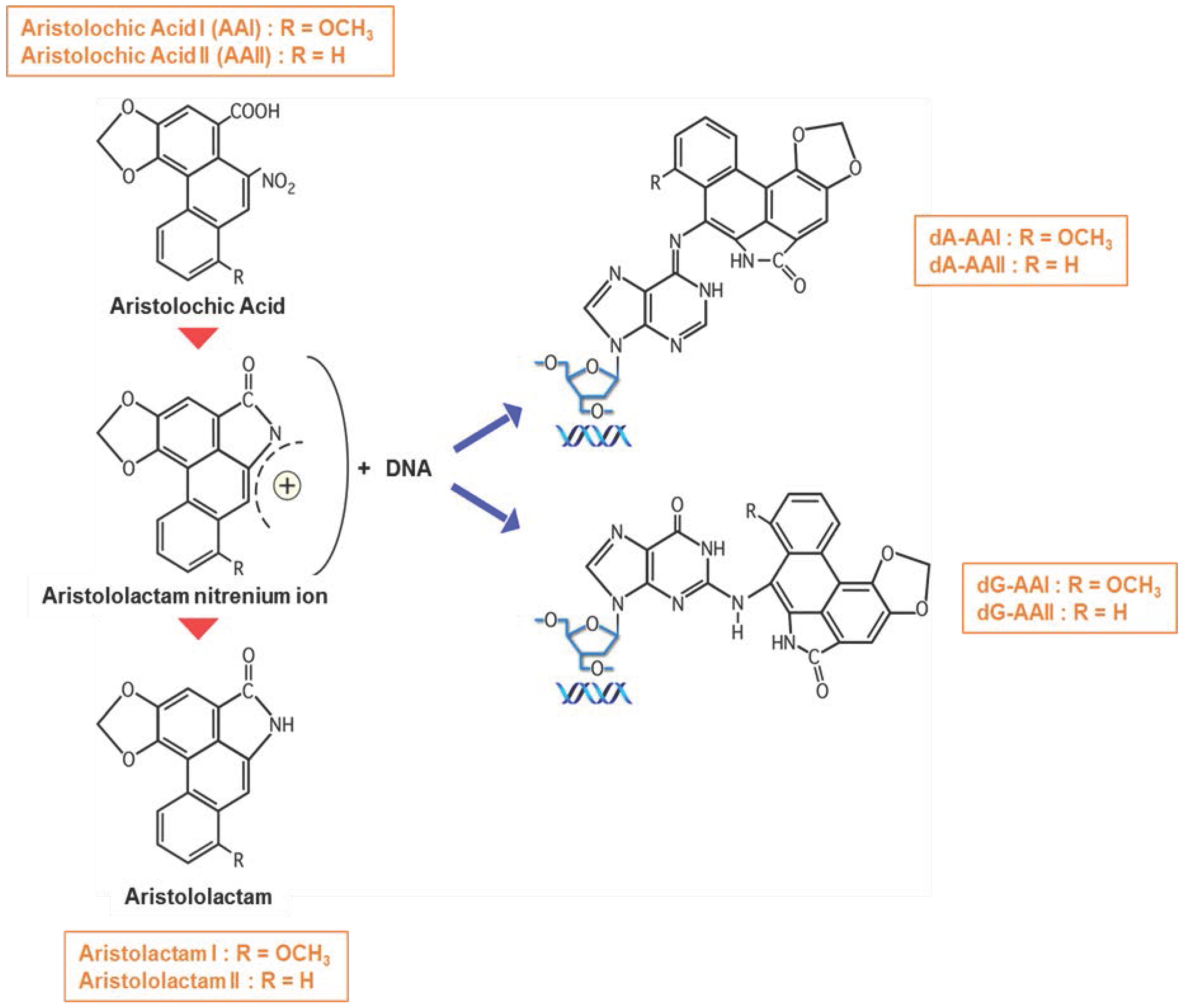
 ) to from DNA adducts. Since AA metabolites are found in the urine, we hypothesize (?) that AA metabolites are secreted through the apical membrane in the proximal tubular lumen via OAT4. MRP2 and MRP4 could also be involved in the efflux of AA metabolites in the proximal tubule lumen. However, the majority of AA metabolites, the aristolactams, are trapped inside the cell due to the formation of specific DNA adducts.
) to from DNA adducts. Since AA metabolites are found in the urine, we hypothesize (?) that AA metabolites are secreted through the apical membrane in the proximal tubular lumen via OAT4. MRP2 and MRP4 could also be involved in the efflux of AA metabolites in the proximal tubule lumen. However, the majority of AA metabolites, the aristolactams, are trapped inside the cell due to the formation of specific DNA adducts.
) to from DNA adducts. Since AA metabolites are found in the urine, we hypothesize (?) that AA metabolites are secreted through the apical membrane in the proximal tubular lumen via OAT4. MRP2 and MRP4 could also be involved in the efflux of AA metabolites in the proximal tubule lumen. However, the majority of AA metabolites, the aristolactams, are trapped inside the cell due to the formation of specific DNA adducts.
) to from DNA adducts. Since AA metabolites are found in the urine, we hypothesize (?) that AA metabolites are secreted through the apical membrane in the proximal tubular lumen via OAT4. MRP2 and MRP4 could also be involved in the efflux of AA metabolites in the proximal tubule lumen. However, the majority of AA metabolites, the aristolactams, are trapped inside the cell due to the formation of specific DNA adducts.
 ) forming AA-DNA adducts.
) forming AA-DNA adducts.
) forming AA-DNA adducts.
) forming AA-DNA adducts.
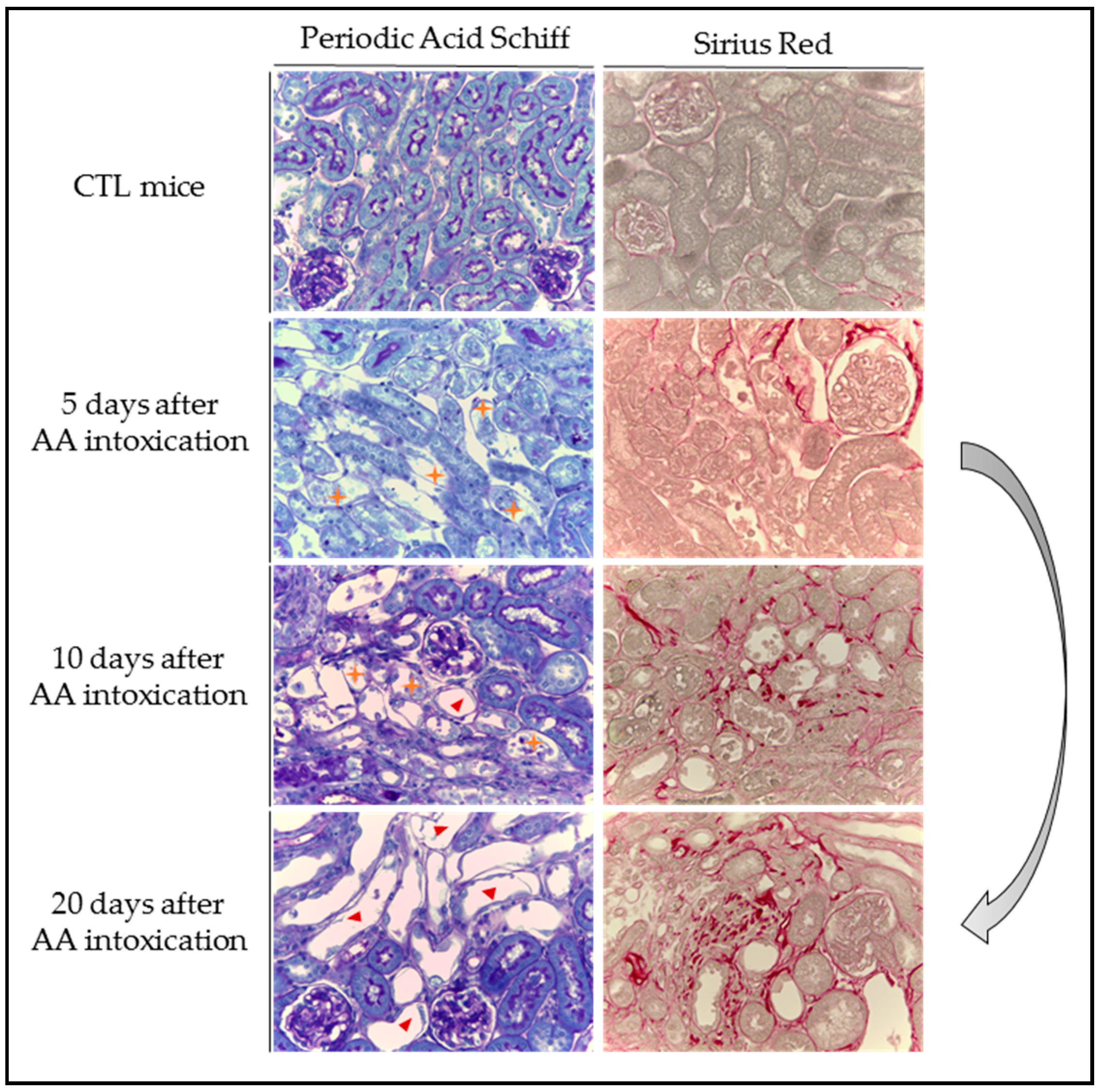
 ) with cell debris in tubular lumens are visible in mice treated with AA at Days 5 and 10 and cystic tubules (
) with cell debris in tubular lumens are visible in mice treated with AA at Days 5 and 10 and cystic tubules (  ) are visible in mice at Days 10 and 20. Collagen I and III, highlighted by Sirius Red staining, accumulate in the interstitium of the kidney of AA-treated mice from Day 10 and even more at Day 20 reflecting the progression to CKD.
) with cell debris in tubular lumens are visible in mice treated with AA at Days 5 and 10 and cystic tubules ( ) are visible in mice at Days 10 and 20. Collagen I and III, highlighted by Sirius Red staining, accumulate in the interstitium of the kidney of AA-treated mice from Day 10 and even more at Day 20 reflecting the progression to CKD.
) are visible in mice at Days 10 and 20. Collagen I and III, highlighted by Sirius Red staining, accumulate in the interstitium of the kidney of AA-treated mice from Day 10 and even more at Day 20 reflecting the progression to CKD.
) with cell debris in tubular lumens are visible in mice treated with AA at Days 5 and 10 and cystic tubules ( ) are visible in mice at Days 10 and 20. Collagen I and III, highlighted by Sirius Red staining, accumulate in the interstitium of the kidney of AA-treated mice from Day 10 and even more at Day 20 reflecting the progression to CKD.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year of Publication | Authors | Country | Numb of Cases | Purpose of AA Ingestion Suspected Aristolochia Species |
|---|---|---|---|---|
| 1993 | Vanherweghem et al. [1] | Belgium | 9 | Slimming pills containing Chinese herbs. Aristolochia fangchi |
| 1996 | Peña et al. [22] | Spain | 1 | Infusion made with a mixture of herbs. Aristolochia pistolochia |
| 1997 | Tanaka et al. [35] | Japan | 1 | Health food for atopic dermatitis. |
| 1998 | Stengel and Jones [23] | France | 2 | Slimming pills containing Chinese herbs. |
| 1998 | Vanherweghem et al. [21] | Belgium | 100 | Slimming pills containing Chinese herbs. Aristolochia fangchi |
| 1999 | Lord et al. [24] | UK | 2 | Herbal preparation for treatment of eczema. Aristolochia manshuriensis |
| 1999 | Lee et al. [33] | Taiwan | 1 | Chinese herbal medicine for peripheral extremities weakness and numbness. |
| 2000 | Meyer et al. [26] | USA | 1 | Chinese herbal medicine for pain relief. |
| 2000 | Tanaka et al. [36] | Japan | 2 | Not described. Aristolochia manshuriensis |
| 2000 | Yang et al. [32] | Taiwan | 12 | Chinese herbal medicine for weight control, nutritional supplements, treatment of arthralgia, hypertension or hepatitis. |
| 2001 | Krumme et al. [25] | Germany | 1 | Chinese herbal medicine for hyperuricaemia. |
| 2001 | Chen et al. [28] | China | 58 | Not described. |
| 2004 | Lo et al. [29] | China | 1 | Chinese herbal medicine as a “tonic herbal remedy”. |
| 2004 | Lee et al. [39] | Korea | 1 | Chinese herbs mixture for slimming purposes. |
| 2004 | Kazama et al. [37] | Japan | 1 | Chinese herbal medicine for sterility. |
| 2006 | Hong et al. [34] | Taiwan | 1 | Chinese herbal medicines for “health improvement”. |
| 2007 | Yang et al. [30] | China | 8 | Chinese herb “Guanmutong” Aristolochia manshuriensis. |
| 2011 | Chau et al. [27] | Australia | 1 | Chinese herbal medicine to treat psoriasis. |
| 2012 | Yang et al. [31] | China | 300 | Not described. |
| 2013 | Michl et al. [38] | Bangladesh | Remedies for snake bites, sexual problems, gastric problems, “tonic remedy”. Aristolochia indica |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jadot, I.; Declèves, A.-E.; Nortier, J.; Caron, N. An Integrated View of Aristolochic Acid Nephropathy: Update of the Literature. Int. J. Mol. Sci. 2017, 18, 297. https://doi.org/10.3390/ijms18020297
Jadot I, Declèves A-E, Nortier J, Caron N. An Integrated View of Aristolochic Acid Nephropathy: Update of the Literature. International Journal of Molecular Sciences. 2017; 18(2):297. https://doi.org/10.3390/ijms18020297
Chicago/Turabian StyleJadot, Inès, Anne-Emilie Declèves, Joëlle Nortier, and Nathalie Caron. 2017. "An Integrated View of Aristolochic Acid Nephropathy: Update of the Literature" International Journal of Molecular Sciences 18, no. 2: 297. https://doi.org/10.3390/ijms18020297
APA StyleJadot, I., Declèves, A.-E., Nortier, J., & Caron, N. (2017). An Integrated View of Aristolochic Acid Nephropathy: Update of the Literature. International Journal of Molecular Sciences, 18(2), 297. https://doi.org/10.3390/ijms18020297