
Anti-Fibrotic Effect of Losartan, an Angiotensin II Receptor Blocker, Is Mediated through Inhibition of ER Stress via Up-Regulation of SIRT1, Followed by Induction of HO-1 and Thioredoxin

Abstract
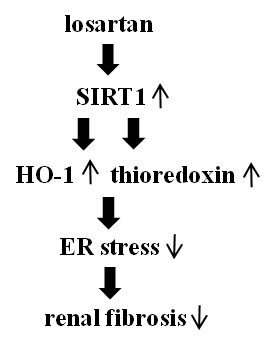
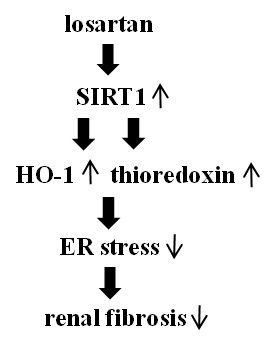
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
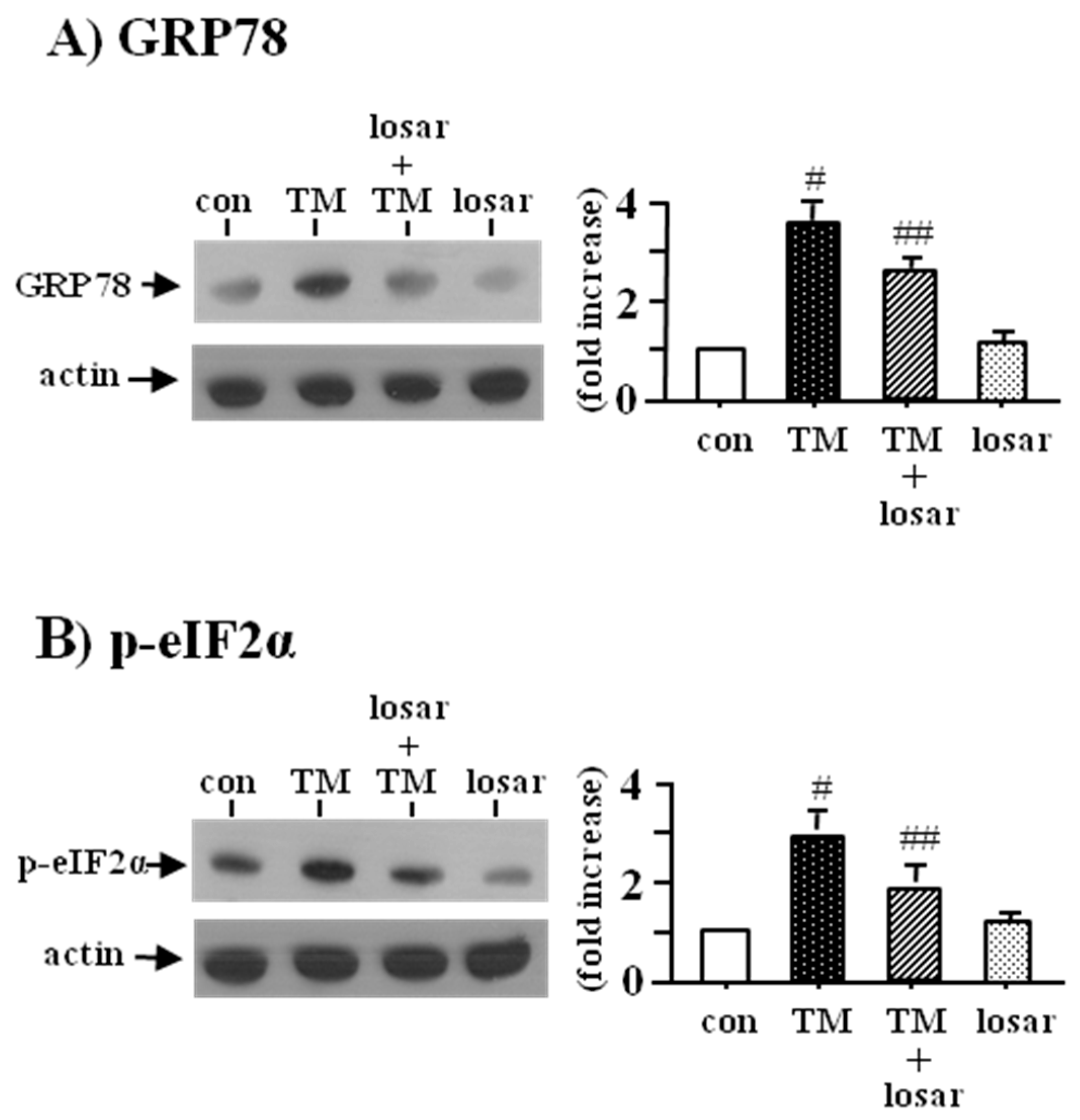
2.1. Losartan Suppressed the ER Stress Induced by Tunicamycinin in Tubular Epithelial Cells
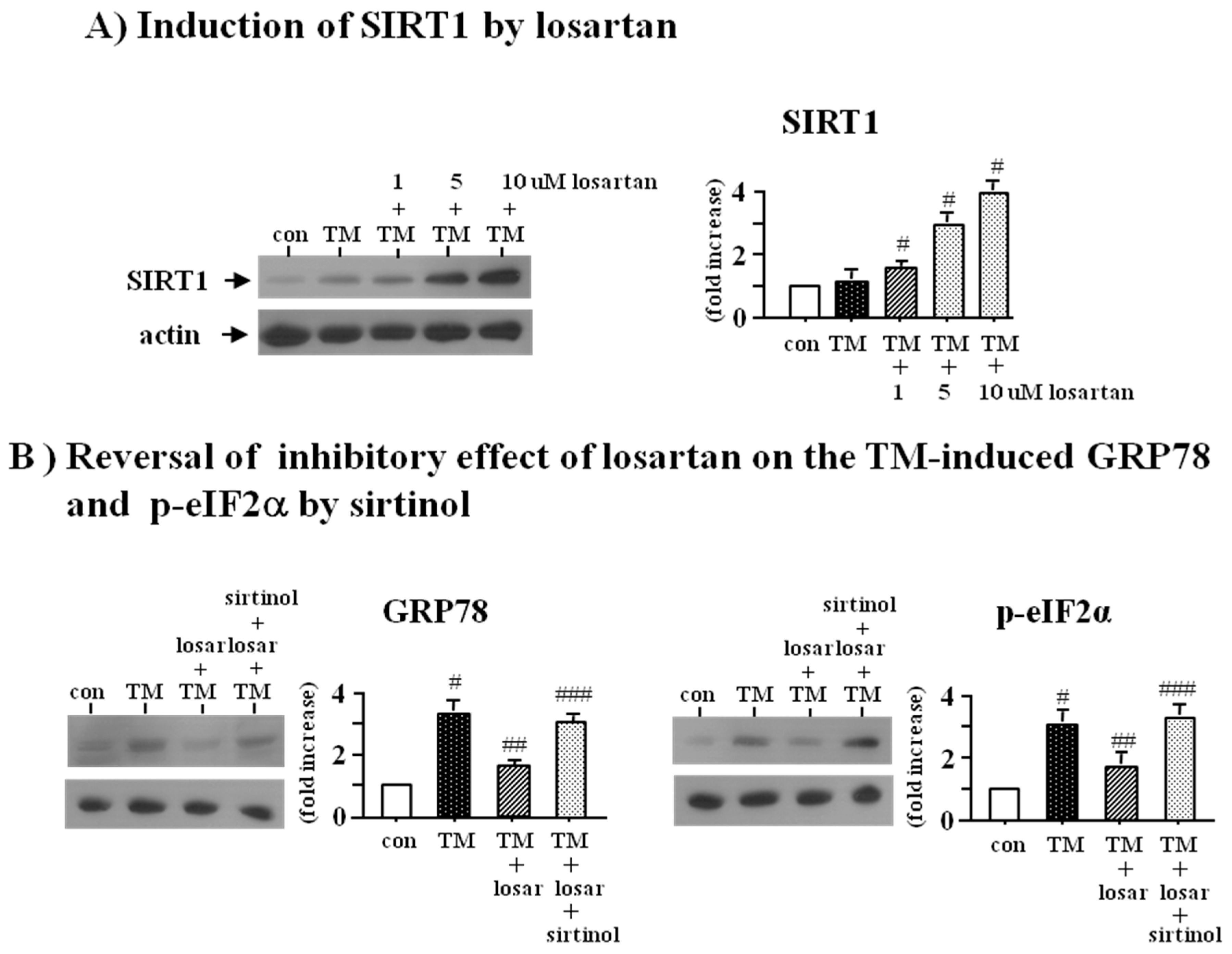
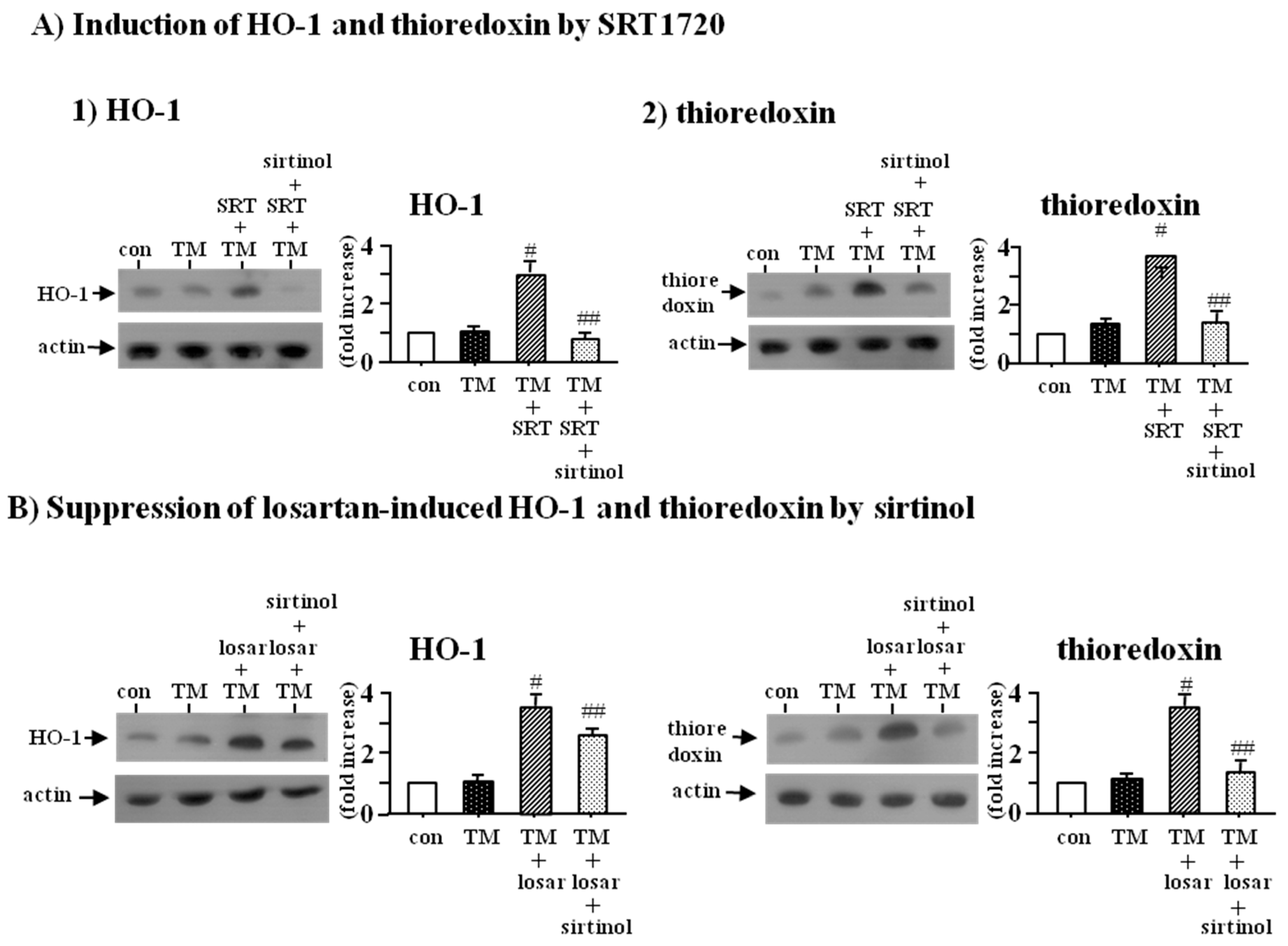
2.2. Inhibitory Effect of Losartan on the Tunicamycin-Induced ER Stress Was Mediated through Up-Regulation of SIRT1
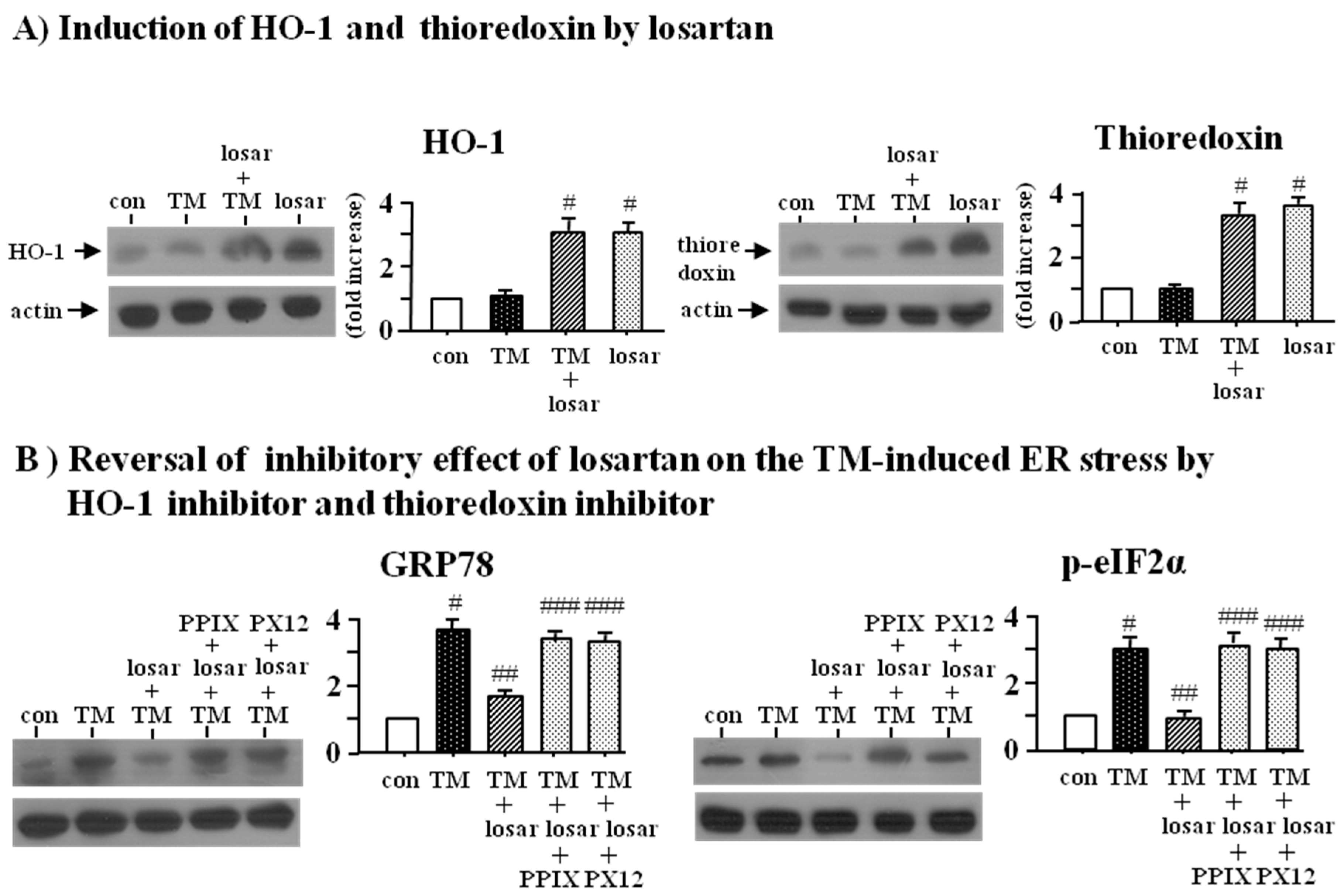
2.3. Inhibitory Effect of Losartan on the Tunicamycin-Induced ER Stress Was Mediated through Induction of Heme Oxygenase-1 and Thioredoxin
2.4. Inhibitory Effect of Losartan on the Tunicamycin-Induced ER Stress Was Mediated through Up-Regulation of SIRT1, Followed by Induction of HO-1 and Thioredoxin
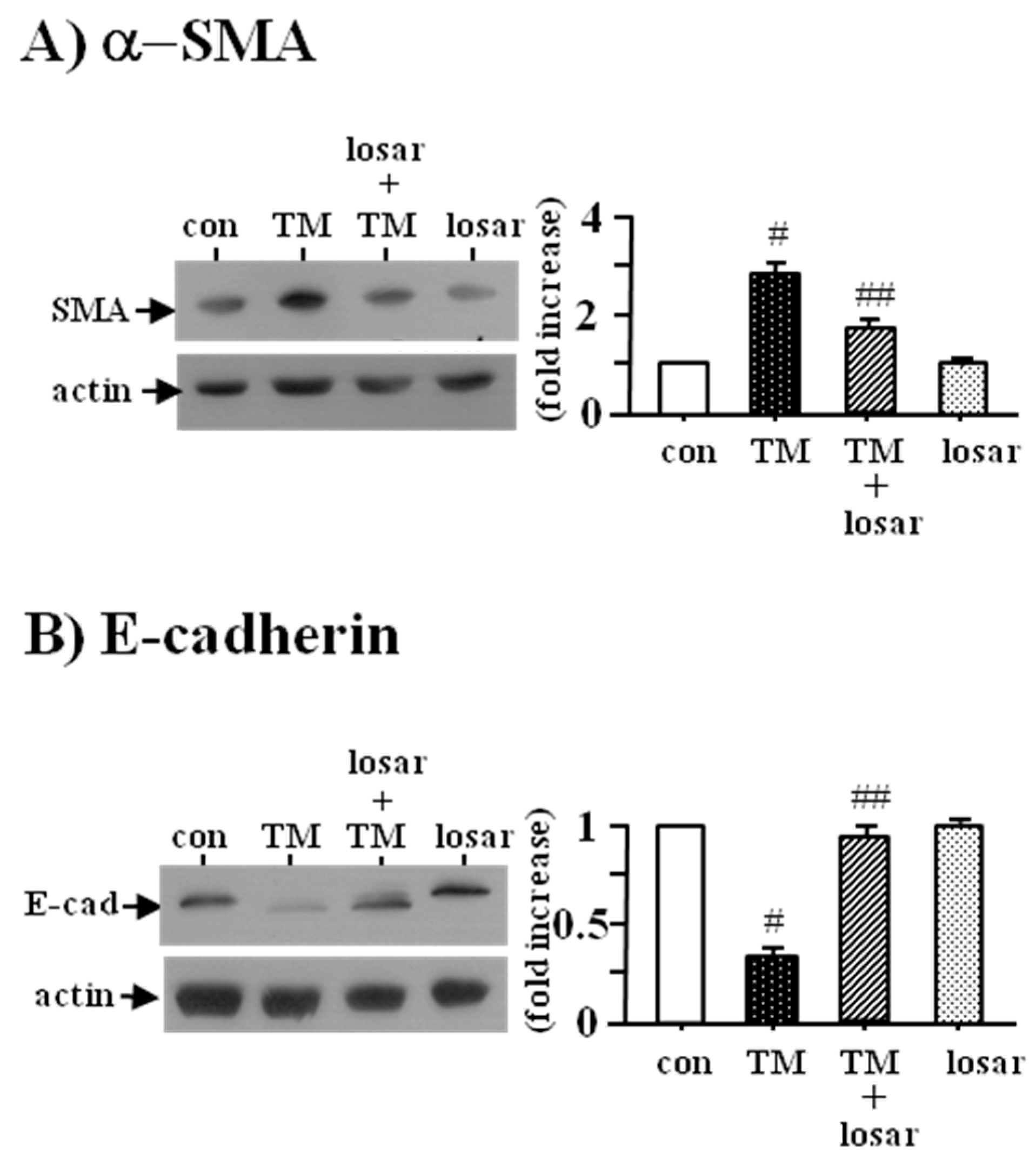
2.5. Losartan also Inhibited the Tunicamycin-Induced Epithelial-Mesenchymal Transition (EMT)
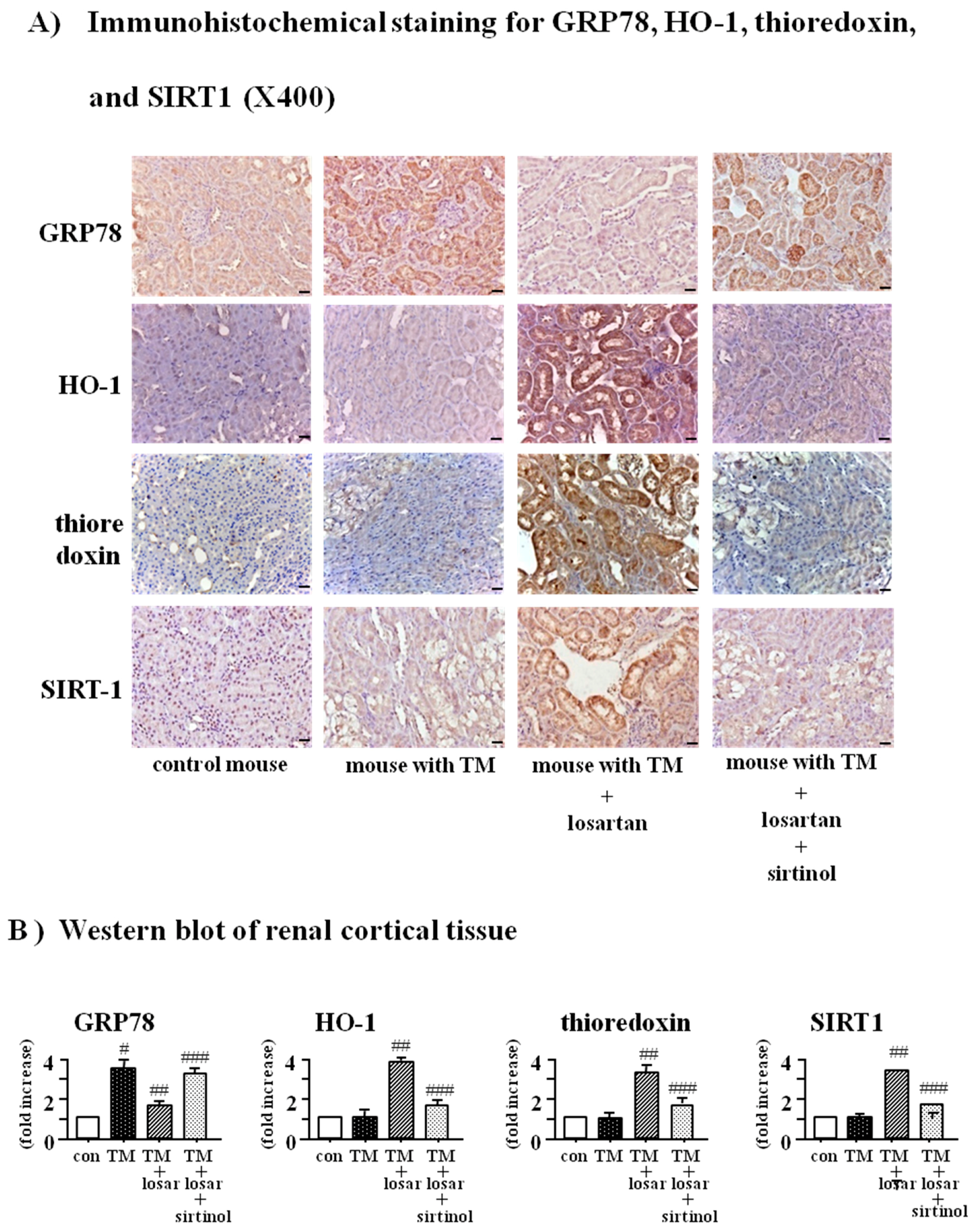
2.6. Treatment with Losartan Reduced the Renal Tubular Expression of GRP78 and Increased the Expression of HO-1 and Thioredoxin through the Up-Regulation of SIRT1 in a Mouse Model of Tunicamycin-Induced ER Stress
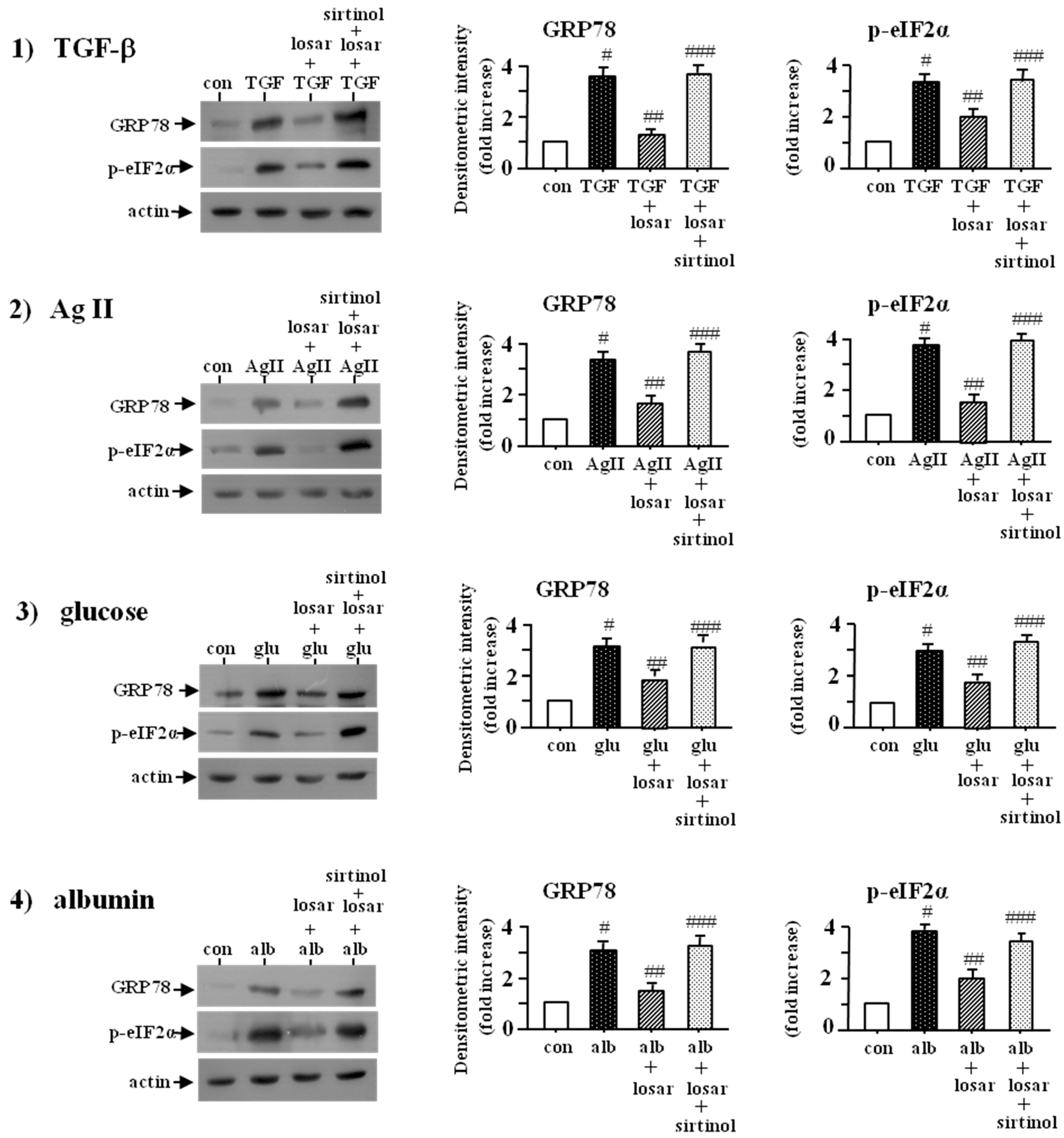
2.7. Losartan also Suppressed the ER Stress Induced by Non-Chemical ER Stress Inducers Such as Tumor Growth Factor-β (TGF-β), Angiotensin II, High Glucose, and Albumin through Induction of SIRT1
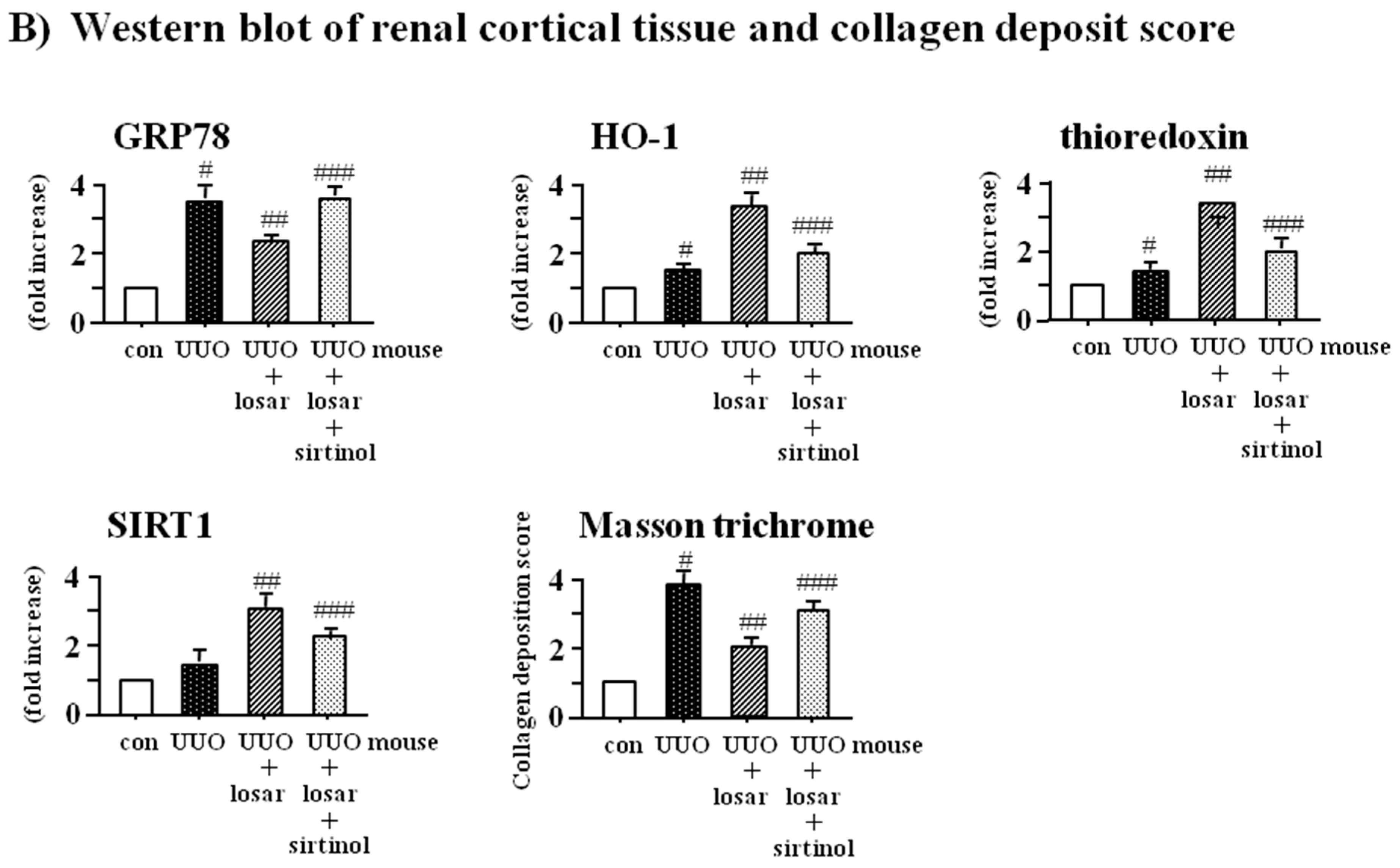
2.8. Treatment with Losartan Reduced the Renal Tubular Expression of GRP78 and Renal Fibrosis through the Up-Regulation of SIRT1 in a Mouse Model of Unilateral Ureteral Obstruction (UUO)
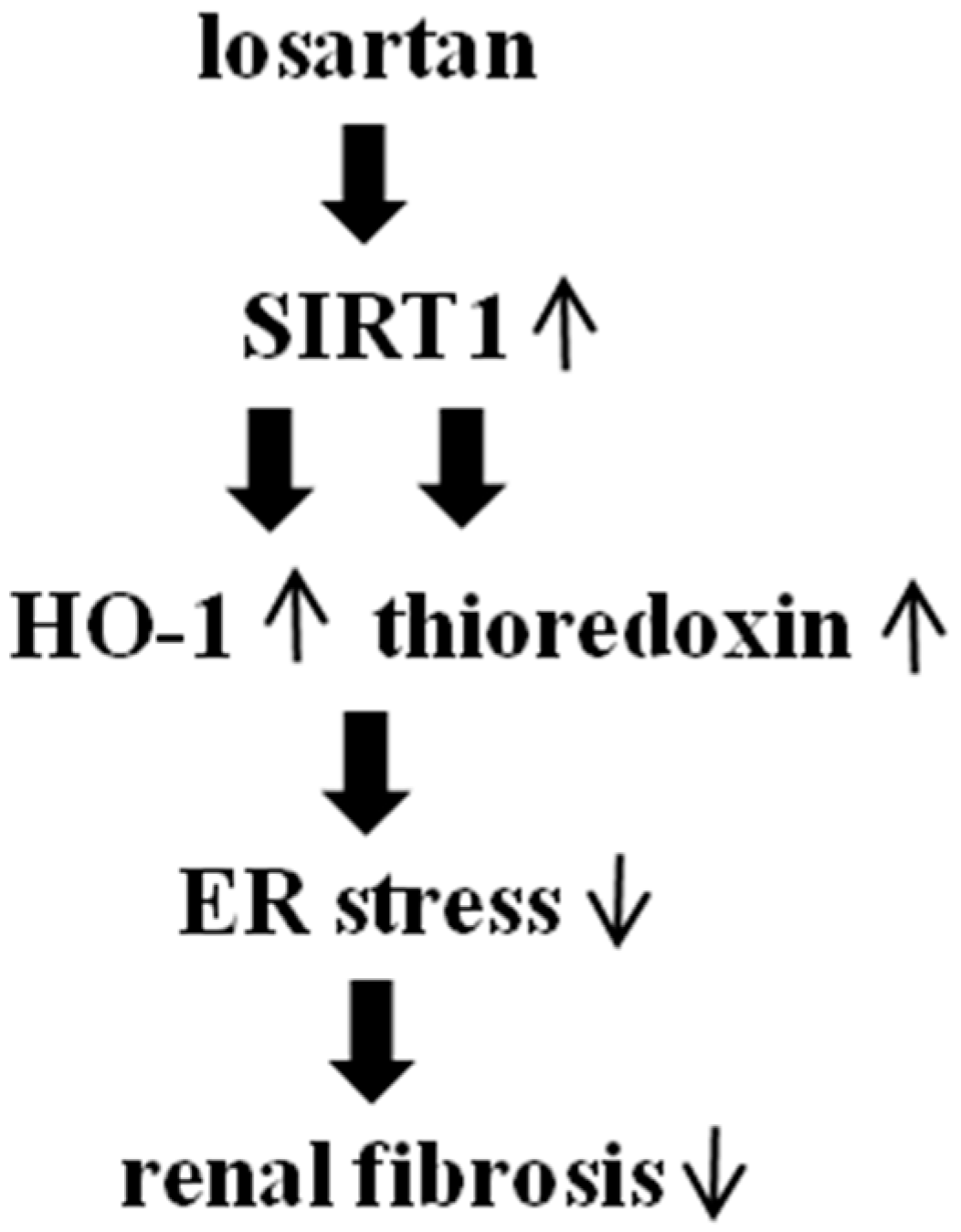
3. Discussion
4. Materials and Method
4.1. Reagent
4.2. Cell Culture and Conditioning
4.3. Western Blot Analysis
4.4. Experimental Mouse Model of Tunicamycin-Induced ER Stress
4.5. Experimental Mouse Model of Unilateral Ureteral Obstruction (UUO)-Induced Progressive Kidney Injury
4.6. Immunohistochemical Staining
4.7. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cybulsky, A.V. Endoplasmic reticulum stress in proteinuric kidney disease. Kidney Int. 2010, 77, 87–193. [Google Scholar] [CrossRef] [PubMed]
- Schröder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat. Res. 2005, 569, 29–63. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Glimcher, L.H. Regulation of innate immunity by signaling pathways emerging from the endoplasmic reticulum. Curr. Opin. Immunol. 2011, 23, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, L.; Tabas, I. Role of endoplasmic reticulum stress in metabolic disease and other disorders. Annu. Rev. Med. 2012, 63, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Tanjore, H.; Lawson, W.E.; Blackwell, T.S. Endoplasmic reticulum stress as a pro-fibrotic stimulus. Biochim. Biophys. Acta. 2013, 1832, 940–947. [Google Scholar] [CrossRef] [PubMed]
- Lenna, S.; Trojanowska, M. The role of endoplasmic reticulum stress and the unfolded protein response in fibrosis. Curr. Opin. Rheumatol. 2012, 24, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Brasier, A.R.; Recinos, A.; Eledrisi, M.S. Vascular inflammation and the rennin angiotensin system. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1257–1266. [Google Scholar] [CrossRef] [PubMed]
- Donnini, S.; Terzuoli, E.; Ziche, M.; Morbidelli, L. Sulfhydryl angiotensin converting enzyme inhibitor promotes endothelial cell survival through nitric-oxide synthase, fibroblast growth factor-2 and telomerase cross-talk. J. Pharmacol. Exp. Ther. 2010, 332, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Sinclair, D.A. Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. 2010, 5, 253–295. [Google Scholar] [CrossRef] [PubMed]
- Kitada, M.; Kume, S.; Koya, D. Role of sirtuins in kidney disease. Curr. Opin. Nephrol. Hypertens. 2014, 23, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.W.; Kim, H.; Baek, C.H.; Lee, R.B.; Yang, W.S.; Lee, S.K. Up-regulation of SIRT1 reduces endoplasmic reticulum stress and renal fibrosis. Nephron 2016, 133, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Pae, H.O.; Zheng, M.; Park, R.; Kim, Y.M.; Chung, H.T. Carbon monoxide induces hemeoxygenase-1 via activation of protein kinase R-like endoplasmic reticulum kinase and inhibits endothelial cell apoptosis triggered by endoplasmic reticulum stress. Circ. Res. 2007, 101, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Yamawaki, H.; Haendeler, J.; Berk, B.C. Thioredoxin: A key regulator of cardiovascular homeostasis. Circ. Res. 2003, 93, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Moon, S.Y.; Kim, J.S.; Baek, C.H.; Kim, M.; Min, J.Y.; Lee, S.K. Activation of AMP activated protein kinase inhibits ER stress and renal fibrosis. Am. J. Physiol. Renal. Physiol. 2015, 308, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Li, E.; Park, S. Insulin-like growth factor-1 inhibits 6-hydroxydopamine mediated endoplasmic reticulum stress-induced apoptosis via regulation of hemeoxygenase-1 and Nrf2 expression in PC12 cells. Int. J. Neurosci. 2012, 122, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Q.; Chen, X.; Zhang, C.; Tao, L.; Zhang, Z.H.; Liu, X.Q.; Xu, Y.B.; Wang, H.; Li, J.; Xu, D.X. Phenylbutyric acid protects against carbon tetrachloride-induced hepatic fibrogenesis in mice. Toxicol. Appl. Pharmacol. 2013, 266, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Ayala, P.; Montenegro, J.; Vivar, R.; Letelier, A.; Urroz, P.A.; Copaja, M.; Pivet, D.; Humeres, C.; Troncoso, R.; Vicencio, J.M.; et al. Attenuation of endoplasmic reticulum stress using the chemical chaperone 4-phenylbutyric acid prevents cardiac fibrosis induced by isoproterenol. Exp. Mol. Pathol. 2012, 92, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.K.; Hsu, S.P.; Wu, C.T.; Huang, J.W.; Cheng, H.T.; Chang, Y.W.; Hung, K.Y.; Wu, K.D.; Liu, S.H. Endoplasmic reticulum stress implicated in the development of renal fibrosis. Mol. Med. 2011, 17, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Kaufman, R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Goodall, J.C.; Wu, C.; Zhang, Y.; McNeill, L.; Ellis, L.; Saudek, V.; Gaston, J.S. Endoplasmic reticulum stress-induced transcription factor, CHOP, is crucial for dendritic cell IL-23 expression. Proc. Natl. Acad. Sci. USA 2010, 107, 17698–17703. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Turner, M.J.; de Lay, M.L.; Klenk, E.I.; Sowders, D.P.; Colbert, R.A. Endoplasmic reticulum stress and the unfolded protein response are linked to synergistic IFN-β induction via X-box binding protein 1. Eur. J. Immunol. 2008, 38, 1194–1203. [Google Scholar] [CrossRef] [PubMed]
- Lenna, S.; Townsend, D.M.; Tan, F.K.; Kapanadze, B.; Markiewicz, M.; Trojanowska, M.; Scorza, R. HLA-B35 upregulates endothelin-1α and downregulates endothelial nitric oxide synthase via endoplasmic reticulum stress response in endothelial cells. J. Immunol. 2010, 184, 4654–4661. [Google Scholar] [CrossRef] [PubMed]
- Menu, P.; Mayor, A.; Zhou, R.; Tardivel, A.; Ichijo, H.; Mori, K.; Tschopp, J. ER stress activates the NLRP3 inflammasome via an UPR-independent pathway. Cell Death Dis. 2012, 3, e261. [Google Scholar] [CrossRef] [PubMed]
- Kassan, M.; Galán, M.; Partyka, M.; Saifudeen, Z.; Henrion, D.; Trebak, M.; Matrougui, K. Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1652–1661. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, S.; Thandavarayan, R.A.; Palanivandi, S.S.; Giridharan, V.V.; Arozal, W.; Sari, F.R.; Soetikno, V.; Harima, M.; Suzuki, K.; Kodama, M.; et al. Candesartan cilexetil protects from cardiac myosin induced cardiotoxicity via reduction of endoplasmic reticulum stress and apoptosis in rats: Involvement of ACE2-Ang (1–7)-mas axis. Toxicology 2012, 291, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Lakshmanan, A.P.; Thandavarayan, R.A.; Palanivandi, S.S.; Sari, F.R.; Meilei, H.; Giridharan, V.V.; Soetikno, V.; Suzuki, K.; Kodama, M.; Watanabe, K. Modulation of AT-1R/CHOP-JNK-Caspase12 pathway by olmesartan treatment attenuates ER stress-induced renal apoptosis in streptozotocin-induced diabetic mice. Eur. J. Pharm. Sci. 2011, 44, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Shiota, A.; Shimabukuro, M.; Fukuda, D.; Soeki, T.; Sato, H.; Uematsu, E.; Hirata, Y.; Kurobe, H.; Maeda, N.; Sakaue, H.; et al. Telmisartan ameliorates insulin sensitivity by activating the AMPK/SIRT1 pathway in skeletal muscle of obese db/db mice. Cardiovasc. Diabetol. 2012, 11, 139–417. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Qian, J.; Castillo, A.C.; Perez-Polo, J.R.; Birnbaum, Y. Aliskiren and valsartan reduce myocardial AT1 receptor expression and limit myocardial infarct size in diabetic mice. Cardiovasc. Drugs Ther. 2011, 25, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.S.; Jia, J.J.; Kwon, Y.; Wang, S.D.; Bai, J. The role of thioredoxin-1 in suppression of endoplasmic reticulum stress in Parkinson disease. Free Radic. Biol. Med. 2014, 67, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Olmos, Y.; Sánchez-Gómez, F.J.; Wild, B.; García-Quintans, N.; Cabezudo, S.; Lamas, S.; Monsalve, M. SIRT1 regulation of antioxidant genes is dependent on the formation of a FoxO3a/PGC-1α complex. Antioxid. Redox Signal. 2013, 19, 1507–1521. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Epithelial to mesenchymal transition in renal fibrogenesis: Pathologic significance, molecular mechanism, and therapeutic intervention. J. Am. Soc. Nephrol. 2004, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 2002, 110, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.Y.; Kim, H.S.; Nho, K.W.; Jang, Y.J.; Lee, S.K. Endoplasmic reticulum stress induces epithelial-mesenchymal transition through autophagy via activation of c-Src kinase. Nephron. Exp. Nephrol. 2014, 126, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Xue, H.; Yuan, P.; Ni, J.; Yu, C.; Huang, Y.; Lu, L.M. Angiotensin AT1 receptor activation mediates high glucose-induced epithelial-mesenchymal transition in renal proximal tubular cells. Clin. Exp. Pharmacol. Physiol. 2010, 37, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Wang, Y.; Jin, Z.; Wang, H.; Cheng, W.; Zhou, H.; Yin, P.; Peng, W. Losartan alleviates renal fibrosis by down-regulating HIF-1α and up-regulating MMP-9/TIMP-1 in rats with 5/6 nephrectomy. Ren. Fail. 2012, 34, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Li, D.; Zhang, B. Losartan attenuates renal interstitial fibrosis and tubular cell apoptosis in a rat model of obstructive nephropathy. Mol. Med. Rep. 2014, 10, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jia, Z.; Liu, S.; Downton, M.; Liu, G.; Du, Y.; Yang, T. Combined losartan and nitro-oleic acid remarkably improves diabetic nephropathy in mice. Am. J. Physiol. Ren. Physiol. 2013, 305, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- Barrilli, A.; Molinas, S.; Petrini, G.; Menacho, M.; Elías, M.M. Losartan reverses fibrotic changes in cortical renal tissue induced by ischemia or ischemia-reperfusion without changes in renal function. Mol. Cell. Biochem. 2004, 260, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Burdmann, E.A.; Andoh, T.F.; Nast, C.C.; Evan, A.; Connors, B.A.; Coffman, T.M.; Lindsley, J.; Bennett, W.M. Prevention of experimental cyclosporine-induced interstitial fibrosis by losartan and enalapril. Am. J. Physiol. 1995, 269, 491–499. [Google Scholar]
- Kitamura, M. Endoplasmic reticulum stress and unfolded protein response in renal pathophysiology: Janus faces. Am. J. Physiol. Ren. Physiol. 2008, 295, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.J.; Johnson, G.; Kirk, J.; Fuerstenberg, S.M.; Zager, R.A.; Torok-Storb, B. HK-2: An immortalized proximal tubule epithelial cell line from normal adult human kidney. Kidney Int. 1994, 45, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Hodeify, R.; Megyesi, J.; Tarcsafalvi, A.; Mustafa, H.I.; HtiLarSeng, N.S.; Price, P.M. Gender difference control the susceptibility to ER stress-induced acute kidney injury. Am. J. Physiol. Renal Physiol. 2013, 304, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, J.L., Jr.; Sundell, C.L.; Wang, S.; Kopp, J.B.; Montero, A.; Martínez-Maldonado, M. Role of angiotensin II in the expression and regulation of transforming growth factor β in obstructive nephropathy. Kidney Int. 1995, 48, 1233–1246. [Google Scholar] [CrossRef] [PubMed]










© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.; Baek, C.H.; Lee, R.B.; Chang, J.W.; Yang, W.S.; Lee, S.K. Anti-Fibrotic Effect of Losartan, an Angiotensin II Receptor Blocker, Is Mediated through Inhibition of ER Stress via Up-Regulation of SIRT1, Followed by Induction of HO-1 and Thioredoxin. Int. J. Mol. Sci. 2017, 18, 305. https://doi.org/10.3390/ijms18020305
Kim H, Baek CH, Lee RB, Chang JW, Yang WS, Lee SK. Anti-Fibrotic Effect of Losartan, an Angiotensin II Receptor Blocker, Is Mediated through Inhibition of ER Stress via Up-Regulation of SIRT1, Followed by Induction of HO-1 and Thioredoxin. International Journal of Molecular Sciences. 2017; 18(2):305. https://doi.org/10.3390/ijms18020305
Chicago/Turabian StyleKim, Hyosang, Chung Hee Baek, Raymond Bok Lee, Jai Won Chang, Won Seok Yang, and Sang Koo Lee. 2017. "Anti-Fibrotic Effect of Losartan, an Angiotensin II Receptor Blocker, Is Mediated through Inhibition of ER Stress via Up-Regulation of SIRT1, Followed by Induction of HO-1 and Thioredoxin" International Journal of Molecular Sciences 18, no. 2: 305. https://doi.org/10.3390/ijms18020305